Introduction
Hepatocellular carcinoma (HCC) currently represents
the sixth most common cancer in the world. Its incidence, very high
in Asian countries, is steadily increasing in occidental countries.
HCC is often refractory to radiation therapy (RT) and chemotherapy
leaving surgery (followed by liver transplantation) as the sole
alternative to delay disease progression. As with other forms of
cancer, the radioresistance of HCC cells stems, in part, from their
intrinsic inability to undergo an apoptotic process in response to
ionizing radiation (IR). The toxicity of IR on surrounding healthy
hepatic tissue is another source of limitation for RT. Among other
therapeutic strategies, two promising approaches to overcome the
resistance of HCC cells to IR and conventional chemotherapy have
been proposed: i) the utilization of high linear energy transfer
(LET) radiation instead of low-LET radiation (1) and ii) the implementation of
molecularly targeted drugs (2).
High-LET based RT, or hadrontherapy, principally involves the use
of accelerated carbon ions. The main advantage of charged high-LET
particles is that they can deposit their energy within the tumor
with extreme accuracy (3,4). Although only a handful of
hadrontherapy facilities currently exist worldwide, their number is
slowly, but steadily, growing. The use of chemotherapeutic drugs
that can be concomitantly associated with radiation offer another
possibility of treatment for HCC. However, few such
radiosensitizing drugs are currently available and their efficacy
is limited. There is, therefore, an obvious need for novel
radiosensitizing agents, more potent and molecularly targeted
(5,6). In this context, RAD001 (everolimus),
an inhibitor of the mammalian target of rapamycin (mTOR) could
offer new perspectives in the treatments of HCC. Indeed, mTOR is a
key downstream protein kinase in the PI3K/AKT pathway, which is
commonly involved in tumorigenesis, including cancer cell growth
and angiogenesis (7,8). Aberrant hyperactivation of mTOR has
been related to cancer progression (9) and is found in up to 60% of HCCs,
which led to the recommendation of RAD001 for the treatment of this
disease (10). Thus, it was
interesting to evaluate the consequences of a combination between
two modern, but non-standard treatments of HCC: high-LET radiation
on the one hand, and a molecularly targeted anticancer drug on the
other hand.
The present study was designed to assess the effect
of RAD001 combined with low- and high-LET radiation upon the growth
of SK-Hep1, a hepatic mesenchymal tumor cell line. For this
purpose, we used fast neutrons as high-LET radiation. Although
neutrons are now rarely utilized in therapy, they remain helpful as
models for investigating the biological action of high-LET
particles (11). We report here
that the association of RAD001 with fast neutron irradiation
resulted in greater antiproliferative and cytotoxic effects than
the association of RAD001 with γ-irradiation. This indicates that
high-LET radiation combined with RAD001 may provide an approach for
HCC treatment that merits further evaluation.
Materials and methods
Reagents
RAD001 was a generous gift of Novartis (Basel,
Switzerland). It was dissolved in DMSO at 40 μM and stored at −20°C
and final dilutions were prepared extemporaneously in culture
medium. Sulforhodamine B (SRB) and trichloroacetic acid (TCA) were
purchased from Sigma-Aldrich (Saint-Quentin Fallavier, France).
Cell culture
The human HCC SK-Hep1 cell line was purchased from
the American Type Culture Collection (ATCC, USA). Cultures were
maintained at 37°C in a humid atmosphere of 5% CO2.
Cells were grown in Dulbecco’s modified Eagle’s medium (PAN Biotech
GmbH) supplemented with 10% fetal bovine serum (PAN Biotech GmbH),
1 mM sodium pyruvate, 1 mM non-essential amino acids and 50 μg
penicillin-streptomycin (Life Technologies, Grand Island, USA).
Disaggregation was carried out using 5 min incubation at 37°C with
a solution of trypsin-EDTA (PAN Biotech GmbH).
Irradiation procedure and treatment
schedule
Asynchronous, exponentially growing cells were
exposed at room temperature to low- or high-LET radiation. Cells
were contained in 6-well plates filled with 4 ml culture medium or
in 96-flat bottomed microplates, filled with 0.2 ml culture medium.
Cells were treated with solvent control or RAD001 1 h before
irradiation. Irradiations with low-LET radiation were carried out
with a 137Cs γ irradiator (Biobeam GM8000, GSM Gmbh,
Leipzig, Germany). Dose rate was 3.4 Gy/min and doses ranged from 2
to 16 Gy. For high-LET radiation, we used p(65) + Be neutrons
produced by a cyclotron at the Cyclotron Resources Center (CRC) of
Louvain-la-Neuve (Belgium) and at iThemba LABS (Faure, South
Africa). Dose rate was usually 0.2 Gy/min in both facilities, and
doses ranged from 1 to 8 Gy. Dose determination was performed using
ionization chambers and thermo luminescent dosimeters (TLD). Each
experiment was repeated at least three times.
Cell proliferation assay
The effects of the combined treatments on the growth
of SK-Hep1 were investigated using the sulforhodamine B (SRB)
colorimetric assay. Cells were seeded at a density of
5×103 cells/wells in 100 μl in 96-flat bottomed well
plates (Falcon 3072). Various dilutions of RAD001 (100 μl) were
added 24 h later to quintuplicate wells. Cells were then irradiated
and incubated at 37°C for 2, 6 or 9 days. They were then fixed with
10% TCA for 1 h at 4°C, washed 5-fold with tap water, air dried and
stained with 0.4% SRB in 1% acetic acid for 30 min. SRB-stained
cells were then dissolved in 200 μl 10 mM Tris-base (pH 10.5) and
the absorbance of each well was measured at 565 nm using an MRX
microplate reader (Labtek, Issy-les-Moulineaux, France). Results
are expressed in optical density (OD), after subtractions of the
blank (no cells).
