Introduction
In Taiwan, 33.5 per 100,000 people die from liver
cancer each year according to the Department of Health, Executive
Yuan in 2010s (http://www.doh.gov.tw/EN2006/indexEN.aspx/).
Hepatocellular carcinoma (HCC) is one of the most common primary
cancers of the liver in the world and in Taiwan, and accounts for
more than 90% of all primary liver tumors (1). HCC is the sixth most common cancer
and the third most common cause of death from cancer worldwide
(2,3). HCC is a high degree of malignancy but
poor prognosis disease, and most patients with HCC are not curable
(4). Chemotherapy is one of the
treatment options in HCC, but these outcomes are not fully
satisfactory (5). Multidrug
resistance gene is overexpressed in HCC, which results in
un-sensitizing chemotherapeutic agents (6–8).
Therefore, discovery to more effective chemotherapeutic agents for
HCC is urgently needed.
The most effective strategy for curing HCC is to
induce cell cycle arrest and cell death (9,10).
The promotion of cell cycle progression at G2/M phase
has been intensively investigated. Cyclin-dependent protein kinase
1 (CDK1)/cyclin B complex plays a major role in the regulation of
the cell cycle in G2/M phase. When the cells progress
from S phase into G2/M phase, the CDK1/cyclin B1 complex
becomes active, suggesting that CDK1 has undergone an activating
phosphorylation (on Thr161), and the inhibitory phosphorylation (on
Thr14 and Tyr15) removed by active Cdc25c phosphatase (11,12).
The Weel kinase is regulating G2/M phase transition by
phosphorylation of CDK1 at Tyr15. In addition, Chk1 is the
essential kinase in the G2/M checkpoint by
phosphorylating Cdc25c in response to cellular damaging and
antimitotic agents (13–15).
Three major morphologically processes lead to cell
death, apoptosis, necrosis and autophagic cell death (16,17).
Autophagic cell death, or cellular self-digestion, occurs in
multi-cellular organisms and plays an important role in normal
physiology in animals (18). When
the cells undergo nutrient starvation, cellular damage, pathogen
infection, aging and degenerative processes, autophagic cell death
is required for the promotion of cellular survival (19,20).
Autophagy (self-eating), causes specific morphological and
biochemical modification, and is considered as programmed cell
death type II (apoptosis, programmed cell death type I), which
occurs in some situations and then induces cell death (21,22).
The cytoplasm and double smooth membrane (a phagophore) of various
organelles such as mitochondria, endoplasmic reticulum (ER) and
peroxisomes are sequestered by a membrane to form an autophagosomes
and then the autophagosome fuses with the lysosome, forming
autophagolysosome, finally resulting in degradation of the captured
proteins/organelles by lysosomal enzymes (18,23–25).
Many studies have demonstrated that a group of autophagy-related
proteins (Atg) is involved in autophagic cell death. The membrane
nucleation is mediated by a class III phosphatidylinositol 3-kinase
and Beclin 1. The production of autophagosome is necessary for the
recruitment of Atg12-Atg7-Atg5 complex and microtubule-associated
protein 1 light chain 3 (LC3) (26–29).
There are two major forms of LC3, type I is cytosolic and type II
is membrane-bound. When cells undergo autophagic cell death,
LC3-II, an autophagosomal marker, increases from the conversion of
LC3-I (30). Promotion of
autophagic cell death from cancer cells is one of the best
strategies in chemotherapy (31–33).
Bufalin, a digoxin-like chemical reagent, is one of
the major Chinese tradition medicine Chan-Su components extracted
from dried toad venom from the skin glands of Bufo bufo
gargarizan or Bufo melanostictus. It has been
demonstrated that bufalin exhibits significant anticancer
activities against many human tumor cells in vitro and in
vivo. The anticancer activities by bufalin are involved in the
induction of cell differentiation, cell cycle arrest, apoptosis and
inhibition of cell metastasis. Xie et al demonstrated that
bufalin induces autophagic cell death through reactive oxygen
species (ROS) generation and JNK activation in HT29 human colon
cancer cells (34). The anticancer
activities of bufalin have been reported, however, no comprehensive
studies have been reported on the effects of bufalin on human
hepatocellular carcinoma cells. The goal of this study is to
explore whether the antitumor activity of bufalin mediates through
the direct cytotoxic effect and to understand the molecular
mechanisms in human hepatocellular carcinoma SK-HEP-1 cells. The
present study is focused on the cell cycle arrest and
autophagy-induced by bufalin in the SK-HEP-1 cells. Our results
demonstrate that bufalin inhibits cells viability, induces
autophagic cell death, and simultaneously causes cell cycle arrest
in G2/M phase through the AKT/mTOR signaling pathway in
SK-HEP-1 cells.
Materials and methods
Chemicals and reagents
Acridine orange (AO), agarose, bafilomycin, bufalin,
dimethyl sulfoxide (DMSO), 3-methyladenine (3-MA), propidium iodide
(PI), RNase A,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide
(MTT), proteinase K, and Triton X-100 were obtained from Sigma
Chemical Co. (St. Louis, MO, USA). Fetal bovine serum (FBS),
L-glutamine, LC3B antibody kit for autophagy,
penicillin/streptomycin, RPMI-1640 medium, trypan blue solution and
Trypsin-EDTA were purchased from Invitrogen/Life Technologies
(Carlsbad, CA, USA). AKT kinase assay Kit was purchased from Cell
Signaling Technology (Danvers, MA, USA). Caspase-3 activity assay
kit was purchased from R&D Systems Inc. (Minneapolis, MN, USA).
CDK1 kinase activity kit was purchased from Medical &
Biological Laboratories International (Nagoya, Japan). Tdt-mediated
deoxyuridine triphosphate nick end labeling (TUNEL) assay kit was
purchased from Roche Diagnostics (GmBH, Mannheim, Germany). Primary
antibodies (anti-cyclin A, anti-cyclin B, anti-CDK1, anti-Cdc25c,
anti-Chk1, anti-Weel, anti-PARP, anti-mTOR and anti-GAPDH), and
second antibodies for western blot analysis were obtained from
Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). The primary
antibodies [anti-phospho-CDK1 (Thr161), phospho-Cdc25c (Ser198),
anti-phospho-AKT (Thr308), anti-phospho-AKT (Ser473),
anti-phospho-mTOR (Ser2481) and anti-caspase-3, anti-LC3-II,
anti-Beclin-1, anti-Atg 5, anti-Atg 7 and anti-Atg 12] were
obtained from Cell Signaling Technology. Antibody against β-actin
was purchased from Sigma Chemical Co. All peroxidase-conjugated
secondary antibodies were purchased from Santa Cruz Biotechnology
Inc. The enhanced chemiluminescence (ECL) detection kit was
purchased from Pierce Chemical (Rockford, IL, USA).
Cell culture
The human hepatocellular carcinoma cell line
(SK-HEP-1) was obtained from the Food Industry Research and
Development Institute (Hsinchu, Taiwan). Cells were cultured in
75-cm2 tissue culture flasks at 37°C under a humidified
5% CO2 atmosphere in RPMI-1640 medium containing 10%
FBS, 2 mM of L-glutamine, 100 U/ml of penicillin and 100 μg/ml of
streptomycin.
Cell viability and morphology
Cell viability was evaluated using the MTT assay
(35). SK-HEP-1 cells
(1×104 cells) in 96-well plates were incubated with 0,
25, 50, 100 and 200 nM of bufalin or 0.1% DMSO as a vehicle control
for 24 h. For incubation with the inhibitors, cells were pretreated
with 3-MA (10 mM) or bafilomycin (10 nM) for 1 h, followed by
treatment with or without bufalin (100 nM). After washing the
cells, RPMI-1640 medium containing MTT (0.5 mg/ml) of was added to
each well. The cells were incubated for 4 h at 37°C the supernatant
was removed. The formed formazan crystals in viable cells were
dissolved with isopropanol in 0.04 N HCl. The absorbance of each
well was measured at 570 nm with ELISA reader with a reference
wavelength of 620 nm. All experiments were performed in triplicate
and the cell viability was expressed as percentage of the control
as previously described. Cell morphological examination of
autophagic vacuoles was determined utilizing a phase-contrast
microscope.
Assessment for cell death
Cell death was evaluated using a trypan blue assay
(36,37). SK-HEP-1 cells (2.5×105
cells) in 24-well plates were incubated with 0, 25, 50, 100 and 200
nM of bufalin for 24 h. At the end of incubation, cells were
stained in the 0.25% trypan blue solution and then determined cell
number by Countess Automated Cell Counter (Invitrogen/Life
Technologies) as previously described.
Cell cycle progression
The 2.5×105 cells of SK-HEP-1 cells in
24-well plate were exposed to bufalin (50 and 100 nM) for 24 h. At
the end of incubation, cells were then collected, fixed in 70%
ethanol overnight, then washed in PBS once, and re-suspended in 500
μl of Na2HPO4 (192 mM), citric acid (4 mM) pH
7.8 at 25°C for 30 min. The cells were stained with 0.5 ml of PBS
containing RNase (1 mg/ml), PI (10 μg/ml) for 30 min in the dark,
and then analyzed by flow cytometry as previously described
(38,39).
CDK1 kinase activity
CDK 1 kinase activity was performed according to the
manufacturer’s protocols (CycLex® Cdc2-Cyclin B Kinase
Assay Kit; MBL International Corporation, Japan) (40). SK-HEP-1 cells were seeded into 75-T
flasks (1×107/each). Bufalin (100 nM) were added and
incubated for 0, 6, 12 and 24 h. At the end of incubation, cells
were harvested, washed twice with ice-cold PBS, and then
re-suspended the cell pellet with 500 μl of extraction buffer (20
mM Tris-HCl, pH 8.5, 150 mM NaCl, 0.2% NP-40, 1 mM DTT, 1 mM EDTA,
1 mM EGTA, 0.2 mM PMSF, 1 μg/ml pepstatin, 0.5 μg/ml leupeptin, 5
mM β-glycerophosphate, 5 mM NaF, 1 mM Na3VO4,
5 mM β-mercaptoethanol). Cell extracts were diluted in a 1:5 ratio
with Q-buffer (20 mM Tris-HCl, pH 8.5, 0.2 mM EDTA, 1 mM EGTA, 1
μg/ml pepstatin, 0.5 μg/ml leupeptin, 0.2 mM
Na3VO4, 5 mM β-mercaptoethanol). The CDK1
activity was measured absorbance using ELISA reader at OD450. All
results were performed in three independent experiments.
