Introduction
Notch signaling pathway is important for many types
of cell fate determinations and essential for proper embryonic
development. Notch pathway comprises a family of transmembrane
receptors and their ligands, negative and positive modifiers, and
transcription factors. Notch receptors (Notch 1–4) and two groups
of ligands, Jagged and Delta-like, have been identified in mammals
(1). During maturation, Notch
precursors are cleaved to produce an extracellular subunit (NEC)
and a transmembrane subunit (NTM) (2). Binding of Notch ligands to receptors
leads to two cleavages involving a metal-loprotease and a
presenilin-dependent protease and a release of the receptor
intracellular region (ICN), which translocates to nucleus to
activate transcription of a group of downstream genes and thus
controls various biological events (3).
Since Notch signaling affects cell differentiation,
proliferation and survival, recent studies have focused on its
effects on malignant tumors and revealed its complex roles in
cancer. In most malignant tumors, Notch plays an oncogenic role.
Notch1 is identified as an oncogene responsible for acute T cell
lymphoblastic leukemia (T-ALL) (4), and its oncogenic effects are also
found in glioma, primary melanoma and pancreatic cancer (5–7).
Notch2 is involved in the aberrant expression of CD23 in B-cell
chronic lymphocytic leukemia and related to the failure of
apoptosis (8). Notch3 contributes
to the growth of human lung cancers by its effect on
mitogen-activated protein kinase pathway (9). Notch4 is found to correlate with
Ki67, and GSI treatment arrests the growth of breast cancer cells
(10). However, in some
circumstances, Notch is found to play a suppressive role. In mouse
skin, Notch1 functions as a tumor suppressor (11). A recent report further demonstrates
that inactivation of Notch1 and Notch2 simultaneously induces the
development of a severe form of atopic dermatitis, which is
accompanied by an increase in immature myeloid populations in the
bone marrow and spleen, and the increase is revealed to be cell
non-autonomous and caused by dramatic microenvironmental
alterations (12). Moreover, in
human breast cancer, Notch2 expression decreases as tumor grade
increases and Notch2 signaling suppresses tumor growth (13).
In research on the liver, reports show that Notch
signaling plays an important role in hepatoblast differentiation
and all four Notch receptors are expressed in adult human liver
(14,15). Most significant upregulation of
Notch1, Notch2, Notch3, Delta1 and Jagged1 is observed in a
hepatectomy (AAF/PHx) model (16).
Notch1, ICN1 and Jagged1 proteins are all upregulated and
Notch1/Jagged1 signaling is activated during rat liver regeneration
(17). Notch3, Notch4, Jagged1,
Delta1 and Hairy Enhancer of Split-1 (HES-1) are all expressed in
HCC (18,19). And the levels of Notch1, Jagged1,
and HES-1 expression are increased in HCC samples relative to the
adjacent HCC-free liver tissue (20). Our previous study also shows that
Notch1, Notch4 and Jagged1 are expressed higher in HCC than in
adjacent nontumor tissue (21,22).
All these studies suggest that Notch signaling might play a role in
the development of HCC, however, investigations are lacking. Since
Notch1 are demonstrated to be upregulated in HCC relative to
adjacent nontumor liver in our previous experiment, we investigated
the effects of Notch1 activation on human HCC HepG2 and SMMC7721
cell growth and proliferation in this study. We found that Notch1
activation contributed to the progression of HCC by enhancing cell
growth activity and promoting cell cycle progression. Our data
define a novel role for Notch1 in HCC progression and indicate that
Notch1 might be a potential therapeutic target for the treatment of
HCC.
Materials and methods
Cell lines and drugs
Five human HCC cell lines, SMMC7721, HepG2, HHCC,
9724, 9204 were obtained from the Cell Bank of the Chinese Academy
of Sciences (Shanghai, China) and the Lab Animal Center of the
Fourth Military Medical University (Xi’an, China). These cells were
cultured at 37°C in Dulbecco’s modified Eagle’s medium (DMEM)
(Gibco, Tulsa, OK, USA) supplemented with 10% fetal calf serum in
5% CO2-humidified air. The plasmid of
pcDNA3.1/ICN1-myc-His(-)C was a gift from Professor Tom Kadesch of
University of Pennsylvania School of Medicine (23). ICN1 included 1760 to 2556 amino
acid positions of full-length human Notch1 and was cloned into
pcDNA3.1/myc-His(-)C at the XhoI and KpnI sites.
Generation of HepG2 cell line that stably expressed ICN1 was
accomplished through transfection with pcDNA3.1/ICN1-myc-His(-)C
using Lipofectamine™ 2000 reagent (Invitrogen, Carlsbad, CA, USA),
following the manufacturer’s protocol. Transgene expressed cells
were selected and maintained with 600 μg/ml G418 (Gibco). Bulk
cultured transfectants were picked and verified by measurement of
myc-tagged ICN1 protein of exact molecular weight (MW), which were
named HepG2-ICN1. Mock transfection was done by transfection of
pcDNA3.1(-) vector (Invitrogen), which was named HepG2-pc. siRNAs
for Notch1 were chemically synthesized (Invitrogen) and the target
sequences were: si1, TATCGACGATTGTCCAGGA; si2, CAACA
ATGAGTGTGAATCC; si3, TCCGAGGACTATGAGAGCT. The siRNAs were ligated
with pSilencer 3.1-H1 neo vector (Ambion, Austin, TX, USA)
respectively, and then pSilencer 3.1-Notch1/RNAi was constructed
and confirmed by DNA sequencing. Control siRNA was processed as
above and pSilencer 3.1-con/RNAi was constructed. Using
Lipofectamine™ 2000 reagent, the pSilencer 3.1-Notch1/RNAi or
pSilencer 3.1-con/RNAi was transfected, respectively, into SMMC7721
cells following the manufacturer’s protocol. Transgene cells were
selected and maintained with 800 μg/ml G418 (Gibco). Bulk cultured
transfectants were picked and verified by measurement of Notch1
protein levels, named 7721-si. Mock transfectants were named
7721-con. For Notch signaling inhibition, HepG2 and SMMC7721 cells
were treated with N-(N-(3,5-difluorophenacetyl-L-alanyl))-S-phenyl
glycine t-butyl ester (DAPT) (Calbiochem, Darmstadt, Germany) at
different final concentrations (2.5, 5 and 10 μM). 5 μM dimethyl
sulfoxide (DMSO) was used as drug control.
Western blot analysis
Immunoblotting of cellular proteins from SMMC7721,
HepG2, HHCC, 9724, 9204 cells was performed using anti-Notch1 goat
polyclonal antibody (pAb) (Santa Cruz Biotechnologies, Santa Cruz,
CA, USA, 1:300). The anti-Notch1 pAb is raised against a peptide
mapping at the C-terminus of Notch1 of human origin (sc-6014).