Apoptotic and autophagic assays
Apoptotic cells were quantified according to a
previous study (12). Briefly,
cells (5×105) were fixed in cold 70% ethanol for at
least 1 h. Then, they were washed in phosphate buffered saline
(PBS) pH 7.2 and resuspended in 100 μl of PBS containing 25 μg of
RNase A, 2 mM EDTA and 10 μg of propidium iodide (PI). Following
incubation in the dark for 30 min at 37°C, the fluorescence of
10,000 cells was analyzed using a FACScan flow cytometer (Becton
Dickinson, San Jose, CA) and Cell Quest software (Becton
Dickinson). Cells with a sub-diploid DNA content were recorded as
apoptotic.
For autophagy determination, we used the Cyto-ID™
Autophagy detection kit (Enzo Life Sciences, Plymouth Meeting, PA)
according to the manufacturer’s instructions. This test measures
autophagic vacuoles and monitors autophagic flux in live cells
using a novel fluorescent cationic amphiphilic dye that selectively
labels autophagic vacuoles. Briefly, cells (5×105) were
washed in PBS pH 7.2 and resuspended in 500 μl of freshly diluted
Cyto-ID® Green Detection Reagent 1 μl to a final volume
of 2 ml with PBS. Following incubation in the dark for 30 min at
37°C, the fluorescence of 10,000 cells was analyzed using a FACScan
flow cytometer (Becton Dickinson). and Cell Quest software (Becton
Dickinson).
Clonogenic survival assay
Twenty-four hours after exposure to fast neutrons or
conventional, low-LET radiations, RAD001-treated and control cells
were trypsinised and counted using a Countess® Cell
Counter (Invitrogen). Unirradiated control cells were submitted to
the same conditions. Irradiated and control cells were resuspended
at an appropriate number in fresh medium and plated at two
different dilutions into 6-well plates. Three wells were used by
experimental point. Fifteen days later, colonies were stained with
0.5% crystal violet and colonies containing more than 50 cells were
scored.
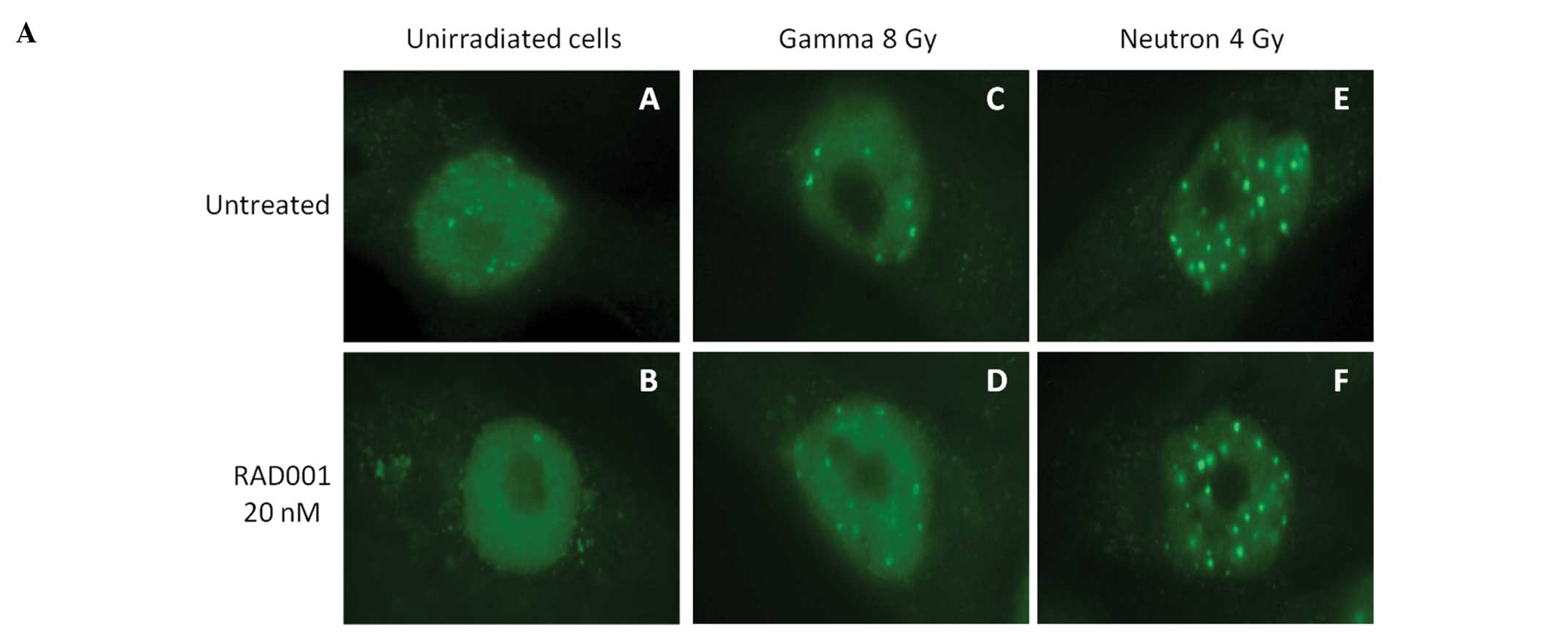
Analysis of γH2AX foci
SK-Hep1 cells were grown on microscopic glass slides
placed in 6-well plates. Twenty-four hours post-irradiation,
culture medium was removed and the slides were washed with PBS.
