Introduction
Lung cancer is one of the most frequent type of
cancer and the leading cause of cancer-related mortality in the
Unites States and developed countries. In the United States,
226,160 new cases of lung cancer and 160,340 deaths from lung
cancer are estimated to occur in 2012 (1). Despite efforts to improve early
diagnosis and treatment, the survival rates of the patients with
lung cancer have not significantly improved over the past 30 years.
The overall five-year survival of lung cancer only increased from
13% between 1987 to 1989, to 16% between 2001 to 2007 in the US
(1,2). The majority (80%) of lung cancer
cases are non-small cell lung cancer (NSCLC) (3). Therefore, the development of novel
antitumor drugs for the treatment of NSCLC is imperative in order
to improve the efficacy of lung cancer therapy and prognosis.
One of the most effective antitumor mechanisms is
the inhibition of uncontrolled cell growth. The G2/M transition is
a crucial step in the cell cycle for controlling cell
proliferation. The agents targeting G2/M phase have been used in
combination with chemotherapy and sensitization to radiotherapy.
However, compared with agents targeting the G1 and S phase, only a
few antitumor agents affecting the G2/M phase have been approved.
Vinca alkaloids and taxanes are the only antitumor drugs affecting
the G2/M phase which have been used in clinical practice for the
treatment of NSCLC. Recently, certain novel anti-NSCLC agents have
been reported to have an inhibitory effect by arresting cells in
the G2/M phase. However, their clinical efficacy and molecular
mechanisms involved are not yet well understood (4–6).
AZ64 (AstraZeneca Pharmaceuticals) is a novel
anticancer agent designed to target tropomyosin-related kinase
(Trk)A, B and C. However, its role and mechanisms in the treatment
of NSCLC remain unclear. In this study, we investigated its
antitumor activity against NSCLC, as well as its mechanism of
action.
Materials and methods
Cell lines and reagents
Six human lung cancer cell lines including three
adenocarcinoma cell lines (A549, HCC827 and H1944), two large cell
carcinoma cell lines (H460 and H1299) and one squamous carcinoma
cell line (Calu-1) were used in this study. All the cell lines were
purchased from the American Type Culture Collection and were
cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented
with 5% fetal bovine serum (FBS), 1% penicillin-streptomycin and 1%
glutamine. The cells were grown in a 37°C incubator in a humidified
atmosphere of 5% CO2. AZ64 compound was synthesized and
supplied by AstraZeneca Pharmaceuticals. It was dissolved in
dimethylsulfoxide (DMSO) with a stock concentration of 10 mM and
stored at −20°C under light-protected conditions. The final
concentrations of DMSO in the experiment did not exceed 0.1%. The
antibodies against cyclin B, Cdc2, phospho-Cdc2, Cdc25C, Polo-like
kinase 1 (Plk1) and Aurora-A were purchased from Cell Signaling
Technology. β-tubulin antibody was purchased from Santa Cruz
Biotechnology Inc.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumtromide (MTT)
reagent was purchased from Sigma-Aldrich Corp. The BD Matrigel
invasion assay kit was purchased from BD Biosciences. The BCA
Protein Assay kit was purchased from Thermo Scientific Pierce Co.
The Vectastain Elite ABC kit was purchased from Vector
Laboratories.
Cell culture and cell proliferation assay
(MTT assay)
A549, HCC827, H1944, H460, H1299 and Calu-1 cells
were plated into 96-well plates at a density of 5×103
cells/well and incubated at 37°C with 5% CO2 for 24 h.
Subsequently, 0 (0.1% DMSO), 0.25, 0.5, 1.0, 2.0, 4.0 and 8.0 μM
concentrations of AZ64 were added to the culture medium. After 72 h
of exposure to AZ64, cell proliferation was evaluated by the MTT
assay. Briefly, the medium was removed and replaced by 200 μl fresh
medium with 500 μg/ml MTT per well. The cells were incubated for 4
h. The medium was then removed and 200 μl of DMSO were added to
each well. The absorbance value of each well was determined
spectrophotometrically at 570 nm on a Microplate ELISA Reader
(Bio-Tek Instruments). The experiment was performed in triplicate.
Each experiment was repeated three times.
Real-time cell growth observation by the
real-time cell electronic sensing (RT-CES) system
ACEA RT-CES, (ACEA Biosciences Inc.), a
microelectronic cell sensor system, was used to measure the cell
index continuously and quantitatively. A549 cells were seeded in
16×E-plates at a density of 5×103 cells/well and
incubated in a 37°C incubator with 5% CO2. The cell
index was measured every 30 min by RT-CES. After 24 h of
incubation, 2.5 and 5.0 μM of AZ64 and 0.1% DMSO (control) were
added to the medium, and the cell growth was then monitored every
30 min for another 72 h by RT-CES. The experiment was performed in
triplicate for each treatment condition and was repeated three
times.
Anchorage-independent colony formation
assay
Six-well plates were pre-coated with 2 ml of bottom
agar of 0.6% low-melting agarose with DMEM. Subsequently, 2 ml of
top agar consisting of 0.3% low-melting agarose, DMEM,
5×103 cells and 2.5 μM AZ64 or 0.025% DMSO were added to
each well. After solidification, each well was covered with 1 ml of
DMEM with 2.5 μM AZ64 or 0.025% DMSO and incubated in a 37°C, 5%
CO2 incubator. The covering medium was refreshed every three days.
After two weeks, the colonies of >100 μm were counted under a
microscope at ×4 magnification. The experiment was performed in
triplicate for each treatment condition and was repeated three
times.
In vitro cell invasion assay
The invasive ability of A549 cell was assayed using
BD BioCoat Matrigel invasion chambers (Becton-Dickinson, San Jose,
CA). Briefly, the chambers were rehydrated with serum-free medium
supplemented with 2.5 μM AZ64 or 0.025% DMSO. The A549 cells were
then seeded in the upper chamber at a density of 2.5×104
cells/well in serum-free medium supplemented with 2.5 μM AZ64 or
0.025% DMSO, whereas the lower chamber contained 10% FBS medium
with 2.5 μM AZ64 or 0.025% DMSO. After 20 h of incubation at 37°C,
the non-invasive cells on the upper surface of the chamber
membranes were removed by a cotton-tipped swab. The invasive cells
on the lower surface of the chamber membrane were stained with
crystal violet and counted at randomly chosen fields under a light
microscope at x10 magnification. The experiment was repeated three
times.
Cell cycle analysis by flow cytometry
(FCM)
The cell cycle analysis was performed by FCM with
DNA propidium iodide (PI) staining. The A549, HCC827, H1944 and
Calu-1 cells were treated with 2.5 μM AZ64 or 0.025% DMSO for 24 h
and then harvested by trypsinization. The cells were fixed with 1
ml of 70% cold ethanol overnight and then stained with staining
buffer containing 0.01% PI, 0.1% sodium citrate, 0.3% Triton X-100
and 0.01% RNase A for 1 h in the dark. The DNA content and cell
cycle distribution were determined using a BD FACSCalibur flow
cytometer and CellQuest software (Becton-Dickinson). The
experiments were repeated three times for the A549 cells and two
times for the other cell lines. Furthermore, the A549 cells were
treated with AZ64 at various concentrations of 0, 0.5, 1, 2.5 and 5
μM for 24 h and then the cell cycles were analyzed by FCM as
described above. The experiments were repeated two times.
Immunofluorescence assay of microtubule
and DAPI staining of chromosome
The A549 cells were cultured on the coverslips and
treated with 0.025% DMSO, 2.5 μM AZ64 or 0.0625 μg/ml (73 nM) of
paclitaxel for 24 h. The cells were fixed with 4% formaldehyde for
15 min and were then permeabilized with 0.5% Triton X-100 in PBS
for 10 min. After blocking with 5% BSA for 30 min, the cells were
incubated with anti-β-tubulin monoclonal antibody (1:100 in the
blocking solution) for 2 h at 37°C followed by incubation with
FITC-conjugated goat anti-mouse IgG and DAPI (1:2000) for 1 h at
37°C. Images of the microtubules and chromosomes were obtained
using a confocal fluorescence microscope. Mitotic index was
calculated by the ratio of the number of cells in mitosis
(including the cell with condensed chromosomes, aligned chromosomes
and segregated chromosomes) to the total number of cells. The
experiment was repeated three times.