DNA gel electrophoresis
SK-HEP-1 cells were seeded into 75-T flasks
(1×107/each). Bufalin (0, 50 and 100 nM) were added and
incubated for 24 h. At the end of incubation, cells were harvested
and washed twice with ice-cold PBS. The cell pellets were
re-suspended in lysis buffer (20 mM Tris-HCl, pH 8.0, 10 mM EDTA,
0.2% Triton X-100). Cell lysates were treated with 0.1 mg/ml
proteinase K at 50°C for 12 h, followed by 50 μg/ml RNase A at 37°C
for 30 min. After precipitation, the DNA was subjected to
electrophoresis in a 1.5% agarose gel (Sigma-Aldrich Corp.). DNA
fragmentation on agarose gel was stained with 1 μg/ml ethidium
bromide (EtBr, Invitrogen/Life Technologies) in 0.5X TBE buffer and
examined in photographs taken under UV light as previously
described (13,41).
TUNEL staining
TUNEL staining was performed according to the
manufacturer’s protocol (in situ cell death detection kit;
Roche Diagnostics) (13,41). The 2.5×105 cells of
SK-HEP-1 cells seeded in each well of 24-well plates were exposed
to bufalin (25, 50 and 100 nM) or paclitaxel (100 nM) for 24 h. At
the end of incubation, cells were collected then fixed in 70%
ethanol then washed twice with ice-cold PBS, and incubated in the
dark for 30 min at 37°C in 100 μl of TdT-containing solution.
Following TUNEL staining, all samples were washed once. TUNEL
positive cells were analyzed by flow cytometry using a FACSCalibur
(Becton Dickinson). The median fluorescence intensity was
quantities by CellQuest software. TUNEL assays were performed in
triplicate on three independent experiments.
Caspase-3 activity assay
SK-HEP-1 cells (1×107 cells/75-T flask)
were treated with bufalin (25, 50 and 100 nM) or paclitaxel (100
nM) for 24 h. At the end of incubation, cells were harvested and
re-suspended in a lysis buffer [50 mM Tris-HCl (pH 7.4), 1 mM EDTA,
10 mM EGTA, 10 mM digitonin and 2 mM DTT]. Cell lysates (50 μg
protein) were incubated with caspase-3 specific substrates
(Ac-DEVD-pNA) (R&D Systems Inc.) for 1 h at 37°C. The caspase-3
activity was determined by measuring OD405 of the
released pNA (42,43).
Transmission electron microscopy
SK-HEP-1 cells (1×106 cells/6-well) were
treated with bufalin (100 nM) for 24 h. At the end of incubation,
cells washing three times with PBS, cells were fixed for 30 min at
room temperature in 2% paraformaldehyde and 2.5% glutaraldehyde in
PBS buffer. The cells were rinsed twice in the same buffer and
subsequently post fixed in 1% osmium tetraoxide. After rinsing
followed by dehydration in graded alcohol series, the cells were
embedded in LR white resin and polymerized at 70°C overnight.
Ultrathin sections were then cut with a diamond knife and loaded
onto TEM grids. The sections were examined by a Philips CM10
electron microscope at accelerating voltage of 120 kV and
micrographs were taken (44).
Detection of acidic vesicular organelles
(AVO) with acridine orange (AO)
SK-HEP-1 cells (1×106 cells/6-well) were
treated with bufalin (100 nM) for 24 h. At the end of incubation,
cells were harvested. To detect and quantify acidic vesicular
organelles (AVO), cells were stained with acridine orange (AO). The
number of acridine orange positive cells was analyzed by flow
cytometry using a FACSCalibur (Becton Dickinson). Cell morphology
was examined using phase-contrast and fluorescence microscopy
(Nikon, Melville, NY, USA). All results were performed in three
independent experiments (45).
Immunofluorescence analysis
LC-3 staining was performed according to the
manufacturer’s protocol (LC3 antibody kit for autophagy;
Invitrogen/Life Technologies) (46,47).
SK-HEP-1 cells (1×106 cells/6-well) in 6-well flask were
treated with bufalin (100 nM) for 24 h. At the end of incubation,
cells were harvested then fixed with 3.7% formaldehyde in PBS for
15 min. Cells were washed three times with PBS, added 0.2% Triton
X-100 in PBS to the cells, and incubated for 15 min at room
temperature. Permeabilized cells were incubated with an anti-LC3
antibody (1:150) at 4°C overnight, and followed by incubation with
an Alexa Fluor 488-conjugated secondary antibody (1:200;
Invitrogen) for 1 h. The images were taken under a fluorescence
microscopy (Nikon). The number of LC-3 positive cells was analyzed
by flow cytometry using a FACSCalibur (Becton Dickinson). All
results were performed in three independent experiments (48).
Western blot analysis
SK-HEP-1 cells (1×107 cells/75-T flask)
were treated with bufalin (100 nM) for 0, 2, 4, 6, 8, 12, 18 and 24
h. At the end of incubation, cytosolic or total proteins were
prepared and determined as previously described. Protein lysates
were sonicated and the supernatants were boiled in SDS sample
buffer for 5 min. The protein concentration was measured by using a
BCA assay kit (Pierce Chemical). Equal amounts of cell lysates were
run on 10 to 12% SDS-polyacrylamide gel electrophoresis and
electro-transferred to a nitrocellulose membrane by using the iBot
Dry Blotting System (Invitrogen/Life Technologies). The transferred
membranes were blocked for 1 h in 5% nonfat dry milk in Tris
buffered saline/Tween-20 and incubated with primary antibodies at
4°C overnight. Membranes were washed three times with Tris-buffered
saline/Tween-20 for 10 min and incubated with secondary
HRP-conjugated antibody. The blots were developed by using an ECL
kit and Kodak Bio-MAX MR film (Eastman Kodak, Rochester, NY, USA)
(35).
In vitro AKT kinase assay
In vitro AKT kinase assay was performed
following the protocol of the manufacturer’s (Cell Signaling
Technology) (46,49). In brief, SK-HEP-1 cells
(1×107 cells/75-T flask) were treated with bufalin (100
nM) for 0, 2, 4, 6 and 8 h. At the end of incubation, cells were
lysed in ice-cold lysis buffer provided by the kit. The 200 μg of
protein from each time point treatment was immuno-precipitated with
2 μg of anti-AKT antibody overnight. Immuno-precipitates were
extensively washed, and then incubated with 1 μg of glycogen
synthase kinase-3 α/β (GSK-3 α/β) fusion protein substrate in 50 μl
of kinase buffer for 30 min at 30°C. Reactions were stopped by SDS
loading buffer and samples were separated on 12% SDS-PAGE. The
phospho-GSK-3 α/β (Ser219) was detected by immunoblotting.
Statistical analysis
All the statistical results were expressed as the
mean ± SEM of triplicate samples. Statistical analyses of data were
done using one-way ANOVA followed by Student’s t-test and
*P<0.05 were considered significant.
Results
Bufalin inhibits cell proliferation,
promotes G2/M phase arrest in SK-HEP-1 cells
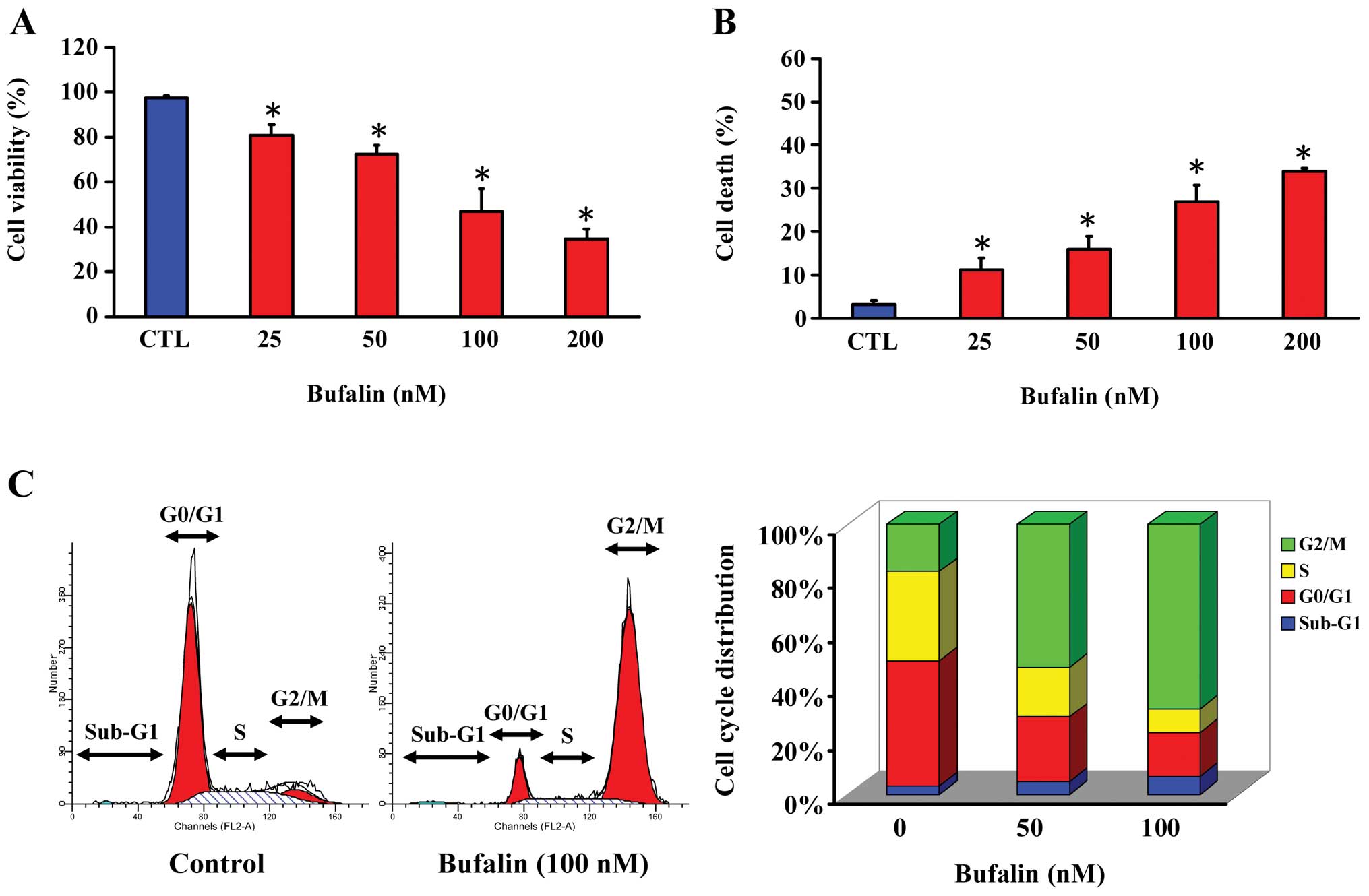
SK-HEP-1 cells were treated with bufalin (0, 25, 50,
100 and 200 nM) for 24 h. Bufalin reduced cell viability of
SK-HEP-1 cells in a concentration-dependent manner as measured by
the MTT assay (Fig. 1A). The half
maximal (50%) inhibitory concentration (IC50) for a 24-h
treatment of bufalin in SK-HEP-1 cell was 110.33±5.32 nM. Fig. 1B shows that bufalin increased cell
death in a concentration-dependent manner by the trypan blue
exclusion assay. We then examined cell cycle distribution in
SK-HEP-1 cells exposed to 100 nM of bufalin for 24 h. These results
showed G2/M accumulation after SK-HEP-1 cells were
treated with 100 nM of bufalin (Fig.