Fixation and permeabilization were carried out using 4%
paraformaldehyde and 0.5% Triton, respectively. Labeling was
performed using a monoclonal mouse anti-γ-H2AX antibody (clone
JBW301, Upstate, Lake Placid, NY). Coverslips were mounted in
4′,6-diamino-2-phenylindole (DAPI)-stained Vectashield (Abcys,
Paris, France). The formation of γH2AX foci in nuclei was monitored
by immunofluorescence microscope imaging. Foci were scored in at
least 40 cells in each experimental condition.
Statistical analysis
Statistical analyses were performed using the
MedCalc statistical software. Differences between the subgroups in
terms of foci number and percentage of viable cell number in SRB
assays with respect to the treatment and irradiation conditions
were pair compared with a Student-Newman-Keuls. Differences were
considered to be significant at p<0.05.
Results
Effect on cell growth and cell
survival
We first evaluated the capacity of RAD001 to
influence cell growth alone or combined with radiation. In a
preliminary series of experiments, serial concentrations of RAD001,
ranging from 1 to 40 nM, were added to SK-Hep1 in the absence of
irradiation. They indicated that 20 nM was sufficient to reduce
cell growth while not affecting cell viability (not shown). Since
this concentration is within the range of serum levels usually
targeted to obtain efficacy with minimal toxicity in the clinic
(13), it was selected for our
subsequent experiments. We then assessed the consequences of a
combination between RAD001 and an irradiation with low- or high-LET
radiation. RAD001 was added to SK-Hep1 cells 1 h before exposure to
either γ-rays or neutrons. SRB assays were performed at different
days afterwards, generally at 6 days as this time interval proved
to be optimal for obtaining a clear-cut difference between control
and experimental groups. In irradiated, untreated cells, a marked
reduction of cell growth was recorded at 8 Gy (photons) and 4 Gy
(neutrons). In RAD001-treated cells, this decrease was slightly,
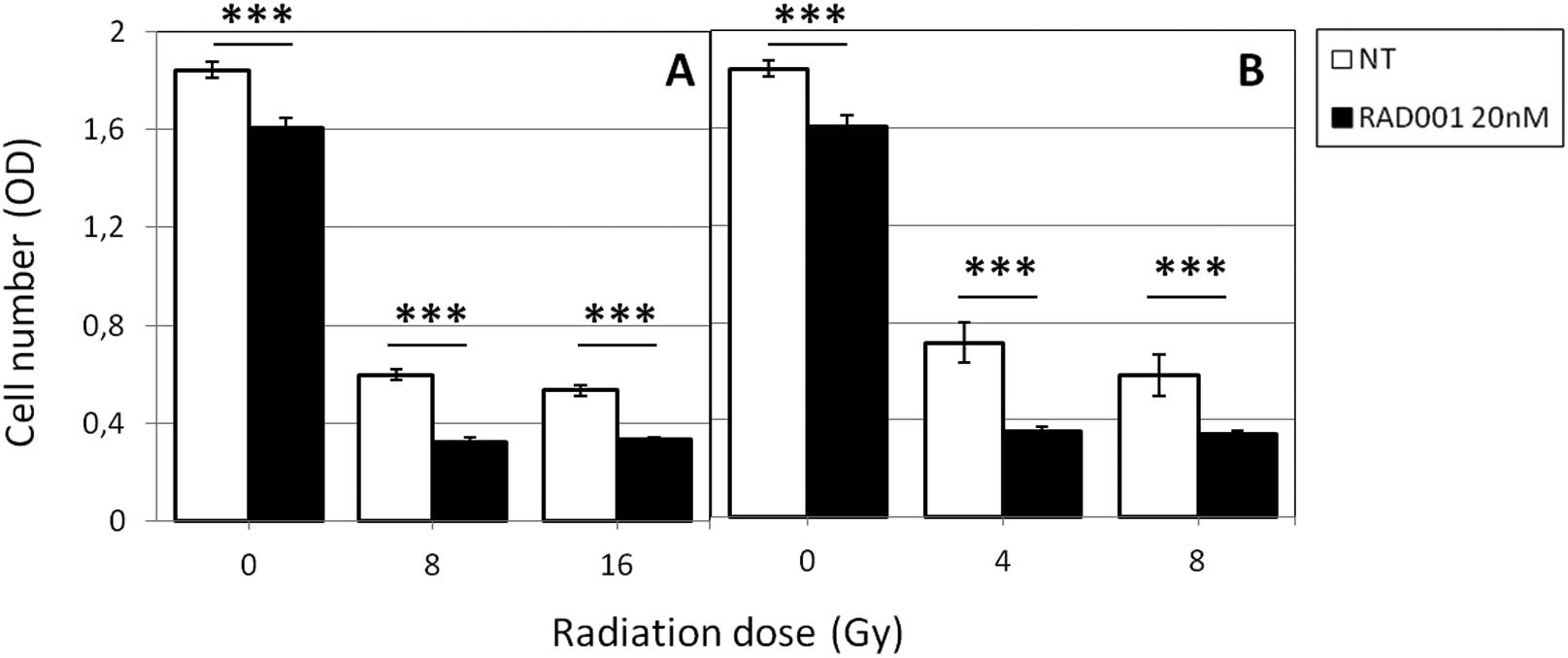
but significantly more pronounced (Fig. 1). Indeed, the ratio: optical
density (OD) of treated cells/OD of untreated cells ×100 was 54% in
8 Gy γ-irradiated cells and 49% in 4 Gy neutrons-irradiated cells.
RAD001 also reduced cell growth in unirradiated cells but at a
lesser extent (87% of control, untreated cells). No clear-cut
decrease of OD was observed according to the irradiation dose,
indicating that 4 Gy (for neutrons) and 8 Gy (for photons) were
sufficient for obtaining a significant reduction of the cell
numbers.