Western blot analysis
The A549 cells were treated with 2.5 μM of AZ64 for
0, 2, 4, 6, 8, 12, 16, 24 and 36 h. The cells were harvested by
scraping into lysis buffer and incubated on ice for 10 min.
Afterwards, the lysates were centrifuged at 14,000 rpm for 20 min
at 4°C and then the supernatants were collected into new tubes. The
protein concentrations were determined using the Pierce BCA protein
assay kit. Equal amounts of protein were then loaded onto a 10%
SDS-PAGE gel. Electrophoresis was run at 150 V for 60–90 min and
then the separated proteins were transferred onto a nitrocellulose
membrane at 100 V for 60–90 min. After blocking with 5% non-fat
milk for 1 h, the membrane was incubated with primary antibodies
against Aurora-A, Plk1, Cdc25C, Cdc2, phospho-Cdc2 (Tyr15), cyclin
B and β-actin (as the control) overnight at 4°C. The membrane was
then incubated with HRP-conjugated secondary antibody (1:10,000)
for 60 min at room temperature. The signal was detected using the
enhanced chemiluminescence (NEN Life Science Products, Boston, MA)
detection system. The experiment was repeated two times.
To further investigate the effect of AZ64 on G2/M
checkpoint proteins in the cell cycle, A549 cells were synchronized
to the S phase by 2.5 μM thymidine treatment for 18 h.
Subsequently, 2.5 μM AZ64 or 0.025% DMSO were added to the medium
at the time of thymidine release. The cells were harvested at 0, 3,
5, 6, 7, 8 and 9 h after treatment. Western blot analysis was
performed with Cdc2, phospho-Cdc2 (Tyr15) and β-actin antibodies.
The cell cycle was examined by FCM assay. The experiment was
repeated two times.
Immunocytochemistry (ICC) staining for
phospho-Cdc2 (Tyr15)
The A549 cells were cultured on the coverslips and
were treated with 2.5 μM AZ64 or 0.025% DMSO alone in triplicate.
At 24 h after exposure, the cells were fixed with 4% formalde-hyde
for 15 min and permeabilized with 0.5% Triton X-100 for 10 min at
room temperature. The ICC staining was performed using a Vectastain
ABC kit according to the supplier’s instructions. Briefly, the
endogenous peroxidase was inactivated with 3%
H2O2 for 15 min. Blocking was performed with
avidin for 10 min, biotin for 10 min and normal goat serum for 30
min. Afterwards, the cells were incubated with 1:50
anti-phospho-Cdc2 (Tyr15) antibody overnight at 4°C, and then
incubated with biotinylated secondary antibody solution for 30 min
followed by incubation with ABC Reagent for 30 min. After washing,
the cells were incubated with 3,3’-diaminobenzidine (DAB) solution
for 2 min. The staining results were observed under a light
microscope. The extent of phospho-Cdc2 (Tyr15) staining was
calculated using the H-score semi-quantitative method. Based on the
staining intensity (0, negative; 1+, weak; 2+, moderate; and 3+,
strong) and the percentages of the cells stained at each intensity
level, the H-score = (% at 0) x 0 + (% at 1+) x 1 + (% at 2+) x 2 +
(% at 3+) x 3 was calculated.
Antitumor activity in vivo
To investigate the antitumor activity of AZ64 in
vivo, a human tumor xenograft model was established. Athymic
nude mice were obtained from the National Cancer Institute. Sixteen
male athymic Swiss nude (nu/nu) mice aged 5–6 weeks with a weight
of 20–22 g were used. All the experimental procedures and care for
the mice were approved by the Institutional Animal Care and Use
Committee and the Department of Veterinary Medicine of the MD
Anderson Cancer Center. A total of 100 μl of A549 cells
(2×106) were injected subcutaneously (s.c.) into a
single dorsal site of each mouse. When the tumors reached 60–110
mm3 in volume, the mice were randomly divided into the
AZ64-treated group and the control group (n=8 per group). AZ64
dissolved in hydroxypropyl methylcellulose (HMPC) was administered
by gavage to the mice in the AZ64-treated group (100 mg/kg body
weight) and the vehicle was administered to the control group. The
tumor size and mouse weight were measured every four days for a
total of 36 days. Tumor volume (TV) was calculated as TV
(mm3) = length (L) x width (W)2/2. Tumor size
was expressed by relative tumor volume (RTVn) =
TVn/TV0.
Statistical analysis
The data are presented as the means ± standard
deviation (SD). The Student’s t-test was used to analyze the
statistical significance of the mean differences between two groups
for normal distributed variables. Two-sided P-values ≤0.05 were
considered to indicate statistically significant differences.
Results
AZ64 inhibits proliferation of NSCLC
cells in a dose- and time-dependent manner
To evaluate the effect of AZ64 on NSCLC cell growth,
three adenocarcinoma cell lines (A549, HCC827 and H1944), two large
cell carcinoma cell lines (H460 and H1299), and one squamous
carcinoma cell line (Calu-1) were treated with various
concentrations of AZ64 or 0.1% DMSO (control). MTT assay was
performed to determine the cell proliferation of the cells after 72
h of treatment with AZ64. The results indicated that the viability
of the cells treated with AZ64 was reduced in all the cell lines
compared with the control cells and that the inhibition was
enhanced with the increased concentration of AZ64 (Fig. 1A and B). The IC50 values (the drug
concentration inhibiting the growth of cell lines by 50%) were
2.22, 1.32, 2.11, 4.75, 5.94 and 5.95 μM in the A549, HCC827,
H1944, H460, H1299 and Calu-1 cell lines, respectively, which
indicated that the adenocarcinoma cell lines were more sensitive to
AZ64 than the large cell carcinoma cell lines and the squamous
carcinoma cell line. Furthermore, the time-response effect of AZ64
on the A549 cells was observed. The cells were treated with AZ64
for 72 h and the cell index was measured every 30 min by RT-CES. It
was found that the reduction in cell viability was increased with
the increased treatment time (Fig. 1C
and D). Taken together, these results suggest that AZ64 exerts
a potent anti-proliferative effect on NSCLC cells in a dose- and
time-dependent manner.
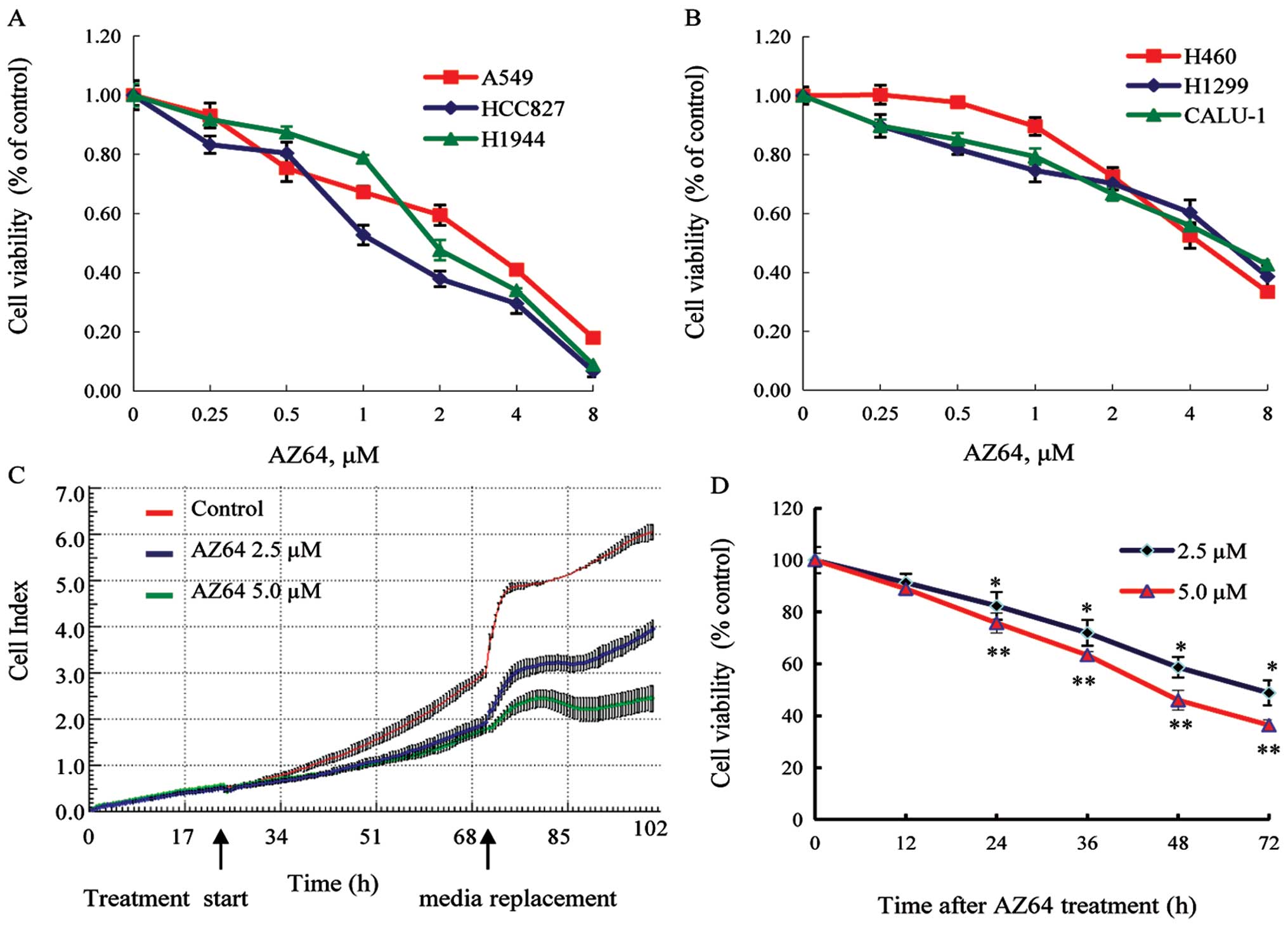 | Figure 1.AZ64 inhibited NSCLC cell
proliferation in a dose- and time-dependent manner. (A) Three
adenocarcinoma cell lines (A549, HCC827 and H1944). (B) Two large
cell lung cancer cell lines (H460 and H1299), and one squamous
cancer cell line (Calu-1). All six cell lines were treated with
AZ64 at various concentrations of 0, 0.25, 0.5, 1.0, 2.0, 4.0 and
8.0 μM for 72 h. The cell viability was measured by the MTT assay.