1C). Interestingly, SK-HEP-1 cells exposed to 50 and 100 nM of
bufalin have undergone cell death, but did not detect significant
sub-G1 DNA content, a typical apoptosis. Our results
suggested that bufalin effectively caused G2/M arrest
and then induced cell death.
Effects of bufalin on G2/M
phase-associated protein levels in SK-HEP-1 cells
We investigated the G2/M phase specific
protein expression levels by western blot analysis. As shown in
Fig. 2A, bufalin (100 nM) caused
an increase in the protein level of Chk1 and Wee1, and a decrease
in the protein levels of Cyclin A, Cyclin B, CDK1, phospho-CDK1
(Thr161), Cdc25c, phospho-Cdc25c (Ser198) for 6, 12 and 24 h
treatment in SK-HEP-1 cells. We examined the CDK1 activity in
bufalin-treated SK-HEP-1 cells. Fig.
2B depicted that bufalin (100 nM) caused a significant decrease
in CDK1 activity for 0, 6, 12 and 24-h treatment in SK-HEP-1 cells.
Our data indicated that bufalin-treated SK-HEP-1 cells
downregulated CDK1 activity and caused G2/M phase
arrest.
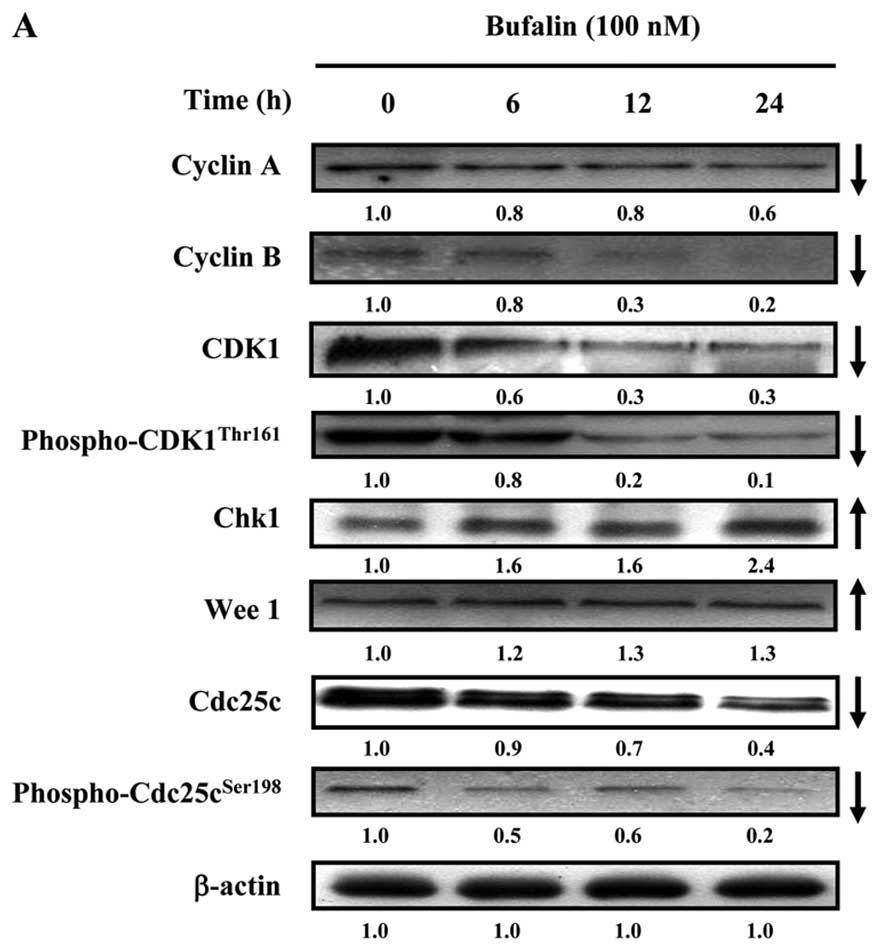 | Figure 2G2/M phase-associated
protein levels and CDK1 activity in bufalin treated SK-HEP-1 cells.
(A) Cells were exposed to bufalin (100 nM) and then incubated for
0, 6, 12 and 24 h. The protein levels of Cyclin A, Cyclin B, CDK1,
phospho-CDK1 (Thr161), Chk1, Wee1, Cdc25c, phospho-Cdc25c (Ser198)
and β-actin in bufalin-treated SK-HEP-1cells were determined by
western blot analysis. (B) CDK1 activity was examined by an in
vitro kinase activity assay. Data are presented as the mean ±
SEM of three independent experiments. *P<0.05,
significantly different compared with control treatment. |
Bufalin induces caspase-independent cell
death in SK-HEP-1 cells
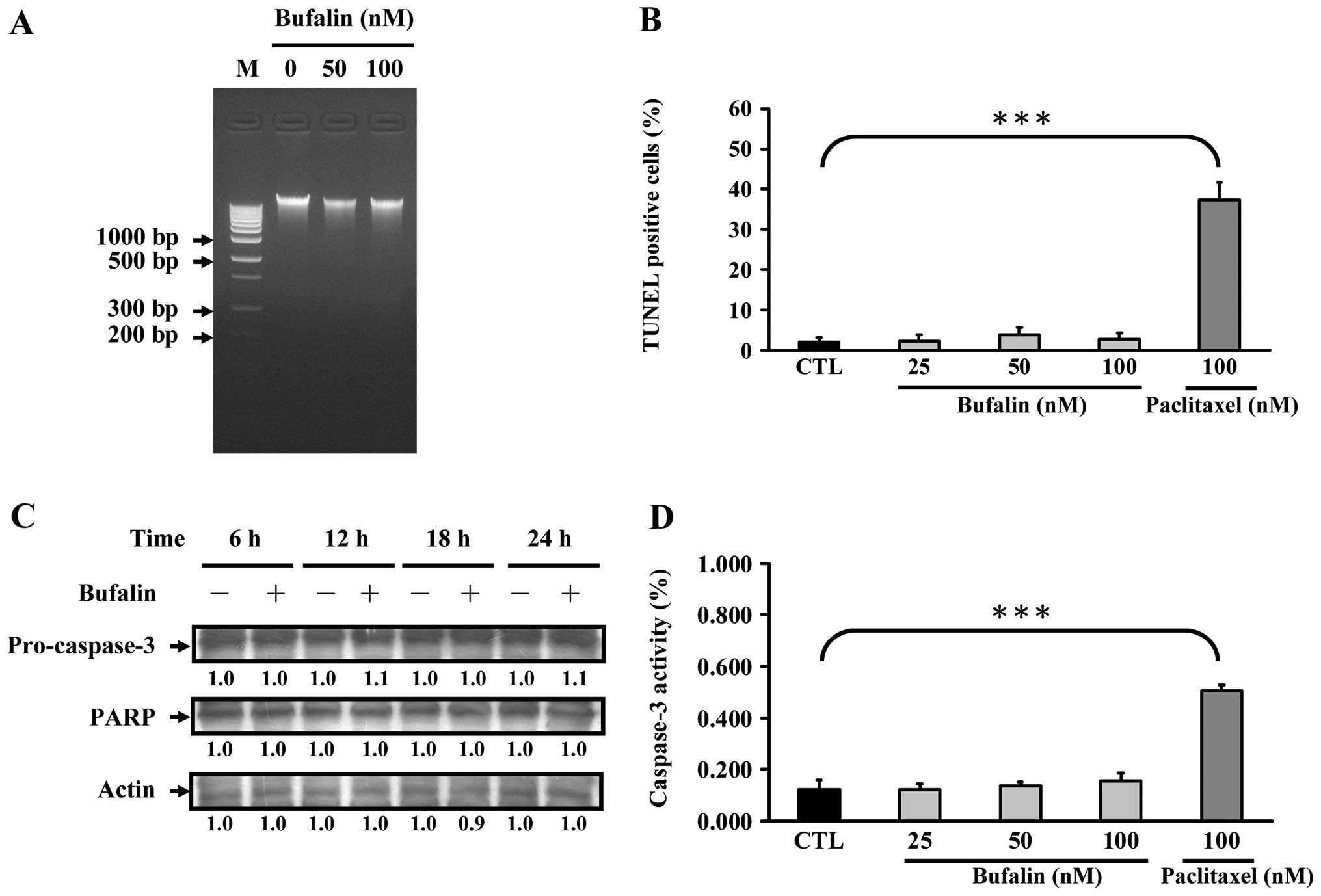
To examine whether bufalin induced apoptosis in
SK-HEP-1 cells, cells were treated with bufalin (0, 50 and 100 nM)
for 24 h, and analyzed DNA fragmentation by DNA gel
electrophoresis. Fig. 3A showed
that no DNA fragmentation was observed in SK-HEP-1 cells treated
with bufalin for 24 h. This result suggested that bufalin did not
induce apoptosis in SK-HEP-1 cells. Similar result was obtained
from TUNEL staining (Fig. 3B) when
SK-HEP-1 cells were treated with bufalin (0, 50 and 100 nM) or
paclitaxel (100 nM; an apoptotic agent) for 24 h. Fig. 3B showed that the percentage of
TUNEL positive cells in bufalin-treated SK-HEP-1 cells was less
than 5% compared with the paclitaxel-treated cells (37.26±4.34%).