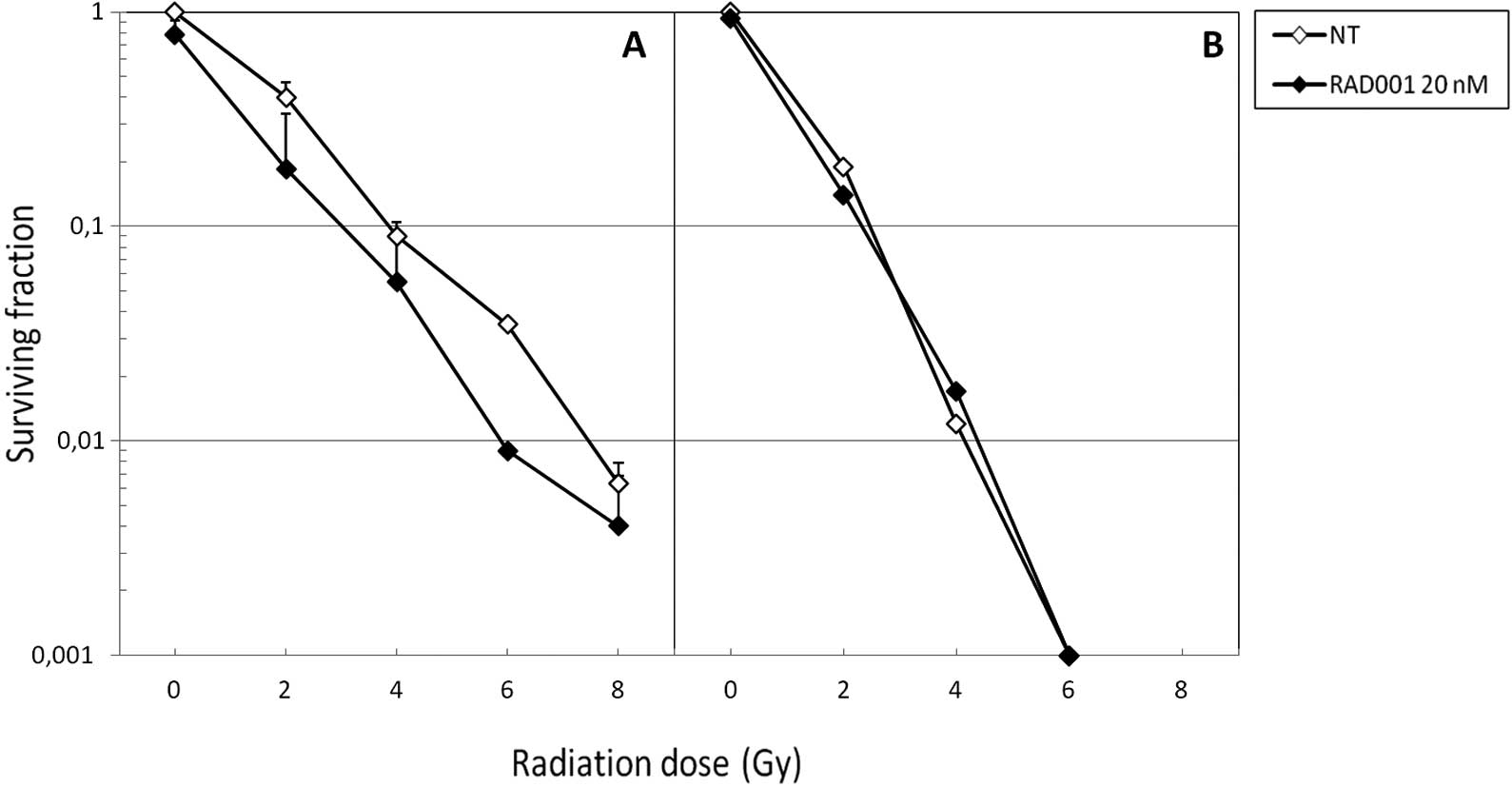
In parallel, we performed clonogenic assays in order
to assess the capacity of the various treatments to affect the
replicative survival of SK-Hep1 cells. As expected, neutrons were
more cytotoxic than photons, since at 6 Gy the former were
sufficient to almost entirely abrogate cell survival in the absence
of RAD001 co-treatment (Fig. 2B).
When associated to photons, RAD001 slightly decreased the survival
fractions at the different irradiation doses (Fig. 2A). Of note, when combined to fast
neutrons, no diminution of the survival fraction was achieved
(Fig. 2B). With both types of
radiation, the slope of the curve was identical for control and
RAD001-treated cells.