Identification of 7721-si or HepG2-ICN1 was performed with the
anti-Notch1 pAb or anti-myc monoclonal antibody (mAb) (1:300, Cell
Signaling Technology, Boston, MA, USA). Nuclear protein extracts of
SMMC7721 or HepG2 treated with 5 μM DAPT or 5 μM DMSO were prepared
with NE-PER Nuclear and Cytoplasmic Extraction Reagents kit (Pierce
Biotechnology, Rockford, IL, USA). The level of ICN1 from the
nuclear protein was detected with the anti-Notch1 pAb. The
incubation of primary antibodies were followed by peroxidase
coupled anti-goat or anti-mouse IgG. Proteins were then visualized
by enhanced chemiluminescence (ECL). Western blot for β-actin or
Histone H3 (1:200, BioLegend, San Diego, CA, USA) was used as
internal sample. Autoradiograms were quantified by densitometry
(software: Bio Image IQ). Relative protein levels were calculated
by referring them to the amount of β-actin or Histone H3 protein.
Mean values from three independent experiments were recorded as the
results.
Luciferase assay
pGa981-6, a reporter gene plasmid which contained a
hexamerized 50 bp Epstein-Barr virus nuclear antigen 2 response
element (EBNA2RE) of the TP-1 promotor in front of the luciferase
gene, was strictly dependent on recombination signal binding
protein-Jκ (RBP-J) (24). pRL-TK
(Promega Corporation, Madison, WI, USA) was co-transfected as an
internal control for transfection efficiency. A negative control
plasmid (neg-pGa981-6) was constructed by replacing EBNA2RE with an
irrelevant DNA segment. For luciferase assay, pGa981-6 and pRL-TK
were co-transfected into the above five HCC cell lines, and also
the HepG2-ICN1, HepG2-pc, 7721-si and 7721-con cell lines, with
Lipofectamine™ 2000 (Invitrogen). Neg-pGa981-6 replacing pGa981-6
was used as negative controls. After 48 h, co-transfected cells
were lysed and luciferase assays were performed in Luminometer
TD-20/20 (Turner Designs, Sunnyvale, CA, USA). All assays were
repeated three times.
MTT assay
The in vitro growth rates of HepG2-ICN1 and
7721-si cells were measured using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
method. Briefly, cells (2×103) were seeded into 96-well
plates, respectively, and cultured in DMEM supplemented with 5%
fetal calf serum. After 1, 2, 3, 4, 5, 6, 7 days, the culture
medium was removed and 20 μl of 5 mg/ml MTT was added. After 4 h,
the supernatant was discarded and 100 μl DMSO was added to each
well. The mixture was shaken at room temperature for 10 min and
measured at 490 nm using a microplate reader (Bio-Rad Laboratories,
Hercules, CA, USA). HepG2-pc and 7721-con cells were used as
controls. The growth inhibitory rate was calculated as: (control A
value-treated A value)/control A value × 100%, where control A
value and treated A value are the average absorbance of three
parallel experiments from treated and control groups, respectively.
DAPT-treated HepG2 and SMMC7721 cells were also performed as above.
DAPT of different concentrations were added to the cells every day.
DMSO was used as control. The cells in this experiment were also
used in the following procedures.
Plate colony-forming assay
Cells mentioned above were seeded at a density of
500 cells/dish in 6-well plates and cultured in DMEM supplemented
with 5% fetal calf serum. The culture medium was replaced by fresh
medium or medium with drug (for DAPT treatment) everyday. After 2
weeks of incubation, cell colonies were washed and fixed with 100%
methanol. Finally, cells were stained with Gimsa dye and colonies
containing more than 50 cells were counted. Each experiment was
done in triplicates.
Soft agar colony-forming assay
Anchorage-independent cell proliferation was
determined by a soft agar assay, 24-well plates were coated with
0.5% of bottom agar solution, and 2×103 cells per well
were embedded into 0.3% top agar gel containing DMEM supplemented
with 10% fetal calf serum, respectively. The cell suspensions were
added over the precoated bottom agar gel in the 24-well plates. The
dishes were examined with a vertical microscope for colony
formation after a 2-week incubation period. For drug treatment,
DAPT of different concentrations were added to the bottom agar and
top agar solution and 5 μM DMSO was used as drug control. Colonies
of >75 mm were counted. Each experiment was done in
triplicates.
Tumorigenicity in nude mice
BALB/c (nu/nu) nude mice (4–6 week old; the Lab
Animal Center of the Fourth Military Medical University) were
injected subcutaneously into the right flank with 4×106
HepG2-ICN1 or 7721-si cell suspension in DMEM. Five animals were
used in each group. After 4 weeks, mice were euthanized, and the
tumors were removed and weighted. HepG2-pc and 7721-con cells were
inoculated as controls. This experiment was in accordance with
approved ethical standards of the responsible committee of the
University and with the Declaration of Helsinki (1975).
PI staining and flow cytometry (FCM)
Cells (1×106) were seeded in 6-well
plates and incubated overnight. Then cells were harvested, fixed
and resuspended in propidium iodide (PI) solution. Samples were
then analyzed for their DNA content by flow cytometer (FCM)
(Coulter Co., Hialeah, FL, USA). For drug treatment, 5 μM DAPT was
added to the culture solution and 5 μM DMSO was used as control
(this drug concentration and control were employed in the following
experiments). Each experiment was done in triplicates.
Annexin V binding assay
Cells undergoing early apoptosis were identified by
binding of Annexin V to membrane phosphatidylserine and assayed
using fluorescein isothiocyanate (FITC)-conjugated Annexin V
(Pharmingen, San Diego, CA, USA) according to the manufacturer’s
instructions. Briefly, cells were seeded in 6-well plates and
incubated overnight. Then cells were harvested, added with PI
(final concentration 1 μg/ml) and Annexin V (final concentration 1
μg/ml) successively, and then analyzed by flow cytometry. Each
experiment was done in triplicates.
TUNEL assay
Terminal deoxynucleotidyl transferase biotindUTP
nick end labeling (TUNEL) staining was performed using an in
situ apoptosis detection kit (Dingguo Inc, Beijing, China). The
positive cells were detected with nitroterazolium blue chloride
(NBT)/5-bromo-4-chloro-3-indolyl phosphate p-toluidine salt (BCIP)
reagent and counterstained with nuclear fast red dye, and then
examined by light microscopy. The total number of apoptotic cells
in 5 randomly selected fields was counted.
TEM assay
The above cells were fixed, dehydrated and embedded.
Then they were sectioned and stained in 1% uranyl acetate and for 5
min with lead citrate. The cell ultrastructure was assessed by
transmission electron microscopy (TEM) at 4,000-fold
magnification.
Statistical analysis
Statistical analysis was performed using SPSS
software (version 13.0, SPSS, Chicago, IL, USA). Two-sided
Student’s t-test was used. P<0.05 was considered as
statistically significant.