To investigate whether bufalin-induced cell death is mediated
through caspase-3 activation, SK-HEP-1 cells were treated with
bufalin (0, 50 and 100 nM) or paclitaxel (100 nM; an apoptotic
reagent) for 24 h and then analyzed the protein levels of cleaved
caspase-3 and poly (ADP-ribose) polymerase (PARP), downstream
target of caspase-3. As shown in Fig.
3C, the protein levels of cleaved caspase-3 or cleaved PARP
were not increased in bufalin-treated SK-HEP-1 cells. In addition,
the caspase-3 activity showed no change in bufalin-treated SK-HEP-1
cells. Our results indicated that bufalin-induced cell death did
not proceed through apoptosis in SK-HEP-1 cells.
Bufalin induces autophagic cell death in
SK-HEP-1 cells
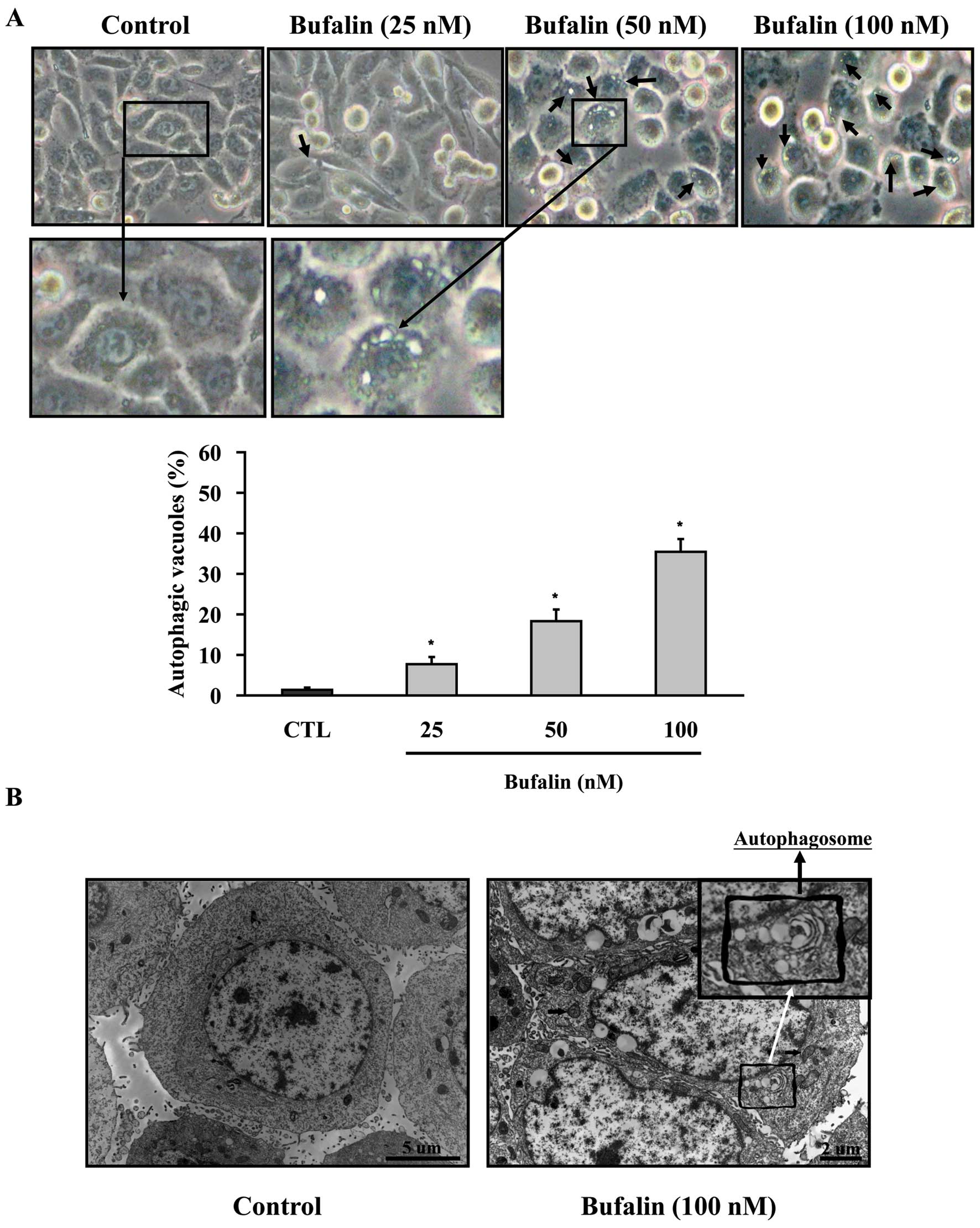
No apoptotic feature was detected in SK-HEP-1 cells.
Next, we examine whether autophagic cell death is involved in
bufalin-induced cell death. SK-HEP-1 cells were treated with
bufalin (0, 25, 50 and 100 nM) for 24 h and the formation of
autophagic vacuoles was examined by the phase microscopy. As shown
in Fig. 4A, bufalin induced the
formation of autophagic vacuoles in SK-HEP-1 cells in
concentration-dependent manner. We further confirmed the formation
of autophagosome vesicles in bufalin-treated SK-HEP-1 cells using
transmission electron microscopic (TEM) analysis (Fig. 4B). Double- or multi-membrane
structure characteristics of autophagosomes and autolysosomes were
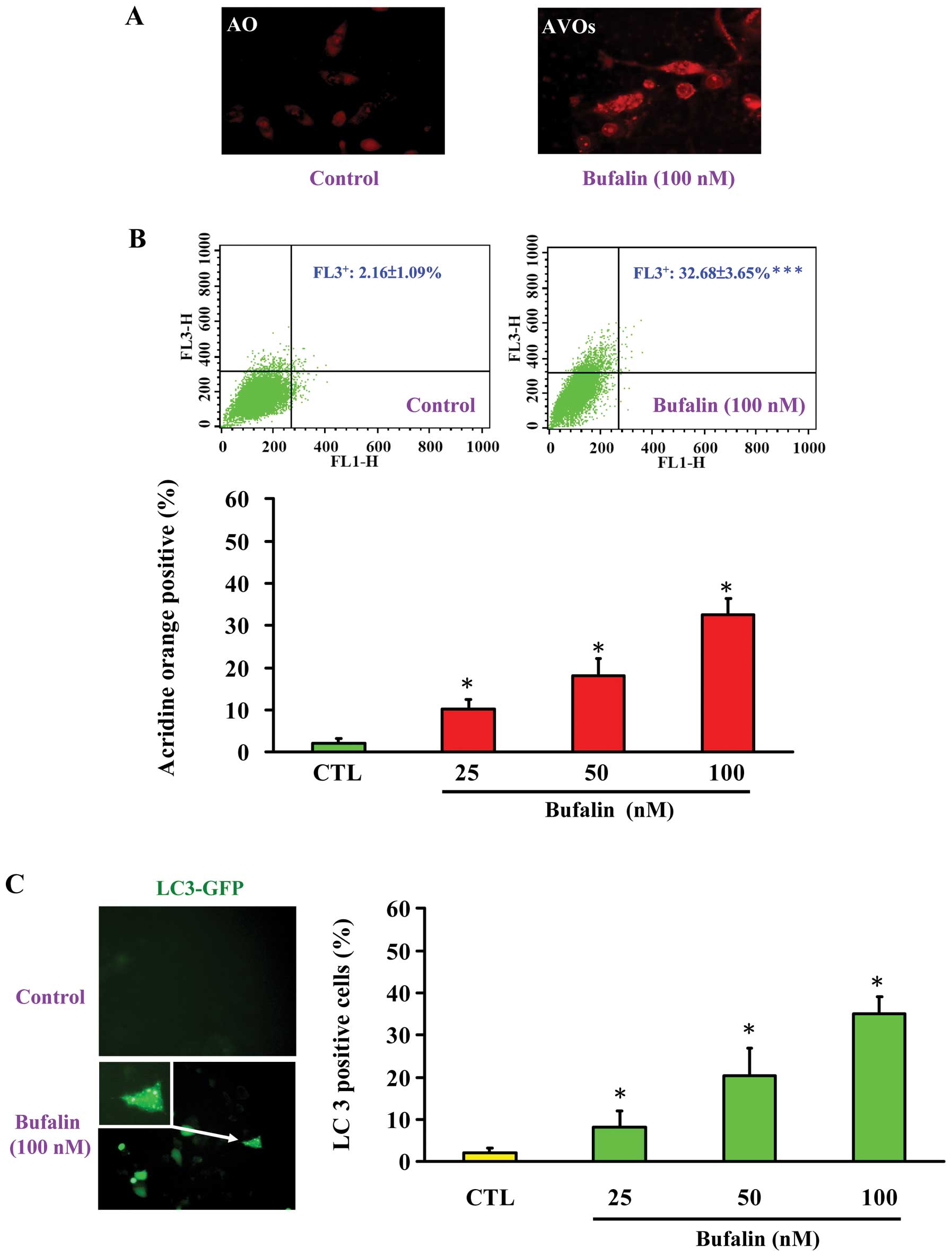
present in bufalin-treated SK-HEP-1 cells. The cytosolic acidic
vesicular organelles (AVOs) are one of the hallmarks of autophagic
cell death. In Fig. 5A, acridine
orange (AO) staining of bufalin-treated SK-HEP-1 cells clearly
showed AVOs within the cytoplasm compared to control by
fluorescence microscopy. This observation was confirmed by FACS
analysis, which showed a clear increase in red fluorescence (FL-3
positive) in concentration-dependent manner on bufalin-treated
SK-HEP-1 cells (Fig. 5B).