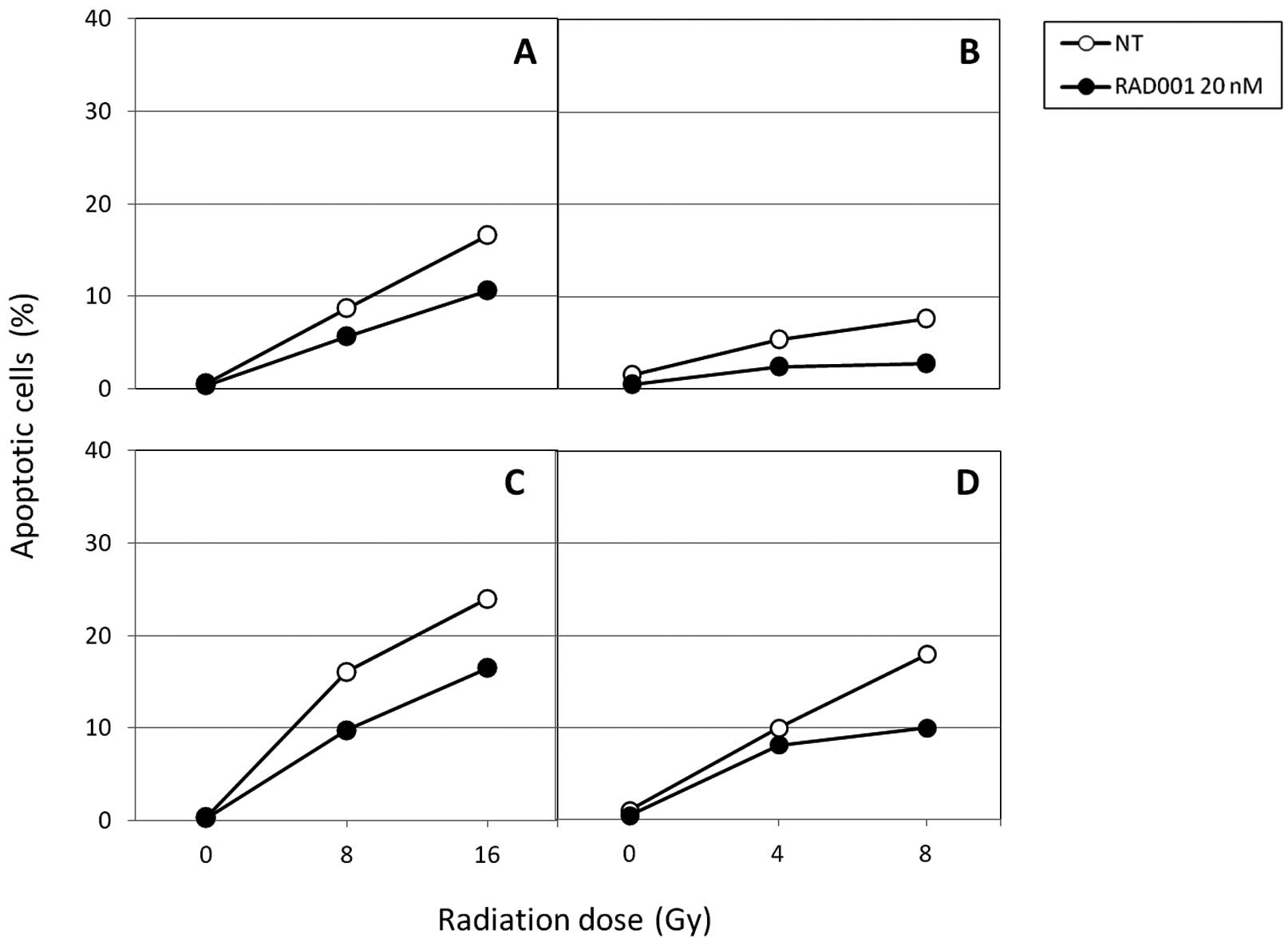
Effect on cell death induction
In further experiments, we attempted to identify the
types of cell death caused by the association between RAD001 and
radiation, and to quantify them. The percentage of cells undergoing
apoptosis and autophagy were determined over a period of 6 days
following irradiation. Results shown in Fig. 3 indicate that apoptosis can be
detected 2 days following irradiation provided by either fast
neutrons or photons but at low levels. Six days after exposure to
the radiation, the percentage of apoptotic cells was higher, but
did not exceed 25% in the control cells. Neutrons were no more
efficient than photons at inducing apoptosis in SK-Hep1, confirming
a previous study (14). In
RAD001-treated cells, regardless of the radiation, apoptosis was
reduced at different doses.
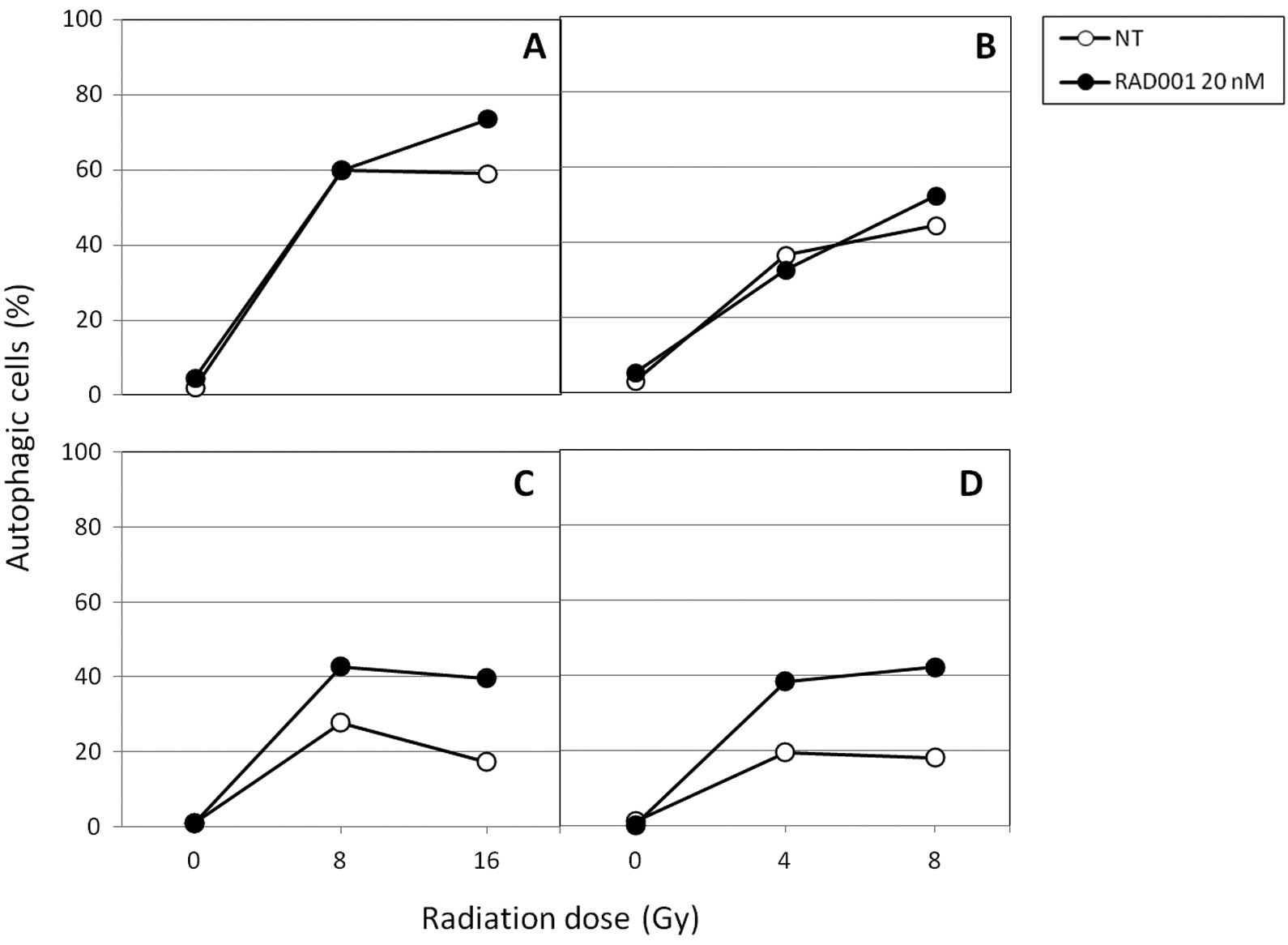
Furthermore, we tested the capacity of RAD001 to
modify autophagy. We used a 488 nm excitable fluorescent reagent
(Cyto-ID™ Autophagy detection kit) that allows a convenient
quantification of autophagic cells by flow cytometry. In
preliminary experiments, we validated the ability of this assay to
record autophagy, by comparing results with other methods, such as
GFP-LC3 expression in autophagosomes and electron microscopy. As
shown in Fig. 4A, photon-induced
autophagy levels were high 2 days following irradiation in
untreated cells, since 60% of cells were positive at 8 Gy and 16
Gy. Six days following irradiation, these values decreased
appreciably, since at the same doses, only 25 and 18% cells were
autophagic at 8 and 16 Gy respectively. In neutron-irradiated cells
(Fig. 4B), the same patterns were
observed according to the dose, but at doses 2-fold lower. In
RAD001-treated cells, a marked increase of autophagy in both
photon- and neutron-irradiated ones was obtained 6 days
post-irradiation, as compared with untreated ones, suggesting that
RAD001 induced a sustained level of autophagy in irradiated cells.
Markedly, when used alone at 20 nM, RAD001 was unable to induce
autophagy at either 2 or 6 days of culture. This lack of autophagy
induction in RAD001-treated, unirradiated cells was confirmed using
green fluorescence protein (GFP)-LC3 staining (not shown). It may
reflect cell type differences, since the same RAD001 concentration
could induce autophagy in U87 glioblastoma cells (not shown), which
is consistent with other recently published data (15).
H2AX foci determination
Radiation lethality results mainly from the
generation of double-strand breaks (DSBs) in DNA. If irradiated
cells fail to repair such lesions, they undergo a death program.
Apoptosis is generally initiated, but autophagy can also be induced