Results
Identification of 7721-si and HepG2-ICN1
cells and detection of activity of Notch signaling
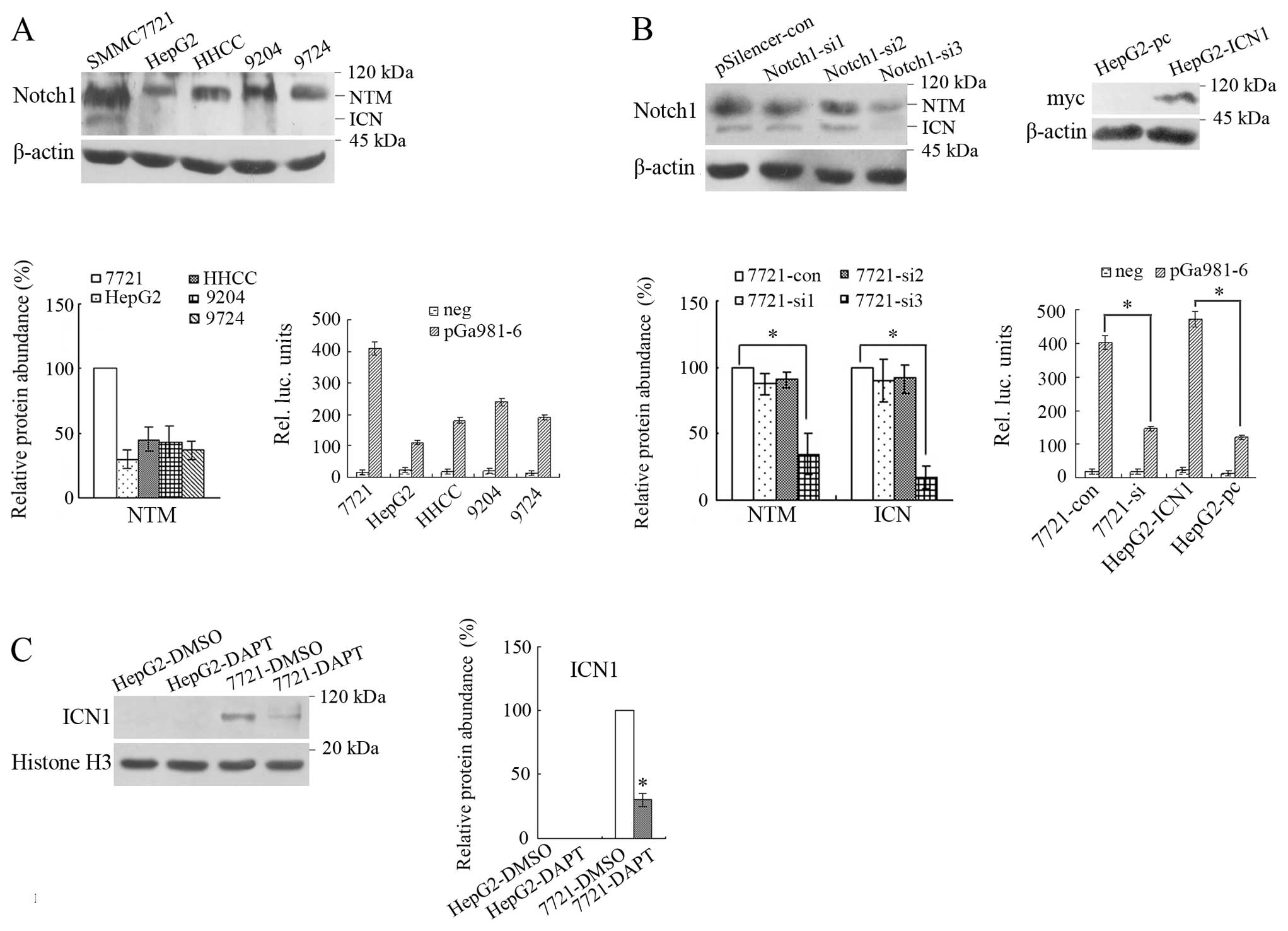
Firstly, we examined the expression of Notch1 in
five human HCC cell lines and found that SMMC7721 had the
relatively high while HepG2 the relatively low expression of this
molecule, and the active ICN1 (the lower molecular band) was most
abundant in SMMC7721 cells while undetectable in the other four
cell lines (25). To further
examine the activity of Notch signaling in the above cells, Notch
signaling reporter plasmid pGa981-6 and the internal control
plasmid pRL-TK were co-transfected into the above five cell lines.
The results from luciferase assay showed that the activity of Notch
signaling was relatively high in SMMC7721 while relatively low in
HepG2 (Fig. 1A). Thus, these two
cell lines were used as experimental targets in this study. After
transfection with three pairs of siRNAs, it was found that Notch1
in SMMC7721 cells was significantly knocked down by the third siRNA
(P<0.05), which showed the 7721-si cells was successfully
achieved. Besides, after transfection with
pcDNA3.1/ICN1-myc-His(-)C, the expression of myc-tagged ICN1 in
transfected HepG2 cells was detected by western blot and HepG2-ICN1
cells were successfully constructed. We also examined the activity
of Notch signaling in these transfectants and found that Notch
signaling was significantly inhibited by RNAi of Notch1 and was
activated by transfection with ICN1 (Fig. 1B). To compare the inhibition
effects of DAPT on ICN1 between HepG2 and SMMC7721 cells, these two
cell lines were treated with DAPT. The result showed that ICN1 in
the nuclear protein of SMMC7721 treated with 5 μM DAPT was
significantly reduced compared with treated with 5 μM DMSO
(P<0.05), while no ICN1 was detected in HepG2 treated with DMSO
or DAPT (Fig. 1C).