Furthermore, it has been shown that microtubule-associated protein
1 light-chain 3 (LC3) is an autophagic membrane marker for the
detection of early autophagosome formation (50). We examined the LC3 distribution in
bufalin-treated SK-HEP-1 cells by fluorescence microscopy and FACS
analysis. As shown in Fig. 5C,
bufalin treatment enhanced the punctate pattern of LC3-GFP in
autophagic SK-HEP-1 cells. In addition, an increase in the green
fluorescence (FL-1 positive) cells by FACS analysis showed that
bufalin-treated SK-HEP-1 cells underwent autophagic cell death in a
concentration-dependent manner (Fig.
5C, right). Our results suggested that bufalin-induced cell
death in SK-HEP-1 cells is dependent on the induction of
autophagy.
Bufalin upregulates the
autophagy-associated protein levels and blocks the AKT/mTOR
signaling in SK-HEP-1 cells
Induced autophagic cell death is associated with the
elevated protein levels of autophagic proteins LC-3, Atg complex
(Atg 5, Atg 7 and Atg 12) and Beclin-1. We investigated the
autophagy-associated protein levels in bufalin-treated SK-HEP-1
cells by western blot analysis. As shown in Fig. 6A, bufalin (100 nM) increased the
protein levels of LC-3 II, Atg 5, Atg 7 and Atg 12 and Beclin-1 in
SK-HEP-1 cells. It is also reported that the AKT activity
contributed to autophagic cell death. To examine whether
bufalin-induced autophagic cell death is through the inhibition of
AKT in SK-HEP-1 cells, cells were harvested after treatment with
100 nM of bufalin for 2, 4, 6 and 8 h, and then determined the
protein levels by western blot analysis. Fig. 6B showed that bufalin (100 nM)
significantly decreased the phospho-AKT (Thr308), phospho-AKT
(Ser473), phospho-mTOR (Ser2481) protein levels in SK-HEP-1 cells.
Bufalin decreased the AKT activity and this effect is
time-dependent (Fig. 6C). Our
results implied that bufalin induced autophagic cell death in
SK-HEP-1 cells through interfering with the AKT/mTOR signaling
pathway.
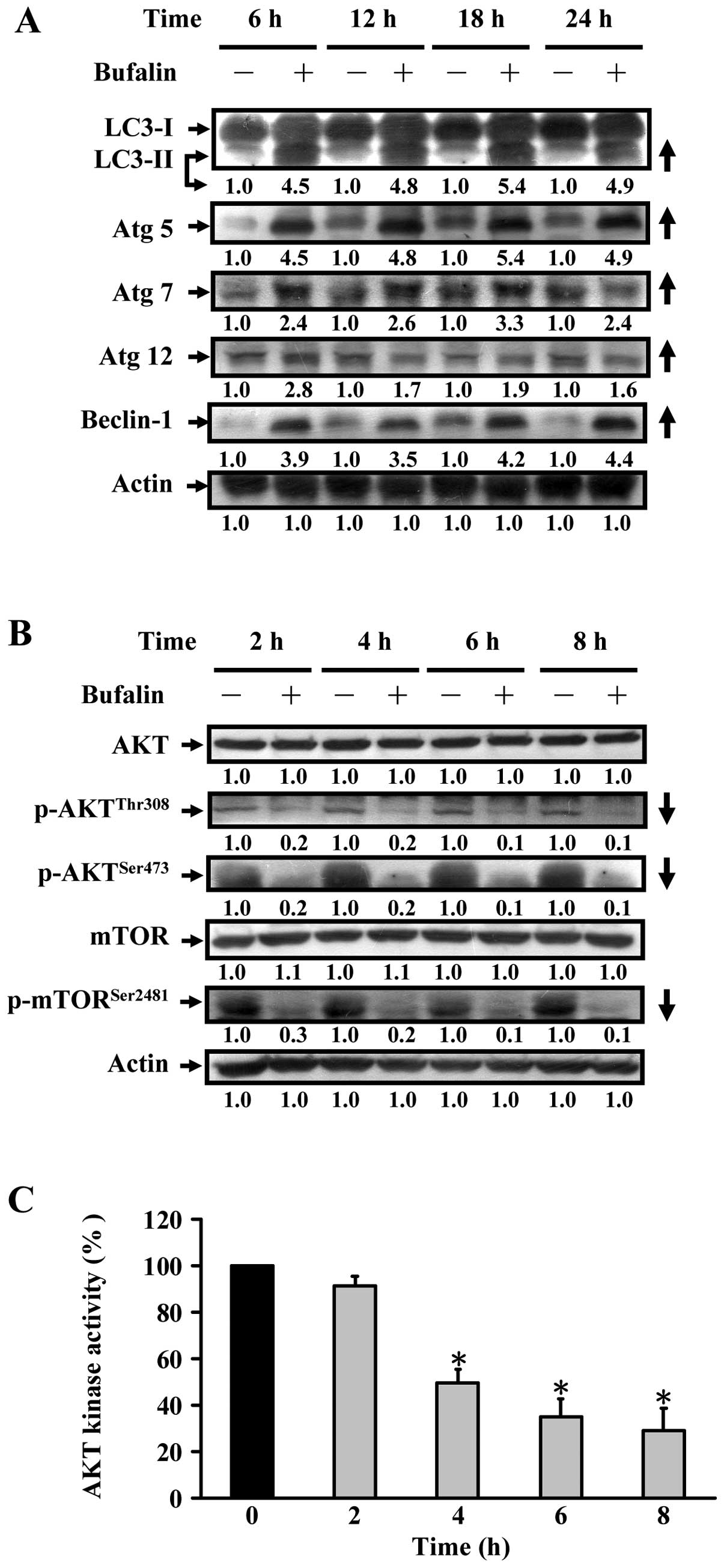 | Figure 6Expression levels of
autophagy-associated proteins and the AKT/ mTOR signaling pathway
in bufalin-treated SK-HEP-1 cells. (A) Cells were treated with 100
nM of bufalin for the indicated time points. The protein levels of
LC-3, Atg 5, Atg 7 and Atg 12, Beclin-1, and β-actin in
bufalin-treated SK-HEP-1 cells were examined by western blot
analysis. (B) Cells were treated as described in (A). The protein
levels of AKT, phospho-AKT (Thr308), phospho-AKT (Ser473),
phospho-mTOR (Ser2481), mTOR, phospho-mTOR (Ser2481) and β-actin in
bufalin-treated SK-HEP-1 cells were examined by western blot
analysis. (C) AKT kinase activity was examined by an in
vitro kinase activity assay. Data are presented as the mean ±
SEM of three independent experiments. *P<0.05,
significantly different compared with control treatment. |
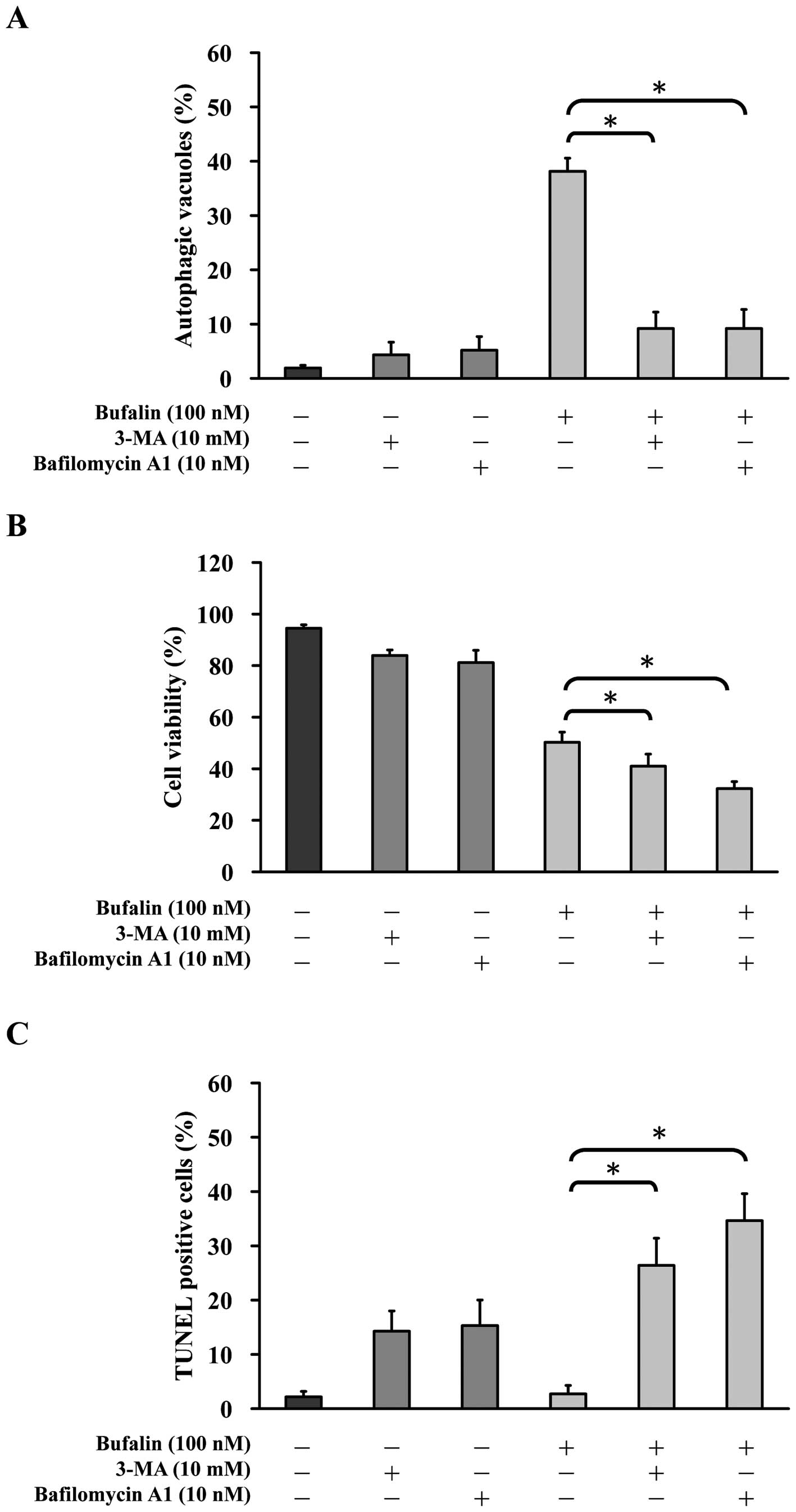
3-Methyladenine and bafilomycin protect
against autophagy and induce apoptosis in bufalin-treated SK-HEP-1
cells
3-Methyladenine (3-MA), an inhibitor of class III
phosphatidylinositol-3 kinase, is a reagent potently inhibiting
autophagy-dependent protein degradation and suppressing the
formation of autophagosomes (51),
whereas bafilomycin A1, an inhibitor of the vacuolar proton pump of
lysosomes and endosomes, appears to block the fusion of
autophagosomes (52). In the
present study, SK-HEP-1cells were pretreated with 3-MA (10 mM) or
bafilomycin A1 (10 nM), and then exposed to 100 nM of bufalin
before harvested for measuring the levels of autophagic vacuoles
and cell viability by MTT assay. Both 3-MA (10 mM) and bafilomycin
A1 (10 nM) inhibit autophagic vacuoles (Fig. 7A) and reduce cell viability
(Fig. 7B) in bufalin-treated
SK-HEP-1 cells. Furthermore, we examined effects of 3-MA (10 mM)
and bafilomycin A1 on apoptosis by TUNEL assay (Fig. 7C). The results showed both 3-MA (10
mM) and bafilomycin A1 (10 nM) increased TUNEL positive apoptotic
cells in bufalin-treated SK-HEP-1 cells. Taken together, inhibiting
autophagic cell death by 3-MA or bafilomycin A1 facilitates
bufalin-induced apoptosis in SK-HEP-1 cells.