by DNA damage (16). We therefore
compared the capacity of RAD001 to interact with the formation and
the repair of DSBs, using the detection of γH2AX foci as an end
point. This method gives valuable information on the persistence of
DSBs, hence their lack of repair. As expected, the number of
persistent foci at 24 h post-irradiation augmented with the
irradiation dose, and was found to be, at the same dose,
approximately 2-fold higher in neutrons irradiated cells than in
low-LET irradiated ones (Fig. 5A).
In RAD001-treated cells, the numbers of foci were lower than in
untreated cells, albeit not significantly (Fig. 5B).
Discussion
We evaluated the cytotoxic consequences of
combinations between the mTOR inhibitor RAD001 and high and low-LET
radiation in SK-Hep1 HCC cells. We report here that this
cotreatment caused a marked decrease of their growth. We also found
that in irradiated SK-Hep1 cells, death occurred by autophagy
instead of apoptosis, confirming previous results obtained in
vitro (14) and in
vivo, after orthotopic transplantation of these cells into nude
mice (17). As previously noted,
high-LET radiation offers several significant advantages over
low-LET radiation. First of all, they provoke more deleterious
damage in DNA than sparsely ionizing radiation resulting from the
fact that high-LET particles produce dense ionization along their
trajectories, causing clustered and complex damage to DNA, known as
‘locally multiple damaged sites’ (LMDS) and also spoiling
intracellular structures (4,18).
Beyond a given threshold of LMDS, the capacity of DNA repair
machinery is overwhelmed, leading to cell death. As a consequence,
high-LET radiation can inactivate or kill malignant cells more
effectively than low-LET radiation, by inducing premature
senescence, apoptosis, necrosis and autophagy (4).
Therefore, it was interesting to compare the effects
of these two types of radiation when combined with RAD001. As
expected, at the same physical dose, neutrons were found to be more
efficient than photons at inducing autophagy and decreasing
proliferation.
In fact, we previously reported that autophagy
principally accounted for the loss of cell survival in HCC
(14,17) and glioblastoma (GBM) (19) neutron-irradiated cell lines alone,
or associated with oxaliplatin. Currently, whether autophagy
constitutes a mode of protection or, conversely, contributes to
cell death is not firmly recognized, and the consequences of its
induction by IR in radiation therapy are still actively debated
(20). However, it must be pointed
out that autophagy implies a series of progressive cellular
processes, and that cell death is induced by an excessive and
extreme form of autophagy (21).