The data represent the means ± SD of triplicate experiments. One
representative derived from three independent experiments is shown.
(C) A549 cells were treated with 2.5 and 5.0 μM of AZ64 and 0.1%
DMSO as the control for 72 h. The real-time cell proliferation was
measured by the RT-CES system. The shadow area represents SD. (D)
The cell viability at time-points of 12, 24, 36, 48, and 72 h of
treatment was quantified using the RT-CES system. The data are
presented as the means ± SD of triplicate experiments. One
representative of three independent experiments is shown.
*P<0.05; **P<0.01, compared with the
previous time-point value, based on a paired t-test. |
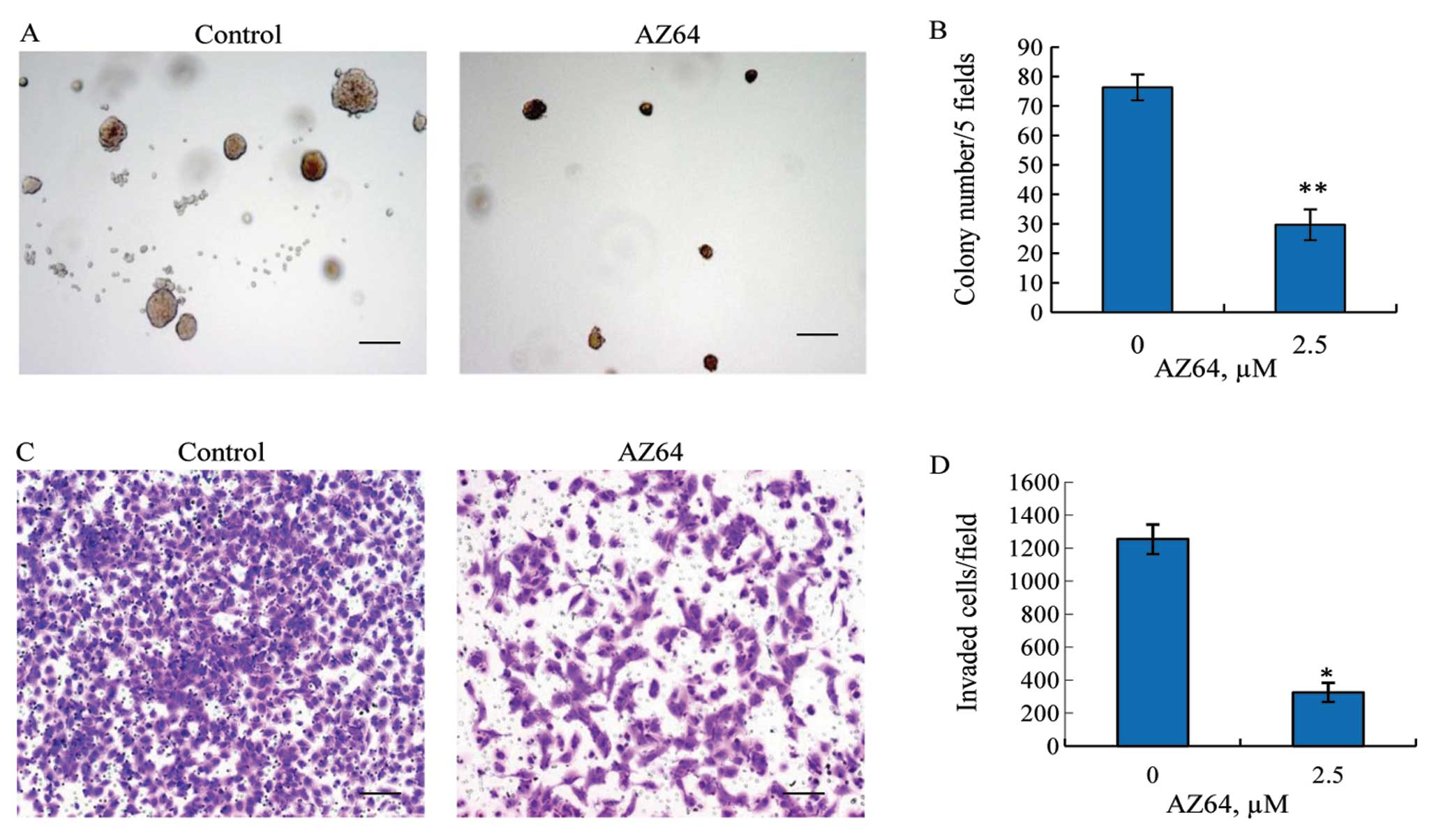
AZ64 inhibits anchorage-independent
growth of A549 cells
To further investigate the antitumor effects of
AZ64, a soft agar colony formation assay was performed. A549 cells
were incubated in soft agar at a low density and treated with
either 2.5 μM AZ64 or DMSO alone for two weeks. The results
revealed that the colonies formed in the cells treated with AZ64
were significantly reduced compared with the untreated cells
(Fig. 2A). The colonies of >100
μm were counted in five fixed fields in each well under a light
microscope at x4 magnification. The colony numbers per five fields
in the AZ64-treated A549 cells were 29.67±5.24 versus 76.33±4.41 in
the control cells (Student’s t-test, P<0.01) (Fig. 2B). This suggests that AZ64 has the
potential to inhibit the anchorage-independent growth of A549
cells.
AZ64 suppresses invasion activity of A549
cells
To explore the anti-metastatic effect of AZ64 on
NSCLC cells, the ability of AZ64 to inhibit A549 cell invasion was
examined by BD Matrigel invasion assay. As shown in the Fig. 2C, the number of cells passing
through the chamber membrane was significantly reduced in the
AZ64-treated group compared with the untreated group. Quantified in
a randomly chosen microscopic field at ×10 magnification, the
number of invasive cells per field was 324.33±57.50 in the
AZ64-treated group, whereas 1,253±90.04 in the untreated group.
There was a significant difference between these two groups
(Student’s t-test, P<0.01) (Fig.
2D). This suggests that AZ64 decreases the invasive capability
of A549 cells.