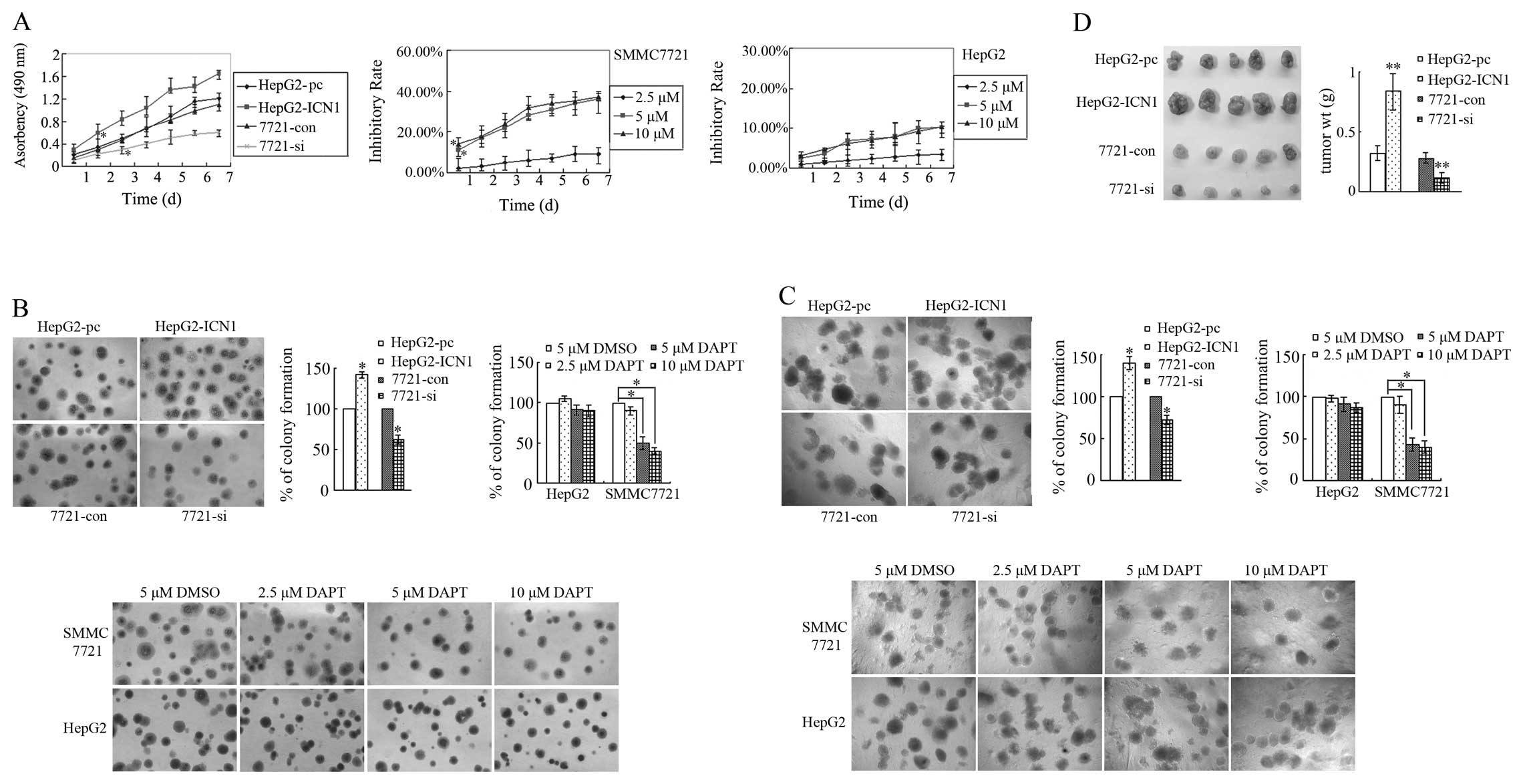
Notch1 activation promotes HCC cell
growth and proliferation
Cell growth and proliferation in vitro was
detected by MTT, plate colony-formation and soft agar assay. The
MTT results showed that the growth of HepG2-ICN1 cells was
significantly faster than that of HepG2-pc cells (since the 2nd
day) (P<0.05), and the growth of 7721-si cells were
significantly slower than that of 7721-con cells (since the 3rd
day) (P<0.05). Since DAPT could significantly reduce the levels
of ICN1 in SMMC7721 cells, cell growth in HepG2 and SMMC7721
treated with DAPT was also examined by the above assays. Compared
with DMSO treatment, no significant cell inhibition was observed
when DAPT was applied at final concentration of 2.5 μM, however,
potent inhibitory effects of DAPT on SMMC7721 cells were observed
at higher concentrations (5 and 10 μM) at the 1st day (P<0.05,
P<0.05, Fig. 2A), and the
degree of inhibition was positively correlated with the exposure
time. It was also observed that the inhibitory rate of 10 μM was
similar with 5 μM DAPT, which showed that 5 μM was a suitable final
concentration. For HepG2 cells, DAPT at all tested concentrations
did not significantly inhibit cell growth compared with DMSO, which
might result from the low activity of Notch signaling in this cell
line.
Cell growth was also detected by colony-formation
and soft agar assay. As showed in Fig.
2B and C, the number of cell colonies of HepG2-ICN1 cells was
much greater than that of HepG2-pc cells (P<0.05), and 7721-si
cell colonies were significantly less than 7721-con cells
(P<0.05). Compared with DMSO, 5 or 10 μM DAPT significantly
reduced capacity of SMMC7721 cells for single cell colony formation
or anchorage-independent cell proliferation (both P<0.05).
Consistent with the MTT result, the inhibitory effect of 10 μM DAPT
was not stronger than 5 μM DAPT. DAPT at all tested concentrations
did not significantly inhibit cell growth of HepG2 cells compared
with DMSO. The above results showed that Notch1 activation promoted
the growth and proliferation of HCC cells, while suppression of
Notch1 activation inhibited HCC cells growth.
Cell growth in vivo was examined by
tumorigenicity in nude mice. All nude mice injected with
HepG2-ICN1, HepG2-pc, 7721-si or 7721-con cells generated tumors in