Discussion
Traditional Chinese medicines (TCM) powerful for
prevention or therapy of cancer are marked with their high
anticancer activity and low toxicity in normal cells (53,54).
Bufalin has been used in clinical trials for cancer therapy in
China (55). Han et al has
demonstrated that bufalin has significant anti-human hepatocellular
carcinoma activity in orthotopic transplantation nude mouse model
in vivo and possesses no marked toxicity (56). Although bufalin upregulates Bax
protein and induces tumor cell apoptosis in vivo, the
signaling pathways underlying bufalin-induced cell death have not
been elucidated. Arsenic trioxide, a TCM, has been reported to
induce autophagic cell death without activation of
caspase-dependent apoptotic cell death (57). The current study focused on
exploring molecular mechanisms of bufalin-induced cell death in
human hepatocellular carcinoma SK-HEP-1 cells. Bufalin induced
autophagic cell death through inhibiting the AKT/mTOR signaling
pathway in SK-HEP-1 cells. Our results suggested that bufalin may
be used as a novel therapeutic reagent for the medical treatment
and/or prevention of human hepatocellular carcinoma.
In addition, we demonstrated that bufalin induced
growth inhibitory effects through G2/M arrest (Fig. 1C) and autophagic cell death in
SK-HEP-1 cells. Our results indicated that bufalin induced
G2/M phase arrest (Fig.
1C). Cyclin A, cyclin B, CDK1, phospho-CDK1 (Thr161), Cdc25c,
phospho-Cdc25c (Ser198) were decreased, while Chk1, Wee1 was
increased by bufalin treatment in a time-dependent manner (Fig. 2A). It was reported in the
G2/M phase progression, which is regulated with CDK1 and
CDK2 kinases that are activated primarily in association with
cyclins A and B. Furthermore, bufalin also inhibited the CDK1
activity (Fig. 2B). Previous
studies have demonstrated that bufalin inhibited cell proliferation
through induction of G0/G1 arrest of T24
human bladder cancer cells (46),
human endometrial stromal cells, and ovarian cancer cells (58), but our findings indicated that
bufalin induced different cell cycle phase arrest dependent on the
cell types. The results are in agreement with previous studies
indicated that bufalin inhibited cell proliferation through
induction of G2/M arrest of endothelial cells (59), leukemia ML1 cells (60) and human osteosarcoma U-2OS and
U-2OS methotrexate 300-resistant cells in vitro (61).
Autophagy plays two physiologic roles: one is
protective role allowing cell survival and generating nutrients and
energy; the other promotes cell death (62). When the cells undergo starvation,
endoplasmic reticulum (ER) stress stimulation, ROS production and
hypoxia, autophagy protects cells from the damage, which leads to
cell survival (63). However,
autophagy is observed to be induced by cytotoxic chemotherapeutic
reagents such as paclitaxel (Taxol) (64), chloroquine, arsenic trioxide
(65,66), sorafenib (67,68)
and imiquimod (69). Bufalin has
been demonstrated to induce apoptosis and cell cycle arrest in
leukemia (70), prostatic cancer
(71), gastric cancer (72), ovarian cancer (58), osteosarcoma (61) and bladder cancer cells (59). In the present study, apoptotic
characteristics such as DNA fragmentation, and cleavages of
caspase-3 and PARP were not found in bufalin-treated SK-HEP-1 cells
(Fig. 3C and D). Our data
indicated that bufalin-induced cell death is caspase-independent
apoptosis in SK-HEP-1 cells. Intriguingly, bufalin-induced
autophagic cell death in SK-HEP-1 cells was demonstrated by several
lines of evidence including autophagic vesicle formation (Fig. 4A), double-membrane vacuoles
(Fig. 4B), acidic vesicular
organelles (Fig. 5A), cleavage of
microtubule-associated protein 1 light chain 3 (LC3) (Fig. 5C) and elevated protein levels of
autophagic proteins, Atg complex (Atg 5, Atg 7 and Atg 12) and
Beclin-1 (Fig. 6). When SK-HEP-1
cells were pre-treated with 3-MA or bafilomycin A1 followed by
treating with bufalin, growth inhibitory effects and apoptosis were
significantly enhanced compared with the bufalin alone treatment
group by TUNEL assay (Fig. 7C).
Our results suggested autophagy protects SK-HEP-1 cells from
undergoing apoptosis in the early stage through antagonizing
bufalin induced apoptosis. Our results did not rule out bufalin
might be involved in apoptotic signaling pathways after longer
exposure (more than 24 h), but showed that SK-HEP-1 cells induced
autophagic cell death after the 24 h treatment with bufalin. This
is in agreement with the finding of Xie et al that human
colorectal cancer HT-29 and Caco-2 cells treated with bufalin for
24 h and induced autophagy (34).
Previous studies demonstrated that autophagic cell
death is triggered by multiple signaling pathways such as the
adenosine monophosphate-activated protein kinase (AMPK) pathway
(73,74), the AKT/mammalian target of
rapamycin (mTOR) pathway (75),
the MAPK (ERK, p38 and JNK) pathway (34,76),
the BCL2 and its family members involved pathway (77), the death-associated protein kinase
(DAPK) pathway and the death-associated related protein kinase 1
(DRP1) pathways (78). The
AKT/mTOR pathway is involved in regulating cell survival and cell
death. In the current study, bufalin inhibited protein levels of
phospho-AKT (Thr308), phospho-AKT (Ser473), phospho-mTOR (Ser2481)
and decreased the activity of AKT in SK-HEP-1 cells (Fig. 6B and C). Liu et al reported
that AKT gene was overexpressed in hepatocellular carcinoma HCC and
suggested that AKT activation participates in the pathogenesis and
progression of HCC (79).
Recently, our preliminary result demonstrated that human oral
squamous cell carcinoma CAL 27 cells stably expressed
constitutively active AKT (CA-AKT) increased AKT activity and
attenuated bufalin-induced growth inhibition and cell death (data
not shown). In the present study, the result showed that the AKT
activity and AKT/mTOR pathway are associated with the induction of
autophagic cell death in bufalin-treated SK-HEP-1 cells.
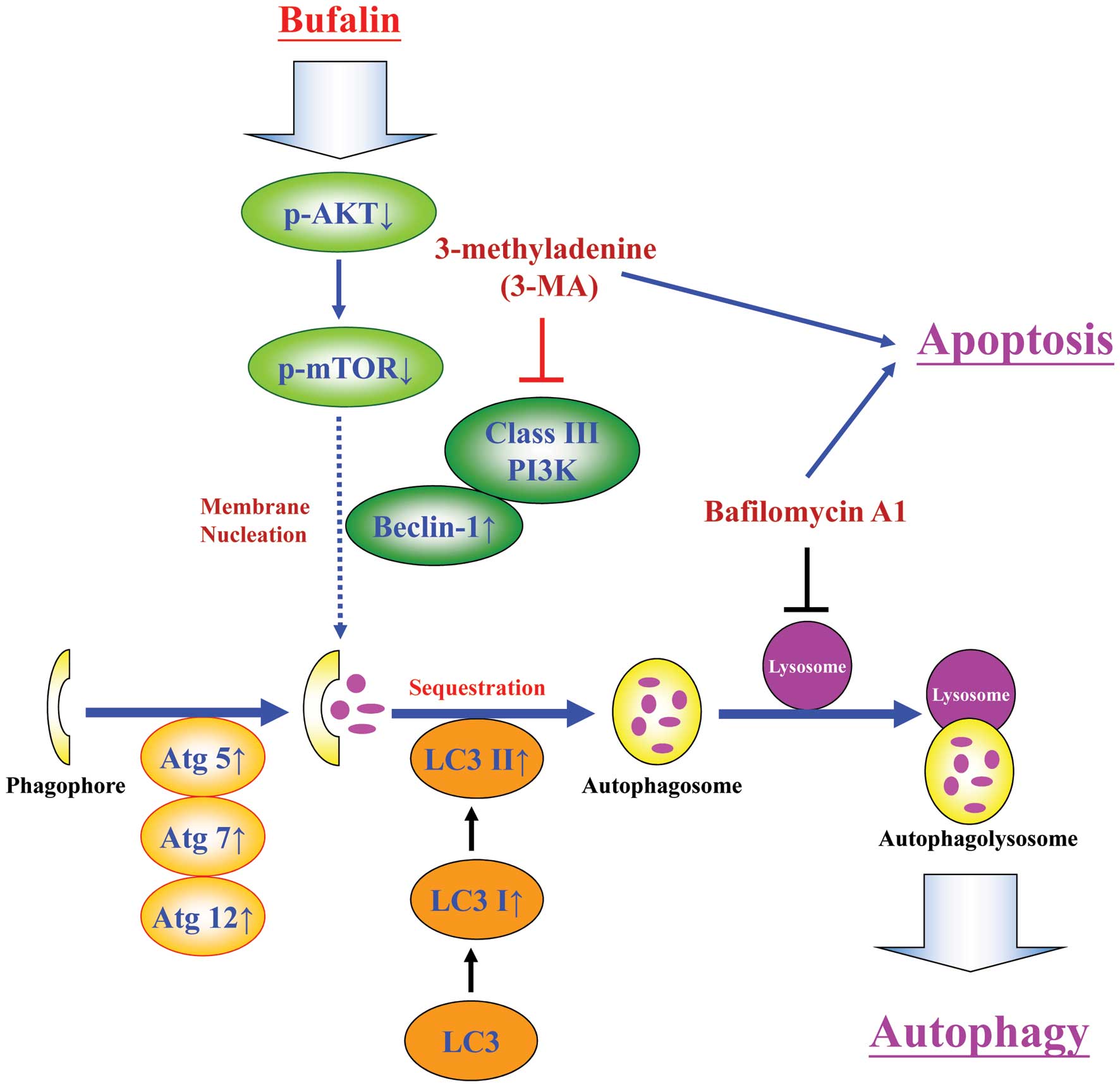
Overall, the molecular signaling pathways are
summarized in Fig. 8. Our results
demonstrated that the AKT/mTOR signaling pathway promotes autophagy
in bufalin-treated SK-HEP-1 cells. These findings implied that
bufalin may be used as a novel therapeutic reagent for the
treatment of hepatocellular carcinoma.