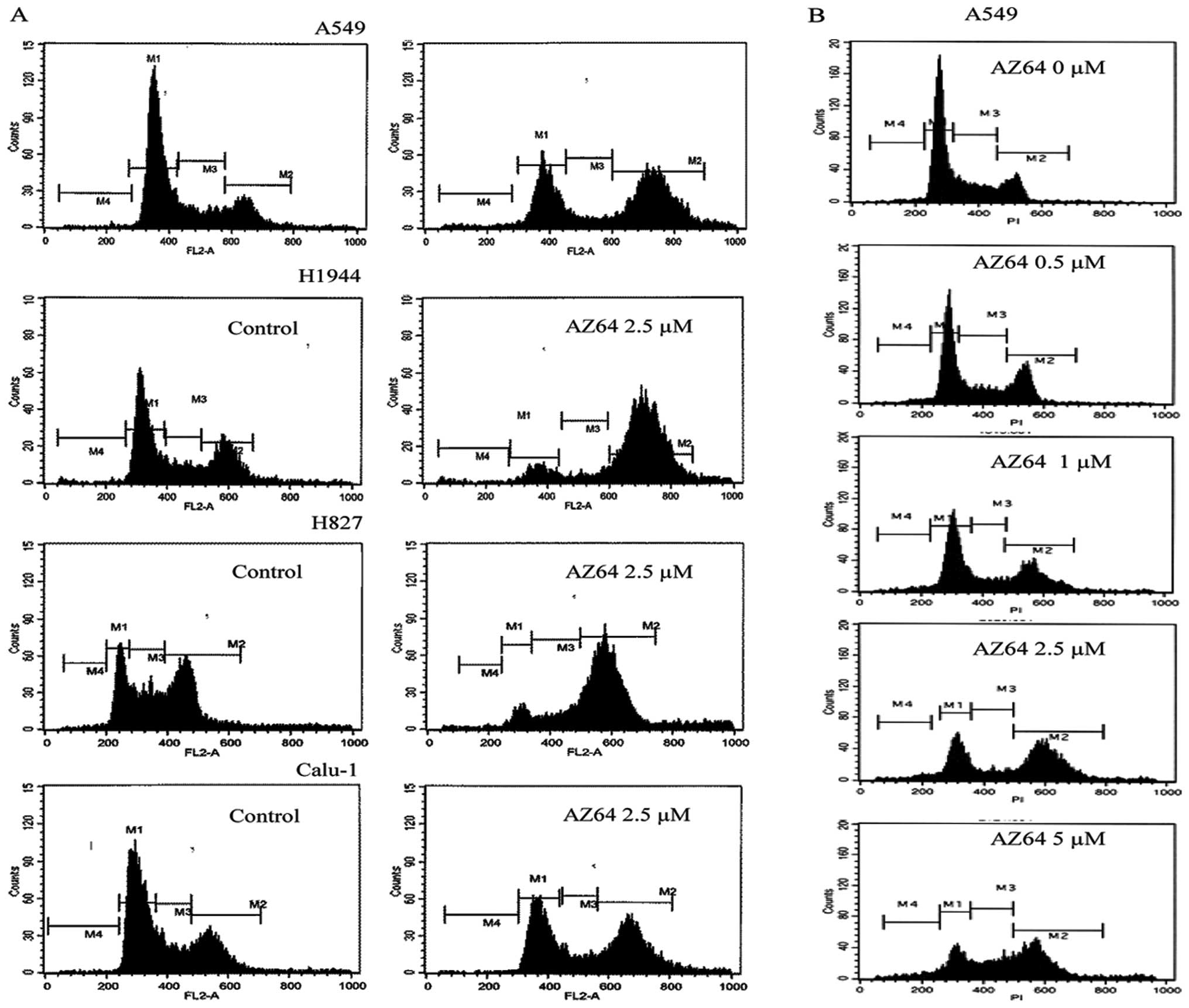
AZ64 induces G2/M arrest of NSCLC
Cells
To identify the influence of AZ64 on the cell cycle
progress of NSCLC cell lines, the cell cycles were analyzed by FCM.
The results showed that the G2/M population in the A549, HCC827,
H1944 and Calu-1 cells significantly increased and that the G0/G1
population decreased following treatment with 2.5 μM of AZ64 for 24
h, compared with control (Fig. 3A
and Table I). To confirm these
results, the analysis of the cell cycle in the A549 cells was
repeated three times. The population of cells in the G2/M was
51.41±3.32% in the AZ64-treated group versus 17.81±0.81% in the
control group (Student’s t-test, P<0.001). In the dose-response
observation, it was found that the population of A549 cells
arrested in the G2/M phase after treatment with AZ64 increased with
the increasing concentration of AZ64 (Fig. 3B and Table II). Taken together, these results
demonstrate that AZ64 induces the G2/M arrest of NSCLC cells in a
dose-dependent manner.
 | Table I.Effect of AZ64 on the cell cycle
distribution of NSCLC cells. |
Table I.
Effect of AZ64 on the cell cycle
distribution of NSCLC cells.
| Cell lines | Treatment | G0/G1 (%) | S (%) | G2/M (%) |
|---|
| A549 | Control | 64.44 | 16.21 | 17.54 |
| AZ64 | 33.17 | 8.48 | 54.55 |
| HCC827 | Control | 24.25 | 27.13 | 43.04 |
| AZ64 | 7.22 | 14.24 | 70.01 |
| H1944 | Control | 50.71 | 13.94 | 30.59 |
| AZ64 | 8.79 | 6.30 | 81.40 |
| Calu-1 | Control | 55.90 | 18.75 | 24.14 |
| AZ64 | 38.78 | 11.78 | 44.66 |
| Table II.Effect of the different
concentrations of AZ64 on cell cycle distribution of A549
cells. |
Table II.
Effect of the different
concentrations of AZ64 on cell cycle distribution of A549
cells.
| AZ64 (μM) | G0/G1 (%) | S (%) | G2/M (%) |
|---|
| 0 | 58.37 | 22.76 | 17.24 |
| 0.5 | 55.80 | 22.23 | 20.46 |
| 1.0 | 49.16 | 13.04 | 33.19 |
| 2.5 | 28.56 | 12.42 | 52.08 |
| 5.0 | 22.21 | 24.26 | 45.41 |
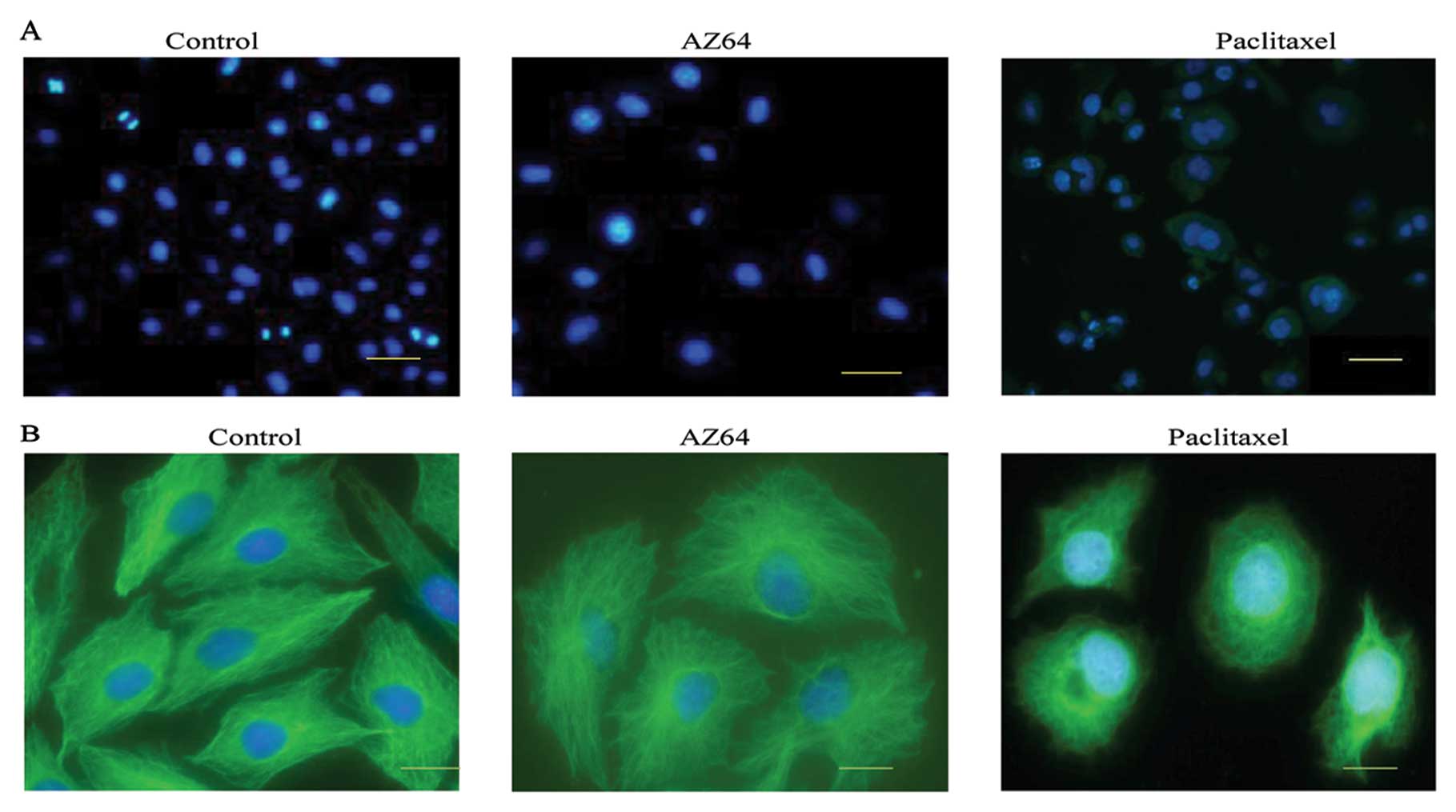
Effect of AZ64 on chromosomes and
microtubules in A549 cells
Following the induction of G2/M phase arrest in the
NSCLC cells after AZ64 treatment, we investigated the impact of
AZ64 on chromosomes and microtububles, which play an important role
in mitosis. The images from the DAPI staining of A549 cells showed
that the nuclei size of tbe AZ64-treated A549 cells was enlarged
compared with the untreated cells. However, there were no increased
proportions of mitotic nuclei characterized as chromosomal
condensation, chromosomal alignment and chromosomal segregation in
the AZ64-treated cells compared with the control cells. No
significant mitotic catastrophe, including multi-nucleation,
micronuclei and chromosome bridges was found in the AZ64-treated
cells compared with those treated with paclitaxel and the control
cells. The proportion of mitotic cells was 2.7±1.09% in the
AZ64-treated cells versus 3.29±1.05% in the DMSO-treated cells.