their right flank after four weeks. The tumors from nude mice
injected with HepG2-ICN1 cells were heavier than those from
HepG2-pc cells (P<0.01), and the mean weight from nude mice
injected with 7721-si cells was less than that from 7721-con cells
(P<0.01, Fig. 2D).
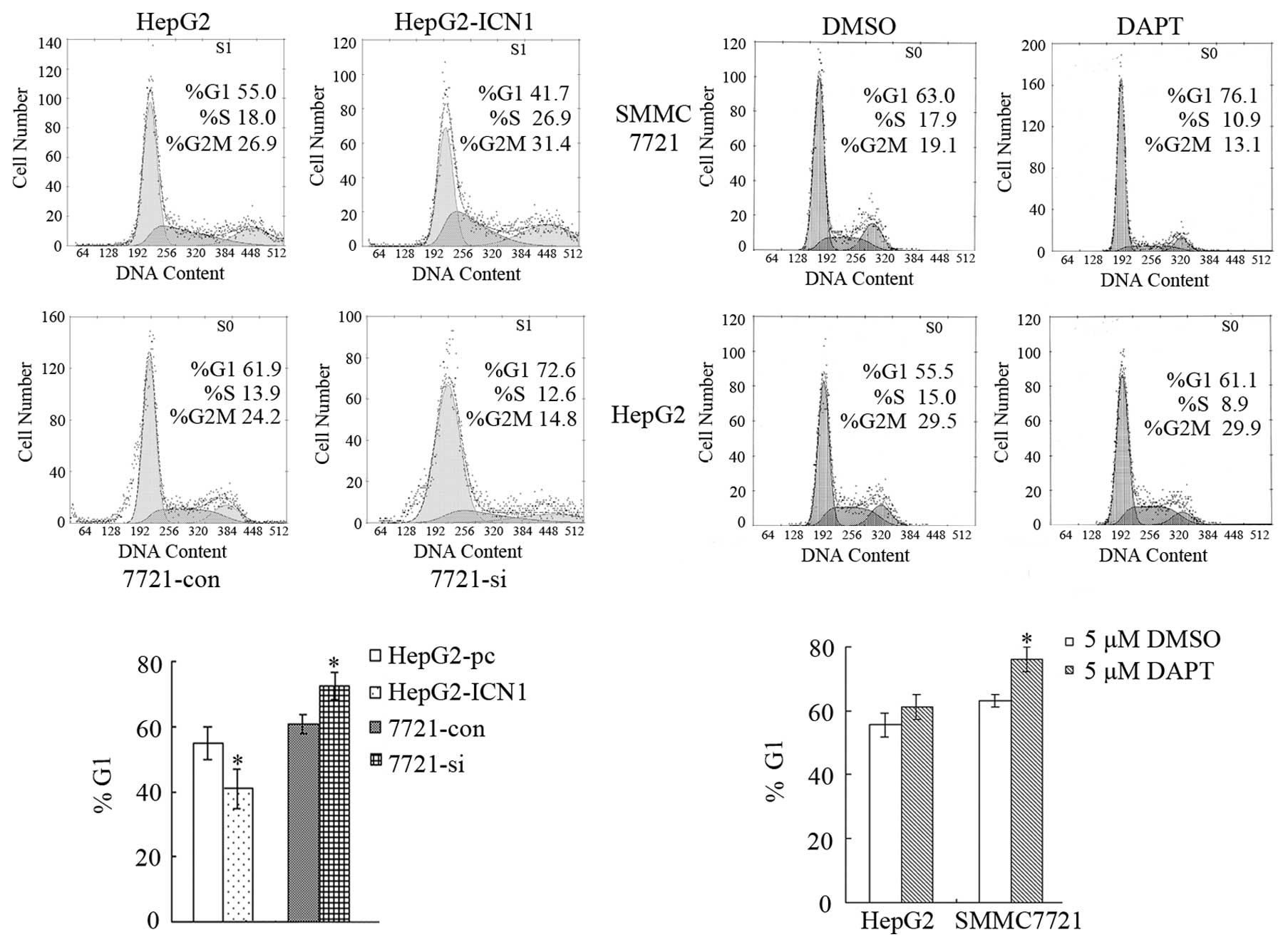
Notch1 activation promotes HCC cell cycle
progression
The effects of Notch1 activation on cell cycle
progression was examined by PI staining and flow cytometric
analysis. DNA histograms of data from FCM analysis showed that the
number of HepG2-pc cells in G1 phase was greater than that of
HepG2-ICN1 cells (P<0.05), and the number of 7721-si cells in G1
phase was greater than that of 7721-con cells (P<0.05). Since 5
μM DAPT could reduce ICN1 and inhibit cell growth in SMMC7721 cells
effectively, this drug condition was used in the following
experiment. Compared with DMSO, the G1 phase cells in SMMC7721
treated with 5 μM DAPT increased significantly (P<0.05), while
neither DAPT nor DMSO could influence HepG2 cell cycle distribution
(Fig. 3). This result showed that
the promotion of cell cycle might be a reason for the stimulation
of cell growth in HepG2 and SMMC7721 cells by Notch1
activation.
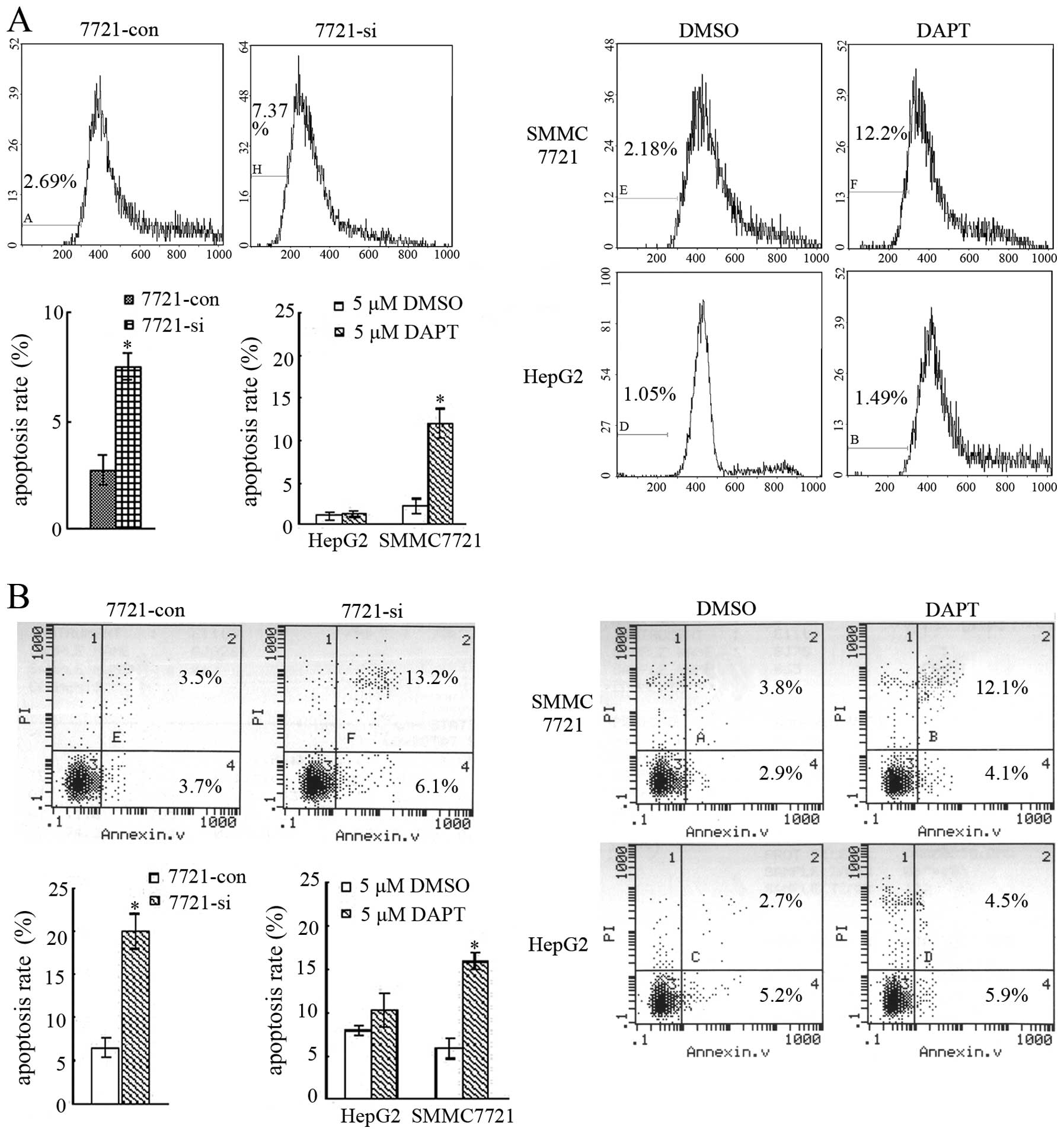
Suppression of Notch1 activation induces
HCC SMMC7721 cell apoptosis
Cell apoptosis was examined after Notch1 activation
was suppressed in SMMC7721 cells. Firstly, apoptotic histograms
from PI staining and FCM were analyzed. The result showed that
apoptosis rate of 7721-si cells (7.37%) were much higher than that
of 7721-con cells (2.69%) (P<0.05). The apoptotic rate of
SMMC7721 increased significantly after treated with 5 μM DAPT
(12.2%) compared with DMSO (2.18%) (P<0.05), whereas, DAPT did
not affect the apoptosis of HepG2 cells significantly (1.05%,
1.49%) (Fig. 4A). Apoptosis was
also assessed by Annexin V and PI double-staining assay. The
percentage of apoptotic cells (Annexin V+/PI-, early stage; Annexin
V+/PI+, late stage) of 7721-si was 19.3%, which was much higher
than 7.2% of 7721-con cells (P<0.05). The percentage of
apoptotic SMMC7721 cells treated with DMSO was 6.7%, and after
treatment with 5 μM DAPT, the percentages of apoptotic cells
increased to 16.2% (P<0.05). For HepG2 cells, drug treatment did
not produce more apoptosis than control (Fig. 4B).