Acknowledgements
This study was supported part by a
research grant from the National Science Council of the Republic of
China (NSC-101-2313-B-039-008) awarded to J.-S. Yang and part by
the grant from China Medical University (CMU100-TC-06) awarded to
W.-W. Huang.
References
|
1
|
Tazi el M, Essadi I, M’Rabti H, Touyar A
and Errihani PH: Systemic treatment and targeted therapy in
patients with advanced hepatocellular carcinoma. N Am J Med Sci.
3:167–175. 2011.PubMed/NCBI
|
|
2
|
Shariff MI, Cox IJ, Gomaa AI, Khan SA,
Gedroyc W and Taylor-Robinson SD: Hepatocellular carcinoma: current
trends in worldwide epidemiology, risk factors, diagnosis and
therapeutics. Expert Rev Gastroenterol Hepatol. 3:353–367. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Simonetti RG, Camma C, Fiorello F, Politi
F, D’Amico G and Pagliaro L: Hepatocellular carcinoma. A worldwide
problem and the major risk factors. Dig Dis Sci. 36:962–972.
1991.PubMed/NCBI
|
|
4
|
Takahashi H and Wands JR: Prognosis of
hepatocellular carcinoma: known to be poor: yet difficult to
predict. J Nucl Med. 32:235–236. 1991.PubMed/NCBI
|
|
5
|
Kim JW, Lee JO, Han SW, et al: Clinical
outcomes of sorafenib treatment in patients with metastatic
hepatocellular carcinoma who had been previously treated with
fluoropyrimidine plus platinum-based chemotherapy. Am J Clin Oncol.
34:125–129. 2011.
|
|
6
|
Luo HY, Yang JY, Liu ZM, Lin QY and Yan
LN: Reversal of multidrug resistance gene MDR1 and MRP of
drug-resistant human hepatocellular carcinoma cells SMMC-7721/ADM
with antisense phosphorothioate oligonucleotides. Zhonghua Gan Zang
Bing Za Zhi. 12:85–87. 2004.(In Chinese).
|
|
7
|
Chang CS, Huang WT, Yang SS, Yeh HZ, Kao
CH and Chen GH: Effect of P-glycoprotein and multidrug resistance
associated protein gene expression on Tc-99m MIBI imaging in
hepatocellular carcinoma. Nucl Med Biol. 30:111–117. 2003.
View Article : Google Scholar
|
|
8
|
Shen DW, Lu YG, Chin KV, Pastan I and
Gottesman MM: Human hepatocellular carcinoma cell lines exhibit
multidrug resistance unrelated to MRD1 gene expression. J Cell Sci.
98:317–322. 1991.PubMed/NCBI
|
|
9
|
Dai R, Chen R and Li H: Cross-talk between
PI3K/Akt and MEK/ERK pathways mediates endoplasmic reticulum
stress-induced cell cycle progression and cell death in human
hepatocellular carcinoma cells. Int J Oncol. 34:1749–1757.
2009.
|
|
10
|
Chen GG, Chan UP, Bai LC, et al: ZBP-89
reduces the cell death threshold in hepatocellular carcinoma cells
by increasing caspase-6 and S phase cell cycle arrest. Cancer Lett.
283:52–58. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu SC and Zhang Y: Cyclin-dependent kinase
1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2)
regulates its stability. J Biol Chem. 286:28511–28519. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hong KU, Kim HJ, Kim HS, et al:
Cdk1-cyclin B1-mediated phosphorylation of tumor-associated
microtubule-associated protein/cytoskeleton-associated protein 2 in
mitosis. J Biol Chem. 284:16501–16512. 2009. View Article : Google Scholar
|
|
13
|
Yang JS, Hour MJ, Huang WW, Lin KL, Kuo SC
and Chung JG: MJ-29 inhibits tubulin polymerization, induces
mitotic arrest, and triggers apoptosis via cyclin-dependent kinase
1-mediated Bcl-2 phosphorylation in human leukemia U937 cells. J
Pharmacol Exp Ther. 334:477–488. 2010. View Article : Google Scholar
|
|
14
|
Rieder CL: Mitosis in vertebrates: the
G2/M and M/A transitions and their associated checkpoints.
Chromosome Res. 19:291–306. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dustin P: Mitosis and differentiation; new
thoughts on the biochemical regulation of growth in the
vertebrates. Arch Anat Histol Embryol. 34:195–201. 1951.PubMed/NCBI
|
|
16
|
Choi KS: Autophagy and cancer. Exp Mol
Med. 44:109–120. 2012. View Article : Google Scholar
|
|
17
|
Naumann P, Fortunato F, Zentgraf H,
Buchler MW, Herr I and Werner J: Autophagy and cell death signaling
following dietary sulforaphane act independently of each other and
require oxidative stress in pancreatic cancer. Int J Oncol.
39:101–109. 2011.
|
|
18
|
Lee J, Giordano S and Zhang J: Autophagy,
mitochondria and oxidative stress: cross-talk and redox signalling.
Biochem J. 441:523–540. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McCormick J, Knight RA, Barry SP, et al:
Autophagy in the stress-induced myocardium. Front Biosci (Elite
Ed). 4:2131–2141. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Y, Zhang Q, Tian R, et al: Lysosomal
transmembrane protein LAPTM4B promotes autophagy and tolerance to
metabolic stress in cancer cells. Cancer Res. 71:7481–7489. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kung HJ: Targeting tyrosine kinases and
autophagy in prostate cancer. Horm Cancer. 2:38–46. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bursch W, Karwan A, Mayer M, et al: Cell
death and autophagy: cytokines, drugs, and nutritional factors.
Toxicology. 254:147–157. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Martinez-Borra J and Lopez-Larrea C:
Autophagy and self-defense. Adv Exp Med Biol. 738:169–184. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Anders HJ and Schlondorff DO: Innate
immune receptors and autophagy: implications for autoimmune kidney
injury. Kidney Int. 78:29–37. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Munafo DB and Colombo MI: A novel assay to
study autophagy: regulation of autophagosome vacuole size by amino
acid deprivation. J Cell Sci. 114:3619–3629. 2001.PubMed/NCBI
|
|
26
|
Yang ZJ, Chee CE, Huang S and Sinicrope
FA: The role of autophagy in cancer: therapeutic implications. Mol
Cancer Ther. 10:1533–1541. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Meschini S, Condello M, Lista P and
Arancia G: Autophagy: molecular mechanisms and their implications
for anticancer therapies. Curr Cancer Drug Targets. 11:357–379.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Karantza-Wadsworth V and White E: Role of
autophagy in breast cancer. Autophagy. 3:610–613. 2007. View Article : Google Scholar
|
|
29
|
Kelekar A: Autophagy. Ann NY Acad Sci.
1066:259–271. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tanida I, Ueno T and Kominami E: LC3
conjugation system in mammalian autophagy. Int J Biochem Cell Biol.
36:2503–2518. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ropolo A, Bagnes CI, Molejon MI, et al:
Chemotherapy and autophagy-mediated cell death in pancreatic cancer
cells. Pancreatology. 12:1–7. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo XL, Li D, Hu F, et al: Targeting
autophagy potentiates chemotherapy-induced apoptosis and
proliferation inhibition in hepatocarcinoma cells. Cancer Lett.
320:171–179. 2012. View Article : Google Scholar
|
|
33
|
Yousefi S and Simon HU: Autophagy in
cancer and chemotherapy. Results Probl Cell Differ. 49:183–190.
2009. View Article : Google Scholar
|
|
34
|
Xie CM, Chan WY, Yu S, Zhao J and Cheng
CH: Bufalin induces autophagy-mediated cell death in human colon
cancer cells through reactive oxygen species generation and JNK
activation. Free Radic Biol Med. 51:1365–1375. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lan YH, Chiang JH, Huang WW, et al:
Activations of both extrinsic and intrinsic pathways in HCT 116
human colorectal cancer cells contribute to apoptosis through
p53-mediated ATM/Fas signaling by Emilia sonchifolia
extract, a folklore medicinal plant. Evid Based Complement Alternat
Med. 2012:1781782012.
|
|
36
|
Lu HF, Lai KC, Hsu SC, et al: Curcumin
induces apoptosis through FAS and FADD, in caspase-3-dependent and
-independent pathways in the N18 mouse-rat hybrid retina ganglion
cells. Oncol Rep. 22:97–104. 2009.
|
|
37
|
Lin HL, Yang JS, Yang JH, et al: The role
of Ca2+ on the DADS-induced apoptosis in mouse-rat
hybrid retina ganglion cells (N18). Neurochem Res. 31:383–393.