Thus, possibly more than with apoptosis, which is an all or nothing
phenomenon, the pharmacological modulation of autophagy might lead
to a differential effect of radiation between normal and malignant
cells. Another aspect of autophagy induction in tumor cells can be
envisioned. Indeed, it has recently been reported that
autophagosomes could play a key role in the presentation of tumor
antigens, and that autophagosomes were efficient carriers for
priming CD8 lymphocytes (22). On
the other hand, it has been pointed out that local radiation
therapy could inhibit tumor growth through the generation of
tumor-specific cytotoxic T lymphocytes (CTL)(23). Thus, we could assume that, by
inducing autophagy instead of apoptosis, radiation could exert its
action by activating the immune defenses of the organism. Clearly,
in cultured cells, such a mechanism cannot account for the
radio-sensitizing effect. However, the importance of mTOR
inhibition in enhancing radiation-induced autophagy has already
been emphasized (24). The
efficacy of RAD001 at augmenting the sensitivity of malignant cells
to radiation has been reported for several types of cancer,
including lymphoma (25) and
pituitary adenoma (26). The
inhibition of mTOR has also been shown to enhance the
radiosensitivity of some, but not all, tumor cell lines. For
instance, an increase of radiosensitivity by mTOR inhibition has
been observed in MCF-7 and MDA-MB-231, two human breast cancer cell
lines (27). In HepG2-R, a
radioresistant subline derived from the HCC HepG2 cell line, the
enhancement of autophagy by rapamycin could also result in
radiosensitization and it was suggested that insufficient
IR-induced autophagy may account for the radioresistance of these
cells (28). However, this
assumption cannot be extrapolated to all cancer cell lines since,
in some cases, the inhibition of mTOR failed to significantly
affect the radiation response. For example, a lack of
radiosensitizing effect of rapamycin was noted in the GBM cell
lines U87 and SKMG-3, as addition of this drug to the cells 24 h
prior to irradiation had no effect on their clonogenic survival
(29). In the mouse GL261 glioma
cell line also, pretreatment with either rapamycin (100 nM) or
RAD001 (5 nM) for 1 h before irradiation did not significantly
enhance cell death over that caused by IR alone, as demonstrated by
clonogenic assays in vitro (30). Recently, evidence was provided that
inhibition of mTOR may also decrease rather than increase the
radiosensitivity of tumor cells in vitro. This was shown in
the case of the HeLa cervical adenocarcinoma cell line, after
pretreatment for 3 or 24 h with rapamycin (50 nM) followed by IR
exposure and evaluation in a clonogenic cell survival assay.
Notably, the increased radioresistance was only observed when
rapamycin was added to the cells before irradiation and not if it
was added at the same time (31).
Whether damage to DNA is involved in the
radiosensitizing effect of RAD001 should also be considered.
Indeed, rapamycin and RAD001 have been demonstrated to enhance the
sensitivity of the HCC cell line Hep3B to cisplatin (32), which is known to exert cytotoxicity
by inducing lesions in DNA. Moreover, rapamycin has recently been
reported to suppress DSB repair in MCF-7 breast cancer cells
(33). However, our results
clearly indicate that the number of persistent DSBs after
irradiation is not substantially altered in RAD001-treated SK-Hep1
cells, indicating that the drug is unlikely to affect the
efficiency of DNA repair in these cells. The balance between the
events leading to a defined cell death program, the PI3K/Akt/mTOR
pathways and the DNA damage repair pathways may also account for
differences between cell lines. As these two pathways can vary
according to the mutational status of the tumor cell lines, further
studies using different tumor cell types are required to clarify
the relationship between mTOR inhibition and the initiation of a
specified form of cellular demise in irradiated cells.
In summary, the present data provide evidence that
the association of RAD001 with high-LET radiation is effective in
reducing the growth of HCC cells. They also suggest that a
reinforcement of autophagy contributes to this effect. Thus, it
should be determined whether RAD001, in combination with high-LET
radiation, induced a sustained level of autophagy in other HCC cell
lines as well as in tumor cells from other origins. Since RAD001 is
well tolerated in patients with advanced HCC (34), its association with high-LET
radiation merits further investigation in order to reach potential
clinical evaluation.
Acknowledgements
We thank Dr Francis J. Dumont for
reviewing the manuscript.
References
|
1
|
Orecchia R, Krengli M, Jereczek-Fossa BA,
Franzetti S and Gerard JP: Clinical and research validity of
hadrontherapy with ion beams. Crit Rev Oncol Hematol. 51:81–90.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Finn RS: Development of molecularly
targeted therapies in hepatocellular carcinoma: where do we go now?
Clin Cancer Res. 15:390–397. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pommier P, Hu Y, Baron MH, Chapet O and
Balosso J: Particle therapy: carbon ions. Bull Cancer. 97:819–829.
2010.PubMed/NCBI
|
|
4
|
Hamada N, Imaoka T, Masunaga S, et al:
Recent advances in the biology of heavy-ion cancer therapy. J
Radiat Res. 51:365–383. 2010. View Article : Google Scholar
|
|
5
|
Dumont F, Altmeyer A and Bischoff P:
Radiosensitising agents for the radiotherapy of cancer: novel
molecularly targeted approaches. Expert Opin Ther Pat. 19:775–799.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Begg AC, Stewart FA and Vens C: Strategies
to improve radiotherapy with targeted drugs. Nat Rev Cancer.