There was no significant difference between the two groups
(Student’s t-test, P=0.398) (Fig.
4A). This suggests that AZ64 led to the arrest of the A549
cells in the G2 phase. Therefore, AZ64 blocks the transition of the
A549 cells from the G2 to the M phase.
Moreover, we performed immunofluorescence staining
of microtubules with anti-β-tubulin monoclonal antibody followed by
FITC-conjugated goat anti-mouse IgG. No aberrant microtubule
depolymerization or disruption was found in the AZ64-treated A549
cells compared with the untreated cells; however, aberrant
microtubule depolymerization was evident in the cells treated with
anti-microtubule drug. The paclitaxel-treated cells exhibited
aberrant microtubule depolymerization or disruption (Fig. 4B). Therefore, we concluded that the
G2/M arrest of A549 cell induced by AZ64 may be a result of the
reduced chromosomal activity, and not from aberrant microtubule
depolymerization.
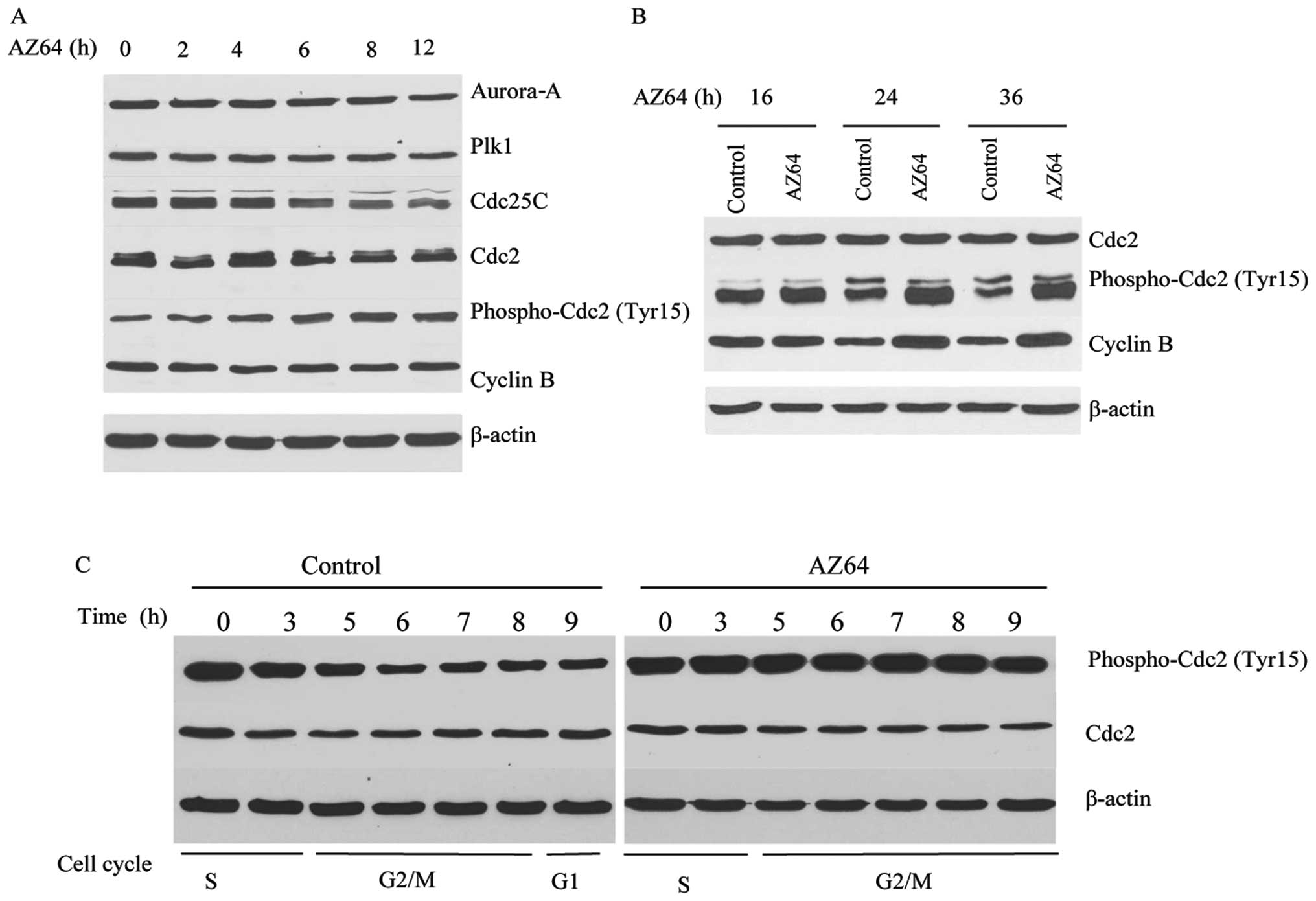
Impact of AZ64 on proteins related to
G2/M transition in A549 cells
Considering the important role of AZ64 in G2/M
transition, the impact of AZ64 on the expression of the proteins
related to the G2/M checkpoint was examined. Following treatment
with 2.5 μM of AZ64 for 0, 2, 4, 6, 8, 12, 16, 24 and 36 h, the
expression of Aurora-A, Plk1, Cdc25C, Cdc2, phospho-Cdc2 (Tyr15)
and cyclin B was measured by western blot analysis in the A549
cells. The results showed that the expression of phospho-Cdc2
(Tyr15) was increased and that of Cdc25C was reduced in the
AZ64-treated A549 cells after 6 h of exposure compared with the
untreated cells. The Aurora-A, Plk1, Cdc2 and cyclin B expression
levels were not altered following treatment with AZ64 (Fig. 5A). This implies that AZ64 inhibits
the dephosphorylation of phospho-Cdc2 (Tyr15) by decreasing the
expression of Cdc25C, which contributes to G2/M arrest. However,
the levels of cyclin B were enhanced after 24 h of treatment with
AZ64 (Fig. 5B). A possible
explanation is that the G2/M arrest may abrogate the degradation of
cyclin B.