Moreover, apoptotic cell morphological assessment
was also carried on. TUNEL method, which labeled fragmented DNA
in situ, was used to detect apoptosis. NBT/BCIP staining in
our TUNEL experiment stained amethyst apoptotic nucleus and red
normal nucleus. Besides, the ultrastructure of cell apoptosis was
investigated by TEM. A characteristic pattern of apoptosis included
chromosome condensation, nuclear fragmentation, cytoplasmic
organelles closely packed, cell blebbing and emergence of apoptotic
bodies. In contrast, normal cells exhibited membrane and nuclei
with evenly distributed chromatin, as well as intact intracellular
organelles. The result showed that 7721-si had more apoptosis than
7721-con cells (P<0.05). Treatment with 5 μM DAPT induced more
cell apoptosis in SMMC7721 than DMSO of the same dose (P<0.05,
Fig. 4C and D). For HepG2, either
DAPT or DMSO could not lead to apparent cell apoptosis. These
results demonstrated that suppression of Notch1 activation could
lead to SMMC7721 cell apoptosis, which might also be a reason for
the retarded growth of SMMC7721 cells by inhibition of Notch1
activation.
Discussion
HCC is one of the most common malignant tumors in
the world. The molecular mechanisms of HCC have been studied for
many years, and many molecules and signaling pathways are found to
be involved in the development of HCC. Recent studies have revealed
that Notch signaling is not only important in embryonic development
and cell fate determination, but also plays crucial roles in the
carcinogenesis, progression, invasion and neurovascular formation
of many malignant tumors. Among Notch, Notch1 is most
comprehensively investigated. For example, activated Notch1
signaling dramatically induces proliferation and inhibition of
apoptosis in Hodgkin and anaplastic large cell lymphoma (26). Constitutive activation of the
Notch1 pathway enhances primary melanoma cell growth and metastatic
capability, which are mediated by β-catenin (27). Downregulation of Notch1 inhibits
cell growth, contributes to apoptosis and suppresses invasion in
pancreatic cancer cells (5,28).
Up to now, there are several studies reporting the roles of Notch1
in HCC. Notch1 signaling could inhibit growth of human HCC through
induction of cell cycle arrest and apoptosis (29), and Notch1 signaling sensitizes
tumor necrosis factor-related apoptosis-inducing ligand-induced
apoptosis in human HCC cells (30), which indicates that Notch1
signaling might play a negative role in HCC. However,
investigations from other teams come up with different results. A
study shows that activation of Notch1 in the pre-neoplastic foci
might be associated with the progression of HCC in the
diethylnitrosamine-induced hepatocarcinogenesis model (31). Downregulation of Notch1 signaling
by a Notch1 inhibitor curcumin, inhibits tumor growth in human HCC
(32). Besides, a research team
newly reports that abnormal Notch1 expression is strongly
associated with HCC metastatic disease and knockdown of Notch1
reverses HCC tumor metastasis in a mouse model (33).
We examined the expression of Notch1 in five human
HCC cell lines and found that SMMC7721 had the relatively high,
while HepG2 the relatively low expression, and the active ICN1 was
most abundant in SMMC7721 cells while undetectable in the other
four cell lines. Since ICN1 is rapidly degraded after it activates
transcription in the nucleus, it is reasonably undetectable even in
cells that require it for survival, due to its short half-life.
Luciferase assay further showed that the activity of Notch
signaling was relatively high in SMMC7721 while relatively low in
HepG2 cells. Ectopic expression of ICN1 activated Notch signaling
in HepG2, while Notch1 knockdown by RNAi led to inhibition of Notch
signaling in SMMC7721. Although it is deemed that Notch1 is vital
for the survival of mammalian cells, and without Notch, cells will
probably die, the protein expression of Notch1 and activity of
Notch signaling still existed in 7721-si cells of our experiments,
which might support the survival of this transfected cell line.
Moreover, our data showed that Notch1 activation by ectopic
expression of ICN1 promoted the growth and proliferation of HepG2
cells, while suppression of Notch1 activation by RNAi of Notch1
inhibited SMMC7721 cell growth. Since our previous study
demonstrates that DAPT reduces the expression of ICN1 and HES-1 in
SMMC7721 cells (34), we also
compared the inhibition effects of DAPT on ICN1 between HepG2 and
SMMC7721 cells in this study. We found that DAPT could reduce the
level of ICN1 in SMMC7721 cells, while no ICN1 was detected in
HepG2 at all. Further, it was demonstrated that DAPT inhibited
tumor cell growth in SMMC7721 while not HepG2 cells. Thus, our
results indicate an oncogenic effect of Notch1 on HCC, which is
consistent with those recent reports. Our data indicate Notch1 as a
critical player in HCC development and progression, while not
except other Notch molecules involved, since other Notch receptors
and ligands are also expressed in HCC (22,35).
The influence of Notch activation on cell growth is
related to its effects on cell cycle and apoptosis, which has been
proved in a series of malignant tumors. For example, in T-ALL
cells, inhibition of the Notch pathway activity leading to
derepression of retinoblastoma protein Rb and subsequent exit from
the cell cycle (36). In
pancreatic cancer, Notch1 downregulation leads to increasing cell
population in the G0-G1 phase (5).
There are also studies showing the relationship of Notch and
apoptosis. It is reported that GSI induces breast cancer cell
apoptosis (37). GSI treatment
also induces apoptosis of myeloma cells via specific inhibition of
Notch signaling, and enhances sensitivity to chemotherapy (38). Since Notch1 activation was found to
promote HCC cell growth and proliferation in our investigation, we
also examined its effect on cell cycle and apoptosis. Indeed, we
found that Notch1 activation increased HepG2 cell number in the S
and G2M phase, while inhibition of Notch1 activation arrested the
cell cycle of SMMC7721 in the G1 phase. Besides, it was
demonstrated that inhibition of Notch1 activation induced SMMC7721
cell apoptosis. The suppression of cell growth, arrest of cell
cycle and induction of cell apoptosis by inhibition of Notch1
activation in our case is consistent with the roles of Notch1 in
the reported other cancers, and the underlying related molecular
mechanisms remain for further study.
To date, Notch has been considered as a new approach
to overcome cancers since RNAi of Notch1 or GSIs was found to
inhibit malignant transformation and development in many tumors
(39,40). For instance, after phase I clinical
trial of MK-0752 in patients with T-ALL and other leukemias in 2006
(41), phase I trial of this novel
Notch inhibitor for children with refractory central nervous system
malignancies is also currently completed (42). The inhibition of cell growth and
induction of cell apoptosis in SMMC7721 in our investigation also
indicate that the siRNA of Notch1 and specific GSIs might be served
as new drugs for HCC therapy. Considering the discrepant expression
of Notch1 and activity of Notch signaling in different HCC cell
lines, we suppose that this kind of novel therapy should be
individualized, which also needs thorough and careful research.