2006.
|
|
38
|
Chen HY, Lu HF, Yang JS, et al: The novel
quinolone CHM-1 induces DNA damage and inhibits DNA repair gene
expressions in a human osterogenic sarcoma cell line. Anticancer
Res. 30:4187–4192. 2010.
|
|
39
|
Chiu YJ, Hour MJ, Lu CC, et al: Novel
quinazoline HMJ-30 induces U-2 OS human osteogenic sarcoma cell
apoptosis through induction of oxidative stress and up-regulation
of ATM/p53 signaling pathway. J Orthop Res. 29:1448–1456. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang SM, Yang JS, Tsai SC, et al: The
novel synthesized
2-(3-(methylamino)phenyl)-6-(pyrrolidin-1-yl)quinolin-4-one (Smh-3)
compound induces G2/M phase arrest and
mitochondrial-dependent apoptotic cell death through inhibition of
CDK1 and AKT activity in HL-60 human leukemia cells. Int J Oncol.
38:1357–1364. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chung JG, Yang JS, Huang LJ, et al:
Proteomic approach to studying the cytotoxicity of YC-1 on U937
leukemia cells and antileukemia activity in orthotopic model of
leukemia mice. Proteomics. 7:3305–3317. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li YC, Lin HJ, Yang JH, et al:
Baicalein-induced apoptosis via endoplasmic reticulum stress
through elevations of reactive oxygen species and mitochondria
dependent pathway in mouse-rat hybrid retina ganglion cells (N18).
Neurochem Res. 34:418–429. 2009. View Article : Google Scholar
|
|
43
|
Yang JS, Chen GW, Hsia TC, et al: Diallyl
disulfide induces apoptosis in human colon cancer cell line (COLO
205) through the induction of reactive oxygen species, endoplasmic
reticulum stress, caspases casade and mitochondrial-dependent
pathways. Food Chem Toxicol. 47:171–179. 2009. View Article : Google Scholar
|
|
44
|
Tseng MT, Lu X, Duan X, et al: Alteration
of hepatic structure and oxidative stress induced by intravenous
nanoceria. Toxicol Appl Pharmacol. 260:173–182. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xiao D, Bommareddy A, Kim SH, Sehrawat A,
Hahm ER and Singh SV: Benzyl isothiocyanate causes FoxO1-mediated
autophagic death in human breast cancer cells. PLoS One.
7:e325972012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Huang WW, Yang JS, Pai SJ, et al: Bufalin
induces G(0)/G(1) phase arrest through inhibiting the levels of
cyclin D, cyclin E, CDK2 and CDK4, and triggers apoptosis via
mitochondrial signaling pathway in T24 human bladder cancer cells.
Mutat Res. 732:26–33. 2012. View Article : Google Scholar
|
|
47
|
Ip SW, Chu YL, Yu CS, et al: Bee venom
induces apoptosis through intracellular Ca2+-modulated
intrinsic death pathway in human bladder cancer cells. Int J Urol.
19:61–70. 2012.PubMed/NCBI
|
|
48
|
Chiou SK, Hoa N and Hodges A: Sulindac
sulfide induces autophagic death in gastric epithelial cells via
survivin down-regulation: a mechanism of NSAIDs-induced gastric
injury. Biochem Pharmacol. 81:1317–1323. 2011. View Article : Google Scholar
|
|
49
|
Enomoto A, Murakami H, Asai N, et al:
Akt/PKB regulates actin organization and cell motility via
Girdin/APE. Dev Cell. 9:389–402. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kabeya Y, Mizushima N, Ueno T, et al: LC3,
a mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Seglen PO and Gordon PB: 3-Methyladenine:
specific inhibitor of autophagic/lysosomal protein degradation in
isolated rat hepatocytes. Proc Natl Acad Sci USA. 79:1889–1892.
1982. View Article : Google Scholar
|
|
52
|
Yamamoto A, Tagawa Y, Yoshimori T,
Moriyama Y, Masaki R and Tashiro Y: Bafilomycin A1 prevents
maturation of autophagic vacuoles by inhibiting fusion between
autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E
cells. Cell Struct Funct. 23:33–42. 1998. View Article : Google Scholar
|
|
53
|
Lee MJ, Chen HM, Tzang BS, et al: Ocimum
gratissimum aqueous extract protects H9c2 myocardiac cells from
H(2) O(2)-induced cell apoptosis through akt signalling. Evid Based
Complement Alternat Med. 2011:5780602011.PubMed/NCBI
|
|
54
|
Robbins D, Gu X, Shi R, et al: The
chemopreventive effects of Protandim: modulation of p53
mitochondrial translocation and apoptosis during skin
carcinogenesis. PLoS One. 5:e119022010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Meng Z, Yang P, Shen Y, et al: Pilot study
of huachansu in patients with hepatocellular carcinoma,
nonsmall-cell lung cancer, or pancreatic cancer. Cancer.
115:5309–5318. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Han KQ, Huang G, Gu W, Su YH, Huang XQ and
Ling CQ: Anti-tumor activities and apoptosis-regulated mechanisms
of bufalin on the orthotopic transplantation tumor model of human
hepatocellular carcinoma in nude mice. World J Gastroenterol.
13:3374–3379. 2007.
|
|
57
|
Ondrouskova E, Soucek K, Horvath V and
Smarda J: Alternative pathways of programmed cell death are
activated in cells with defective caspase-dependent apoptosis. Leuk
Res. 32:599–609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Takai N, Ueda T, Nishida M, Nasu K and
Narahara H: Bufalin induces growth inhibition, cell cycle arrest
and apoptosis in human endometrial and ovarian cancer cells. Int J
Mol Med. 21:637–643. 2008.PubMed/NCBI
|
|
59
|
Hong SH and Choi YH: Bufalin induces
apoptosis through activation of both the intrinsic and extrinsic
pathways in human bladder cancer cells. Oncol Rep. 27:114–120.
2012.PubMed/NCBI
|
|
60
|
Jing Y, Watabe M, Hashimoto S, Nakajo S
and Nakaya K: Cell cycle arrest and protein kinase modulating
effect of bufalin on human leukemia ML1 cells. Anticancer Res.
14:1193–1198. 1994.PubMed/NCBI
|
|
61
|
Yin JQ, Shen JN, Su WW, et al: Bufalin
induces apoptosis in human osteosarcoma U-2OS and U-2OS
methotrexate300-resistant cell lines. Acta Pharmacol Sin.
28:712–720. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Codogno P: Autophagy in cell survival and
death. J Soc Biol. 199:233–241. 2005.(In French).
|
|
63
|
Hoyer-Hansen M and Jaattela M: Autophagy:
an emerging target for cancer therapy. Autophagy. 4:574–580. 2008.
View Article : Google Scholar
|
|
64
|
Eum KH and Lee M: Crosstalk between
autophagy and apoptosis in the regulation of paclitaxel-induced
cell death in v-Ha-ras-transformed fibroblasts. Mol Cell Biochem.
348:61–68. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kanzawa T, Kondo Y, Ito H, Kondo S and
Germano I: Induction of autophagic cell death in malignant glioma
cells by arsenic trioxide. Cancer Res. 63:2103–2108.
2003.PubMed/NCBI
|
|
66
|
Kanzawa T, Zhang L, Xiao L, Germano IM,
Kondo Y and Kondo S: Arsenic trioxide induces autophagic cell death
in malignant glioma cells by upregulation of mitochondrial cell
death protein BNIP3. Oncogene. 24:980–991. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bareford MD, Park MA, Yacoub A, et al:
Sorafenib enhances pemetrexed cytotoxicity through an
autophagy-dependent mechanism in cancer cells. Cancer Res.
71:4955–4967. 2011. View Article : Google Scholar
|
|
68
|
Ullen A, Farnebo M, Thyrell L, et al:
Sorafenib induces apoptosis and autophagy in prostate cancer cells
in vitro. Int J Oncol. 37:15–20. 2010. View Article : Google Scholar
|
|
69
|
Huang SW, Liu KT, Chang CC, et al:
Imiquimod simultaneously induces autophagy and apoptosis in human
basal cell carcinoma cells. Br J Dermatol. 163:310–320. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Chen A, Yu J, Zhang L, et al: Microarray
and biochemical analysis of bufalin-induced apoptosis of HL-60
Cells. Biotechnol Lett. 31:487–494. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Yu CH, Kan SF, Pu HF, Jea Chien E and Wang
PS: Apoptotic signaling in bufalin- and cinobufagin-treated
androgen-dependent and -independent human prostate cancer cells.
Cancer Sci. 99:2467–2476. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Li D, Qu X, Hou K, et al: PI3K/Akt is
involved in bufalin-induced apoptosis in gastric cancer cells.
Anticancer Drugs. 20:59–64. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Mihaylova MM and Shaw RJ: The AMPK
signalling pathway coordinates cell growth, autophagy and
metabolism. Nat Cell Biol. 13:1016–1023. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Hoyer-Hansen M and Jaattela M:
AMP-activated protein kinase: a universal regulator of autophagy?
Autophagy. 3:381–383. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Shi WY, Xiao D, Wang L, et al: Therapeutic
metformin/AMPK activation blocked lymphoma cell growth via
inhibition of mTOR pathway and induction of autophagy. Cell Death
Dis. 3:e2752012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Corcelle E, Djerbi N, Mari M, et al:
Control of the autophagy maturation step by the MAPK ERK and p38:
lessons from environmental carcinogens. Autophagy. 3:57–59. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Levine B, Sinha S and Kroemer G: Bcl-2
family members: dual regulators of apoptosis and autophagy.
Autophagy. 4:600–606. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Inbal B, Bialik S, Sabanay I, Shani G and
Kimchi A: DAP kinase and DRP-1 mediate membrane blebbing and the
formation of autophagic vesicles during programmed cell death. J
Cell Biol. 157:455–468. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Liu L-X, Liu Z-H, Jiang H-C, et al:
Overexpression of Akt-1 gene in human hepatocellular carcinoma.
Chinese J Cancer Res. 14:161–164. 2002. View Article : Google Scholar
|