To further elucidate the effect of the alteration of
phospho-Cdc2 (Tyr15) levels on the cell cycle, we synchronized the
A549 cells with thymidine treatment and treated them with 2.5 μM of
AZ64 or 0.025% DMSO alone after thymidine release. The cells were
harvested at 0, 3, 5, 6, 7, 8 and 9 h after AZ64 treatment and the
levels of phospho-Cdc2 (Tyr15) and Cdc2 were examined by western
blot analysis. The cell cycle activities were measured by FCM. The
results demonstrated that the phospho-Cdc2 (Tyr15) level in the
control cells decreased at the time of the G2/M transition,
although the phospho-Cdc2 (Tyr15) level in the AZ64-treated A549
cells dramatically increased at the time of the G2/M transition,
while there was no significant change in total Cdc2 expression
(Fig. 5C). This demonstrates that
AZ64 inhibits the dephosphorylation of phospho-Cdc2 (Tyr15) at the
time of the G2/M transition, which results in G2/M arrest.
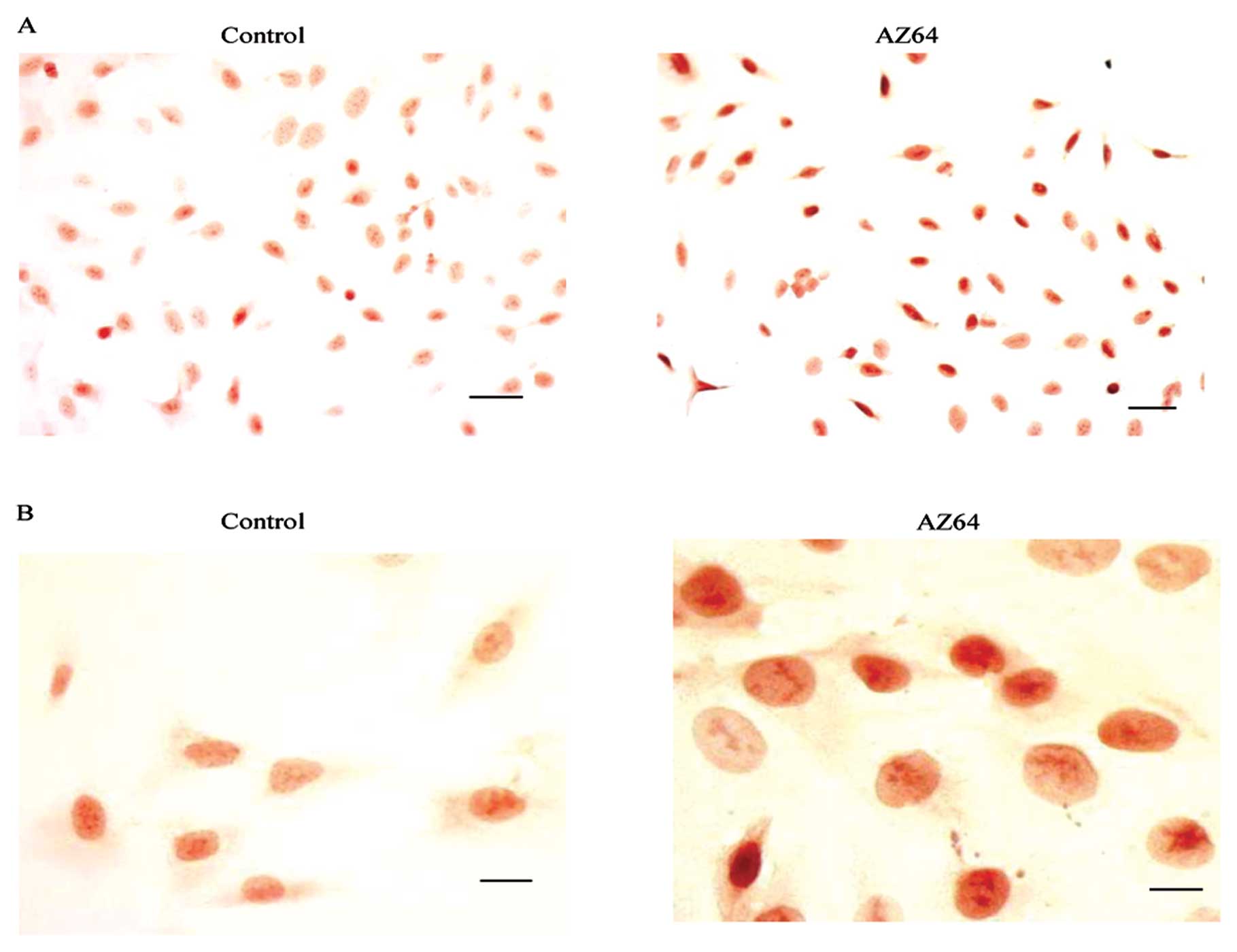
To verify the effect of AZ64 on the
dephosphorylation of phospho-Cdc2 (Tyr15), the ICC staining of
phospho-Cdc2 (Tyr15) was performed on the A549 cells. As shown in
Fig. 6, high levels of
phospho-Cdc2 (Tyr15) accumulated in the nuclei of the AZ64-treated
A549 cells compared with the DMSO-treated control cells. The
H-score of phospho-Cdc2 (Tyr15) was 267±15 in the AZ64-treated
cells versus 157±12 in the untreated cells (Student’s t-test,
P<0.01) (Fig. 6A and B). These
results further demonstrate that AZ64 inhibits the
dephosphorylation of phospho-Cdc2 (Tyr15), which is consistent with
the findings from western blot analysis.
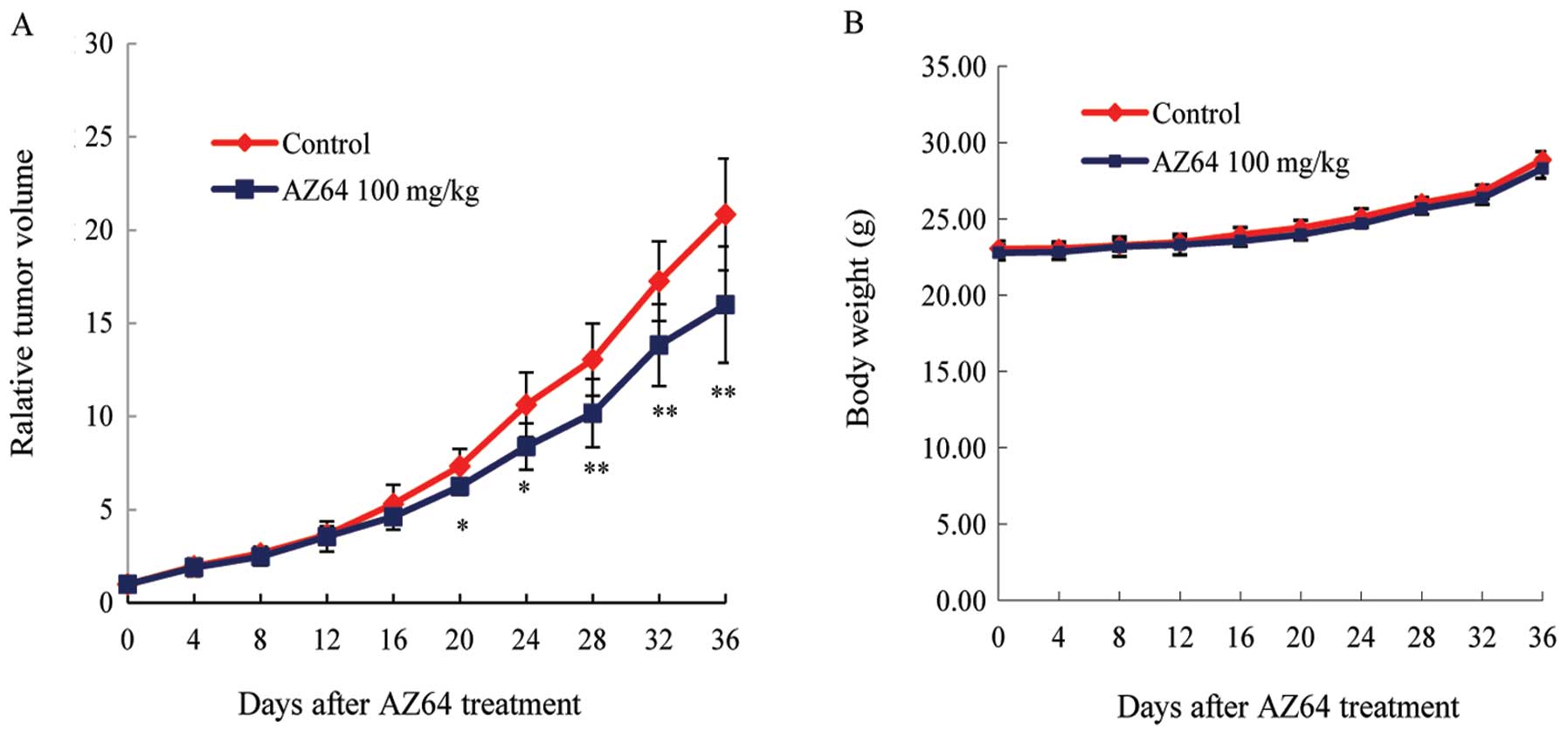
AZ64 inhibits tumor growth in the A549
xenograft model
To determine the antitumor effect of AZ64 in
vivo, A549 cells were xenografted into nude mice. When the
tumors were measurable (approximately 60–100 mm3), AZ64
or vehicle was administrated by gavage, and the tumor size and
mouse weight were measured for a total of 36 days. It was found
that the average tumor volume of the A549 xenografted nude mice was
reduced in the AZ64-treated group compared to the control group
starting from day 20 (Student’s t-test, P<0.05 from day 20 to
24; P <0.01 from day 28 to 36; Fig.