In conclusion, our study demonstrates that Notch1
activation contributes to tumor cell growth and proliferation
whereas suppression of Notch1 activation inhibits cell growth,
induces arrest of cell cycle and cell apoptosis in human HCC cells.
Our findings refer to Notch1 as a new molecular mechanism in the
development of HCC and support a potential oncogenic role of this
molecule in HCC. Further studies clarifying the composition of
Notch1 signaling and its downstream molecules mediating multiple
steps in HCC progression will contribute to make clear how Notch1
activation regulates HCC and will also be helpful for providing
more support for Notch1 used as a valuable approach for HCC
therapy.
Acknowledgements
We thank Professor Tom Kadesch
(University of Pennsylvania School of Medicine, USA) and Professor
Hua Han (the Fourth Miltary Medical University, China) for their
generous gifts of pcDNA3.1/ICN1-myc-His(-)C and pGa981-6 plasmids,
respectively.
References
|
1.
|
Artavanis-Tsakonas S, Rand MD and Lake RJ:
Notch signaling: cell fate control and signal integration in
development. Science. 284:770–776. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Blaumueller CM, Qi H, Zagouras P and
Artavanis–Tsakonas S: Intracellular cleavage of Notch leads to a
heterodimeric receptor on the plasma membrane. Cell. 90:281–291.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Davis RL and Turner DL: Vertebrate hairy
and enhancer of split related proteins: transcriptional repressors
regulating cellular differentiation and embryonic patterning.
Oncogene. 20:8342–8357. 2001. View Article : Google Scholar
|
|
4.
|
Ellisen LW, Bird J, West DC, Soreng AL,
Reynolds TC, Smith SD and Sklar J: TAN-1, the human homolog of the
Drosophila notch gene, is broken by chromosomal
translocations in T lymphoblastic neoplasms. Cell. 66:649–661.
1991.PubMed/NCBI
|
|
5.
|
Wang Z, Zhang Y, Li Y, Banerjee S, Liao J
and Sarkar FH: Down-regulation of Notch-1 contributes to cell
growth inhibition and apoptosis in pancreatic cancer cells. Mol
Cancer Ther. 5:483–493. 2006. View Article : Google Scholar
|
|
6.
|
Purow BW, Haque RM, Noel MW, Su Q, Burdick
MJ, Lee J, Sundaresan T, Pastorino S, Park JK, Mikolaenko I, Maric
D, Eberhart CG and Fine HA: Expression of Notch-1 and its ligands,
Delta-like-1 and Jagged-1, is critical for glioma cell survival and
proliferation. Cancer Res. 65:2353–2363. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Liu ZJ, Xiao M, Balint K, Smalley KS,
Brafford P, Qiu R, Pinnix CC, Li X and Herlyn M: Notch1 signaling
promotes primary melanoma progression by activating
mitogen-activated protein kinase/phosphatidylinositol 3-kinase-Akt
pathways and up-regulating N-cadherin expression. Cancer Res.
66:4182–4190. 2006. View Article : Google Scholar
|
|
8.
|
Hubmann R, Schwarzmeier JD, Shehata M,
lgarth M, Duechler M, Dettke M and Berger R: Notch2 is involved in
the overexpression of CD23 in B-cell chronic lymphocytic leukaemia.
Blood. 99:3742–3747. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Haruki N, Kawaguchi KS, Eichenberger S,
Massion PP, Olson S, Gonzalez A, Carbone DP and Dang TP:
Dominant-negative Notch3 receptor inhibits mitogen-activated
protein kinase pathway and the growth of human lung cancers. Cancer
Res. 65:3555–3561. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Rizzo P, Miao H, D’Souza G, Osipo C, Yun
J, Zhao H, Mascarenhas J, Wyatt D, Antico G, Hao L, Yao K, Rajan P,
Hicks C, Siziopikou K, Selvaggi S, Bashir A, Bhandari D, Marchese
A, Lendahl U, Qin JZ, Tonetti DA, Albain K, Nickoloff BJ and Miele
L: Cross-talk between notch and the estrogen receptor in breast
cancer suggests novel therapeutic approaches. Cancer Res.
68:5226–5235. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Nicolas M, Wolfer A, Raj K, Kummer JA,
Mill P, van Noort M, Hui CC, Clevers H, Dotto GP and Radtke F:
Notch1 functions as a tumor suppressor in mouse skin. Nat Genet.
33:416–421. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Dumortier A, Durham AD, Piazza MD,
Vauclair S, Koch U, Ferrand G, Ferrero I, Demehri S, Song LL, Farr
AG, Leonard WJ, Kopan R, Miele L, Hohl D, Finke D and Radtke F:
Atopic dermatitis-like disease and associated lethal
myeloproliferative disorder arise from loss of Notch signaling in
the murine skin. PloS One. 5:e92582010. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
O’Neill CF, Urs S, Cinelli C, Lincoln A,
Nadeau RJ, León R, Toher J, Mouta-Bellum C, Friesel RE and Liaw L:
Notch2 signaling induces apoptosis and inhibits human MDA-MB-231
xenograft growth. Am J Pathol. 171:1023–1036. 2007.PubMed/NCBI
|
|
14.
|
Tanimizu N and Miyajima A: Notch signaling
controls hepato-blast differentiation by altering the expression of
liver-enriched transcription factors. J Cell Sci. 117:3165–3174.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Nijjar S, Crosby HA, Wallace L, Hubscher
SG and Strain AJ: Notch receptor expression in adult human liver: a
possible role in bile duct formation and hepatic
neovascularisation. Hepatology. 34:1184–1192. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Jensen CH, Jauho EI, Santoni-Rugiu E,
Holmskov U, Teisner B, Tygstrup N and Bisgaard HC:
Transit-amplifying ductular (oval) cells and their hepatocytic
progeny are characterized by a novel and distinctive expression of
Delta-like protein/preadipocyte factor1/fetal antigen1. Am J
Pathol. 164:1347–1359. 2004. View Article : Google Scholar
|
|
17.
|
Kohler C, Bell AW, Bowen WC, Monga SP,
Fleig W and Michalopoulos GK: Expression of Notch-1 and its ligand
Jagged-1 in rat liver during liver regeneration. Hepatology.
39:1056–1065. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Giovannini C, Lacchini M, Gramantieri L,
Chieco P and Bolondi L: Notch3 intracellular domain accumulates in
HepG2 cell line. Anticancer Res. 26:2123–2127. 2006.PubMed/NCBI
|
|
19.
|
Gramantieri L, Giovannini C, Lanzi A,
Chieco P, Ravaioli M, Venturi A, Grazi GL and Bolondi L: Aberrant
Notch3 and Notch4 expression in human hepatocellular carcinoma.