11:239–253. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Abraham RT and Gibbons JJ: The mammalian
target of rapamycin signaling pathway: twists and turns in the road
of cancer therapy. Clin Cancer Res. 13:3109–3114. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chiong E, Lee IL, Dadbin A, et al: Effects
of mTOR inhibitor everolimus (RAD001) on bladder cancer cells. Clin
Cancer Res. 17:2863–2873. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dancey J: mTOR signaling and drug
development in cancer. Nat Rev Clin Oncol. 7:209–219. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kudo M: mTOR inhibitor for the treatment
of hepatocellular carcinoma. Dig Dis. 29:310–315. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gueulette J, Slabbert JP, Bischoff P,
Denis JM, Wambersie A and Jones D: Fast neutrons: Inexpensive and
reliable tool to investigate high-LET particle radiobiology. Rad
Meas. 45:1414–1416. 2010. View Article : Google Scholar
|
|
12
|
Riccardi C and Nicoletti I: Analysis of
apoptosis by propidium iodide staining and flow cytometry. Nature
Protocols. 1:1458–1461. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu B, Wu Y, Shen L, et al: Two-dose-level
confirmatory study of the pharmacokinetics and tolerability of
everolimus in Chinese patients with advanced solid tumors. J
Hematol Oncol. 13:4–3. 2001.PubMed/NCBI
|
|
14
|
Altmeyer A, Jung AC, Ignat M, et al:
Pharmacological enhancement of autophagy induced in a
hepatocellular carcinoma cell line by high-LET radiation.
Anticancer Res. 30:303–310. 2010.PubMed/NCBI
|
|
15
|
Nyfeler B, Bergman P, Triantafellow E, et
al: Relieving autophagy and 4EBP1 from rapamycin resistance. Mol
Cell Biol. 31:2867–2876. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rodriguez-Rocha H, Garcia-Garcia A,
Panayiotidis MI and Franco R: DNA damage and autophagy. Mutat Res.
3:158–166. 2011. View Article : Google Scholar
|
|
17
|
Altmeyer A, Ignat M, Denis JM, Messaddeg
N, Gueulette J, Mutter D and Bischoff P: Cell death after high-LET
irradiation in orthotopic human hepatocellular carcinoma in vivo.
In vivo. 25:1–9. 2011.PubMed/NCBI
|
|
18
|
Hada M and Georgakilas AG: Formation of
clustered DNA damage after High-LET irradiation: a review. J Radiat
Res. 49:203–210. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Benzina S, Altmeyer A, Malek F, et al:
High-LET radiation combined with oxaliplatin induces autophagy in
U-87 glioblastoma cells. Cancer Lett. 264:63–70. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zois CE and Koukourakis MI:
Radiation-induced autophagy in normal and cancer cells: towards
novel cytoprotection and radiosensitization policies? Autophagy.
5:442–450. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Todde V, Veenhuis M and van der Klei IJ:
Autophagy: Principles and significance in health and disease.
Biochim Biophys Acta. 1792:3–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Y, Wang LX, Pang P, et al:
Tumor-derived autophagosomes vaccine: mechanism of
cross-presentation and therapeutic efficacy. Clin Cancer Res.
17:7047–7057. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Takeshima T, Chamoto K, Wakita D, et al:
Local radiation therapy inhibits tumor growth through the
generation of tumor-specific CTL: its potentiation by combination
with Th1 cell therapy. Cancer Res. 70:2697–2706. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jabouin JJ, Shinohara ET, Moretti L, Yang
ES, Kaminski JM and Lu B: The role of mTOR inhibition in augmenting
radiation induced autophagy. Technol Cancer Res Treat. 6:443–447.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Saunders P, Cisterne A, Weiss J, Bradstock
KF and Bendall LJ: The mammalian target of rapamycin inhibitor
RAD001 (everolimus) synergizes with chemotherapeutic agents,
ionizing radiation and proteasome inhibitors in pre-B acute
lymphocytic leukemia. Haematologica. 96:69–77. 2011. View Article : Google Scholar
|
|
26
|
Sukumari-Ramesh S, Singh N, Dhandapani KM
and Vender JR: mTOR inhibition reduces cellular proliferation and
sensitizes pituitary adenoma cells to ionizing radiation. Surg
Neurol Int. 2:222011. View Article : Google Scholar
|
|
27
|
Albert JM, Kim KW, Cao C and Lu B:
Targeting the Akt/mammalian target of rapamycin pathway for
radiosensitization of breast cancer. Mol Cancer Ther. 5:1183–1189.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kuwahara Y, Oikawa T, Ochiai Y, et al:
Enhancement of autophagy is a potential modality for tumors
refractory to radiotherapy. Cell Deat Dis. 2:1–11. 2011.PubMed/NCBI
|
|
29
|
Eshleman JS, Carlson BL, Madek AC, Kastner
AD, Shide L and Sarkaria JN: Inhibition of the mammalian target of
rapamycin sensitizes U87 xenografts to fractionated radiation
therapy. Cancer Res. 15:7291–7297. 2002.PubMed/NCBI
|
|
30
|
Shinohara ET, Cao C, Niermann K, Mu Y,
Zeng F, Hallahan DE and Lu B: Enhanced radiation damage of tumor
vasculature by mTOR inhibitors. Oncogene. 24:5414–5422. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bandhakavi S, Kim YM, Ro SH, et al:
Quantitative nuclear proteomics identifies mTOR regulation of DNA
damage response. Mol Cell Proteomics. 9:403–414. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tam KH, Yang ZF, Lam CT, Pang RW and Poon
RT: Inhibition of mTOR enhances chemosensitivity in hepatocellular
carcinoma. Cancer Lett. 273:201–209. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen H, Vanderwaal RP, Feng Z, et al: The
mTOR inhibitor rapamycin suppresses DNA double-strand break repair.
Radiat Res. 175:214–224. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhu AX, Abrams TA, Miksad R, et al: Phase
1/2 study of everolimus in advanced hepatocellular carcinoma.
Cancer. 117:5094–5102. 2011. View Article : Google Scholar : PubMed/NCBI
|