7A). To evaluate the toxicity of AZ64, the body weights of the
nude mice were measured. There are no difference in body weight
between the AZ64-treated group and the control group after
treatment (Student’s t-test, P>0.05; Fig. 7B).
Discussion
In this study, we found that AZ64 inhibits the
proliferation of all major subtypes of NSCLC cells, including
adenocarcinoma, large cell carcinoma and squamous carcinoma. The
IC50 of these cell lines ranged from 1.32 to 5.95 μM, which is
similar to that obtained in other reports (7,8).
Based on the IC50 values, it was revealed that the adenocarcinoma
cell lines were more sensitive to AZ64 than the large cell
carcinoma cell lines and the squamous carcinoma cell line. It was
also shown that AZ64 exerts an anti-proliferative effect on the
NSCLC cells in a dose- and time-dependent manner. Moreover, the
soft agar colony formation assay demonstrated that AZ64
dramatically inhibited the anchorage-independent growth of A549
cells and Matrigel invasion assay showed that AZ64 significantly
inhibited cell invasion. Collectively, these data suggest that AZ64
has the potential to inhibit the tumor growth and metastasis of
NSCLC cells in vitro.
Our in vivo experiments showed that the tumor
growth in the AZ64-treated mice was slower than the control group.
Of note, there was no significant weight loss in the AZ64-treated
mice compared with the control group. These results imply that AZ64
has the ability to inhibit tumor growth in vivo and is well
tolerated by mice.
The biological mechanisms of antitumor drugs include
the inhibition of proliferation, growth arrest in the cell cycle,
enhanced apoptosis and the modulation of signal transduction
pathways. Trks are a family of receptor tyrosine kinases (RTKs),
including three members, TrkA, TrkB and TrkC, which bind with its
ligand neurotrophins (NTs) to mediate the trophic effects of the
nerve (9). They are expressed in a
variety of human cancers, such as neuroblastomas, glioblastomas,
prostatic adenocarcinomas and papillary thyroid carcinomas
(10). AZ64 as originally
developed as a Trk inhibitor, acting as a kinase inhibitor by
competing with the binding of ATP to the catalytic domain. It acts
as a Trk inhibitor at low concentrations (single nanomole) but acts
as a multiple kinase inhibitor at high concentrations (11). It has been reported to inhibit the
growth of neuroblastoma cells at a nanomole range of IC50 (12), and to inhibit the growth of lung
cancer cells at the micromole range (IC50) (7,8). At
these concentrations, the mechanism of action of AZ64 should to be
further investigated. It is possible that AZ64 targets multiple
kinases.
Our results from FCM analysis indicated that AZ64
dramatically increased the number of A549, HCC827, H1944 and Calu-1
cells in the G2/M phase. During the dose-dependent observation, we
found that the inhibitory effect of AZ64 on the G2/M phase occurred
in dose-dependent manner. These results strongly suggest that AZ64
induces G2/M arrest in NSCLC cells. Moreover, we examined the
morphological alteration of the A549 cells after treatment with
AZ64, focusing on chromosomes and microtubules. We observed that
the cell and nucleus were enlarged after AZ64 treatment. However,
although A549 cells were arrested in the G2/M phase by AZ64, there
was no increase in the mitotic index in the AZ64-treated A549 cells
compared with the untreated cells. This illustrates that A549 cells
are mostly arrested in the G2 phase and do not reach the M phase.
Therefore, it can be concluded the transition of the A549 cells
from the G2 to the M phase is blocked. In addition, no obvious
mitotic catastrophe, such as multi-nucleation, micronuclei and
chromosome bridges was found and no aberrant microtubule
depolymerization or polymerization were observed in the
AZ64-treated cells compared with the untreated cells. Different
results were observed in the paclitaxel-treated cells, in which
aberrant microtubule depolymerization and mitotic catastrophe were
evident. This indicates that AZ64 blocks the cells in the G2/M
phase not via microtubule inhibition but via other potential
mechanisms.
The cell cycle progression at the G2/M phase was
triggered by the checkpoint known as Cdc2/cyclin B. The activation
of Cdc2/cyclin B initiates the entry of the cells from the G2 to
the M phase through the phosphorylation of proteins that correlate
with chromosomal condensation, nuclear envelope breakdown and
spindle assembly (13). In
addition to binding with cyclin B, the activation of Cdc2 is
regulated by a series of phosphorylation and dephosphorylation
events. The dephosphorylation of the Thr14 and Tyr15 amino acids
(14) and the phosphorylation of
the Thr161 amino acid promote the activation of Cdc2. As the
dephosphorylation of the Thr14 and Tyr15 amino acids plays an
important role in the G2/M transition, to elucidate the molecular
mechanism of the antitumor effect of AZ64, we investigated the
influence of AZ64 on the regulatory proteins involved in G2/M
transition in the A549 cells. It was found that phospho-Cdc2
(Tyr15) levels significantly increased in the AZ64-treated A549
cells compared to the untreated cells, while no significant
difference was observed in the Cdc2 and cyclin B levels between the
two groups. For further verification, we synchronized the A549
cells with thymidine and treated them with AZ64. It was also found
that the phospho-Cdc2 (Tyr15) levels dramatically increased in the
AZ64-treated A549 cells, while the phospho-Cdc2 (Tyr15) levels were
decreased in the untreated cells at the G2/M transition. Moreover,
ICC staining was performed and the results revealed that
phospho-Cdc2 (Tyr15) levels increased in the nuclei of the
AZ64-treated cells compared with the control group. Collectively,
these results suggest that AZ64 blocks the G2/M transition by
inhibiting the dephosphorylation of phospho-Cdc2 (Tyr15).
The dephosphorylation of phospho-Cdc2 (Tyr14 and
Tyr15) is facilitated by Cdc25C phosphatase and the phosphorylation
of Cdc2 on Tyr14 and Tyr15 is catalyzed by a protein kinase known
as Wee1 (15). At the end of the
G2 phase, the abrupt dephosphorylation of phospho-Cdc2 (Tyr14 and
Tyr15) by Cdc25C triggers the activation of Cdc2/cyclin B and
promotes the cell cycle progression to the M phase. Cdc25C is
inactivated by the phosphorylation of Ser216 by Chk1 and Chk2
during the interphase, which binds 14-3-3 proteins and maintains
its cytoplasmic location (16).
During the G2/M transition, Cdc25C is activated by the
phosphorylation of Ser198 and enters the nucleus to activate
Cdc2/cyclin B by dephosphorylating phospho-Cdc2 (Tyr14 and Tyr15).
The phosphorylation of Cdc25C on Ser198 and its nuclear
translocation are facilitated by Plk1 (17) which is a serine/threonine kinase
and activated by its upstream protein kinase, Aurora-A (18).
We further determined the levels of Aurora-A, Plk1
and Cdc25C in the A549 cells after AZ64 treatment. The results
showed that the Cdc25C protein level was downregulated; however,
the Aurora-A and Plk1 levels were not affected. This implied that
the reduction in the dephosphorylation of phospho-Cdc2 (Tyr15) in
the AZ64-treated cells resulted from the downregulation of Cdc25C
by AZ64. However, the phosphorylated forms of Aurora-A and Plk1
need to be investigated for the evaluation of the effect of AZ64 on
the Aurora-Plk pathway.