Liver Int. 27:997–1007. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Cantarini MC, de la Monte SM, Pang M, Tong
M, D’Errico A, Trevisani F and Wands JR: Aspartyl-asparagyl β
hydroxylase over-expression in human hepatoma is linked to
activation of insulin-like growth factor and Notch signaling
mechanisms. Hepatology. 44:446–457. 2006.PubMed/NCBI
|
|
21.
|
Gao J, Chen C, Hong L, Wang J, Du Y, Song
J, Shao X, Zhang J, Han H, Liu J and Fan D: Expression of Jagged1
and its association with hepatitis B virus X protein in
hepatocellular carcinoma. Biochem Biophys Res Commun. 356:341–347.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Gao J, Song Z, Chen Y, Xia L, Wang J, Fan
R, Du R, Zhang F, Hong L, Song J, Zou X, Xu H, Zheng G, Liu J and
Fan D: Deregulated expression of Notch receptors in human
hepatocellular carcinoma. Dig Liver Dis. 40:114–121. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ross DA and Kadesch T: The Notch
intracellular domain can function as a coactivator for LEF-1. Mol
Cell Biol. 21:7537–7544. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Kato H, Taniguchi Y, Kurooka H, Minoguchi
S, Sakai T, Nomura-Okazaki S, Tamura K and Honjo T: Involvement of
RBP-J in biological functions of mouse Notch1 and its derivatives.
Development. 124:4133–4141. 1997.PubMed/NCBI
|
|
25.
|
Struhl G and Adachi A: Nuclear access and
action of notch in vivo. Cell. 93:649–660. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Jundt F, Anagnostopoulos I, Förster R,
Mathas S, Stein H and Dörken B: Activated Notch1 signaling promotes
tumor cell proliferation and survival in Hodgkin and anaplastic
large cell lymphoma. Blood. 99:3398–4403. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Balint K, Xiao M, Pinnix CC, Soma A, Veres
I, Juhasz I, Brown EJ, Capobianco AJ, Herlyn M and Liu ZJ:
Activation of Notch1 signaling is required for
beta-catenin-mediated human primary melanoma progression. J Clin
Invest. 115:3166–3176. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Wang Z, Banerjee S, Li Y, Rahman KMW,
Zhang Y and Sarkar FH: Down-regulation of Notch-1 inhibits invasion
by inactivation of nuclear factor-κB, vascular endothelial growth
factor, and matrix metalloproteinase-9 in pancreatic cancer cells.
Cancer Res. 66:2778–2784. 2006.
|
|
29.
|
Qi R, An H, Yu Y, Zhang M, Liu S, Xu H,
Guo Z, Cheng T and Cao X: Notch1 signaling inhibits growth of human
hepatocellular carcinoma through induction of cell cycle arrest and
apoptosis. Cancer Res. 63:8323–8329. 2003.PubMed/NCBI
|
|
30.
|
Wang C, Qi R, Li N, Wang Z, An H, Zhang Q,
Yu Y and Cao X: Notch1 signaling sensitizes tumor necrosis
factor-related apoptosis-inducing ligand-induced apoptosis in human
hepato-cellular carcinoma cells by inhibiting Akt/Hdm2-mediated p53
degradation and up-regulating p53-dependent DR5 expression. J Biol
Chem. 284:16183–16190. 2009. View Article : Google Scholar
|
|
31.
|
Miki A, Yano Y, Kato H, Seo Y, Kuriyama M,
Azuma T and Hayashi Y: Anti-tumor effect of pegylated interferon in
the rat hepatocarcinogenesis model. Int J Oncol. 32:603–608.
2008.PubMed/NCBI
|
|
32.
|
Ning L, Wentworth L, Chen H and Weber SM:
Down-regulation of Notch1 signaling inhibits tumor growth in human
hepatocellular carcinoma. Am J Transl Res. 1:358–366.
2009.PubMed/NCBI
|
|
33.
|
Wang XQ, Zhang W, Lui EL, Zhu Y, Lu P, Yu
X, Sun J, Yang S, Poon RT and Fan ST: Notch1-Snail1-E-cadherin
pathway in metastatic hepatocellular carcinoma. Int J Cancer.
131:E163–E172. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Gao J, Chen Y, Wu KC, Liu J, Zhao YQ, Pan
YL, Du R, Zheng GR, Xiong YM, Xu HL and Fan DM: RUNX3 directly
interacts with intracellular domain of Notch1 and suppresses Notch
signaling in hepatocellular carcinoma cells. Exp Cell Res.
316:149–157. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Giovannini C, Gramantieri L, Chieco P,
Minguzzi M, Lago F, Pianetti S, Ramazzotti E, Marcu KB and Bolondi
L: Selective ablation of Notch3 in HCC enhances doxorubicin’s death
promoting effect by a p53 dependent mechanism. J Hepatol.
50:969–979. 2009.PubMed/NCBI
|
|
36.
|
Rao SS, O’Neil J, Liberator CD, Hrdwick
JS, Dai X, Zhang T, Tyminski E, Yuan J, Kohl NE, Richon VM, van der
Ploeg LH, Carroll PM, Draetta GF, Look AT, Strack PR and Winter CG:
Inhibition of NOTCH signaling by gamma secretase inhibitor engages
the RB pathway and elicits cell cycle exit in T-cell acute
lymphoblastic leukemia cells. Cancer Res. 69:3060–3068. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Rasul S, Balasubramanian R, Filipović A,
Slade MJ, Yagüe E and Coombes RC: Inhibition of induces G2/M arrest
and triggers apoptosis in breast cancer cells. Br J Cancer.
100:1879–1888. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Nefedova Y, Sullivan DM, Bolick SC, Dalton
WS and Gabrilovich DI: Inhibition of Notch signaling induces
apoptosis of myeloma cells and enhances sensitivity to
chemotherapy. Blood. 111:2220–2229. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Weijzen S, Rizzo P, Braid M, Vaishnav R,
Jonkheer SM, Zlobin A, Osborne BA, Gottipati S, Aster JC, Hahn WC,
Rudolf M, Siziopikou K, Kast WM and Miele L: Activation of Notch-1
signaling maintains the neoplastic phenotype in human
Ras-transformed cells. Nat Med. 8:979–986. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Qin JZ, Stennett L, Bacon P, Bodner B,
Hendrix MJ, Seftor RE, Seftor EA, Margaryan NV, Pollock PM, Curtis
A, Trent JM, Bennett F, Miele L and Nickoloff BJ: p53-independent
NOXA induction overcomes apoptotic resistance of malignant
melanomas. Mol Cancer Ther. 3:895–902. 2004.PubMed/NCBI
|
|
41.
|
Deangelo DJ, Stone R, Silverman LB, Stock
W, Attar EC, Fearen I, Dallob A, Matthews C, Stone J, Freedman SJ
and Aster J: A phase I clinical trial of the notch inhibitor
MK-0752 in patients with T-cell acute lymphoblastic
leukemia/lymphoma (T-ALL) and other leukemias. In: Proceedings of
the ASCO Annual Meeting. J Clin Oncol. 24:65852006.
|
|
42.
|
Fouladi M, Stewart CF, Olson J, Wagner LM,
Onar-Thomas A, Kocak M, Packer RJ, Goldman S, Gururangan S, Gajjar
A, Demuth T, Kun LE, Boyett JM and Gilbertson RJ: Phase I trial of
MK-0752 in children with refractory CNS malignancies: a pediatric
brain tumor consortium study. J Clin Oncol. 29:3529–3534. 2011.
View Article : Google Scholar : PubMed/NCBI
|