In general, a cell with a suppressed cyclin B1/Cdc2
activity tends to be arrested in the G2 phase, whereas a cell with
an elevated cyclin B1/Cdc2 activity enters mitosis. However, a
number of reports have presented conflicting results, demonstrating
that cyclin B levels are increased in the cells arrested in G2/M.
Choi et al (19) showed
that cyclin B upregulation and plays an important role in the
prometaphase arrest in nocodazole-treated cells. Wang et al
(20) reported elevated cyclin B,
Cdc2 and Cdc25C levels in cells in G2/M arrest by the knockdown of
Plk1. In this study, the alteration in cyclin B levels was
biphasic, in that no change was observed in the early phase (0–24
h), while an increase was observed in the later phase (24–36 h)
after AZ64 treatment. Based on the knowledge that regulatory
proteins involved in the G2/M transition accumulate to initiate
entry into the M phase and are degraded in the metaphase-anaphase
transition to allow mitotic exit (21,22),
it is speculated that the reduced Cdc2/cyclin B complex activity
cannot activate the anaphase promoting complex (APC), and
therefore, this abrogates the degradation of cyclin B and thereby
enhances the levels cyclin B. However, further studies are required
in order to confirm this hypothesis.
The targeting of the G2/M transition in the cell
cycle is an important strategy of cancer therapy. The only drugs
targeting the G2/M phase currently used in clinical medicine are
microtubule inhibitors, such as vinca alkaloids (vincristine,
vinblastine and vinorelbine), inhibitors of microtubule
polymerization and taxanes (paclitaxel and docetaxel), both leading
to mitotic catastrophe and cell death (23). However, the microtubule inhibitors
destroy microtubules not only in the proliferated cells but also in
the non-proliferated cells, which induces a systemic cytotoxic
effect. Therefore, the development of new drugs targeting the G2/M
phase may provide a potential novel strategy for anti-cancer
therapy.
A number of compounds have been reported to have the
potential to inhibit NSCLC cell growth by downregulatig the
expression of Cdc2 or cyclin B (24,25).
However, to date, no antitumor agent has been reported to suppress
NSCLC cell growth by reducing the dephosphorylation of phospho-Cdc2
(Tyr15). Lee et al reported that 2-methoxyestradiol induced
G2/M cell-cycle arrest via the accumulation of phospho-Cdc2 (Tyr15)
in a nasopharyngeal carcinoma cell line (26). Their findings are similar to the
ones from our study. However, Zhang et al demonstrated that
the reduced phospho-Cdc2 (Tyr15) levels by 8-chloro-adenosine
resulted in mitotic catastrophe in A549 cells (27), which is in conflict with our
results. We found that AZ64 inhibited the dephosphorylation of
phospho-Cdc2 (Tyr15) via the reduction of Cdc25C expression and
therefore enhanced the phospho-Cdc2 (Tyr15) level at the G2/M
transition in the A549 cells. The IC50 of AZ64 was lower than the
other Cdc2/cyclin B inhibitors (24,28).
These results indicate that AZ64 effectively blocks the cells in
the G2/M phase via the inhibition of Cdc2/cyclin B activity at the
G2/M checkpoint.
In conclusion, in this study, we investigated the
antitumor effect of AZ64 on NSCLC cells, as well as its mechanism
of action. Our results demonstrate that AZ64 inhibits the growth of
NSCLC cells in vitro and in vivo and decreases the
invasive ability of the cells. AZ64 treatment leads to cells
arresting in the G2 phase and blocks the cell cycle progression.
The reduced dephosphorylation of phospho-Cdc2 (Tyr15) at the G2/M
transition may be one of the molecular mechanisms involved.
Acknowledgements
The agent AZ64 was supplied by
AstraZeneca Pharmaceuticals via an MD Anderson – AstraZeneca
MTA.
References
|
1.
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
2.
|
McErlean A and Ginsberg MS: Epidemiology
of lung cancer. Semin Roentgenol. 46:173–177. 2011. View Article : Google Scholar
|
|
3.
|
Jemal A, Siegel R, Ward E, Murray T, Xu J,
Smigal C and Tun MJ: Cancer statistics. CA Cancer J Clin.
56:106–130. 2006.
|
|
4.
|
Hsu HF, Huang KH, Lu KJ, Chiou SJ, Yen JH,
Chang CC and Houng JY: Typhonium blumei extract inhibits
proliferation of human lung adenocarcinoma A549 cells via induction
of cell cycle arrest and apoptosis. J Ethnopharmacol. 135:492–500.
2011. View Article : Google Scholar
|
|
5.
|
Xu X, Zhang Y, Qu D, Jiang T and Li S:
Osthole induces G2/M arrest and apoptosis in lung cancer A549 cells
by modulating PI3K/Akt pathway. J Exp Clin Cancer Res. 30:332011.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Wilkinson RW, Odedra R, Heaton SP, Wedge
SR, Keen NJ, Crafter C, Foster JR, Brady MC, Bigley A, Brown E,
Byth KF, Barrass NC, Mundt KE, Foote KM, Heron NM, Jung FH,
Mortlock AA, Boyle FT and Green S: AZD1152, a selective inhibitor
of Aurora B kinase, inhibits human tumor xenograft growth by
inducing apoptosis. Clin Cancer Res. 13:3682–3688. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Harada T, Yatabe Y, Takeshita M, Koga T,
Yano T, Wang Y and Giaccone G: Role and relevance of TrkB mutations
and expression in non-small cell lung cancer. Clin Cancer Res.
9:2638–2645. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Yilmaz T, Jiffar T, Garza G, Lin H, Milas
Z, Takahashi Y, Hanna E, MacIntyre T, Brown JL, Myers JN, Kupferman
ME and Michael E: Theraputic targeting of Trk supresses tumor
proliferation and enhances cisplatin activity in HNSCC. Cancer Biol
Ther. 10:644–653. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Brodeur GM, Minturn JE, Ho R, Simpson AM,
Iyer R, Varela CR, Light JE, Kolla V and Evans AE: Trk receptor
expression and inhibition in neuroblastomas. Clin Cancer Res.
10:3244–3250. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Koizumi H, Morita M, Mikami S, Shibayama E
and Uchikoshi T: Immunohistochemical analysis of TrkA neurotrophin
receptor expression in human non-neuronal carcinomas. Pathol Int.
2:93–101. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Thress K, Macintyre T, Wang H, Whitston D,
Liu ZY, Hoffmann E, Wang T, Brown JL, Webster K, Omer C, Zage PE,
Zeng L and Zweidler-McKay PA: Identification and preclinical
characterization of AZ-23, a novel, selective, and orally
bioavailable inhibitor of the Trk kinase pathway. Mol Cancer Ther.
7:1818–1827. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Iyer R, Varela CR, Minturn JE, Ho R,
Simpson AM, Light JE, Evans AE, Zhao H, Thress K, Brown JL and
Brodeur GM: AZ64 inhibits TrkB and enhances the efficacy of
chemotherapy and local radiation in neuroblastoma xenografts.
Cancer Chemother Pharmacol. May 24–2012.(Epub ahead of print).
|
|
13.
|
Castedo M, Perfettini JL, Roumier T and
Kroemer G: Cyclin-dependent kinase-1: linking apoptosis to cell
cycle and mitotic catastrophe. Cell Death Differ. 9:1287–1293.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Lindqvist A, Rodríguez-Bravo V and Medema
RH: The decision to enter mitosis: feedback and redundancy in the
mitotic entry network. J Cell Biol. 185:193–202. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Telles E, Gurjar M, Ganti K, Gupta D and
Dalal SN: Filamin A stimulates Cdc25C function and promotes entry
into mitosis. Cell Cycle. 10:776–782. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Peng CY, Graves PR, Thoma RS, Wu Z, Shaw
AS and Piwnica-Worms H: Mitotic and G2 checkpoint control:
regulation of 14-3-3 protein binding by phosphorylation of Cdc25C
on serine-216. Science. 277:1501–1505. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Toyoshima-Morimoto F, Taniguchi E and
Nishida E: Plk1 promotes nuclear translocation of human Cdc25C
during prophase. EMBO Rep. 4:341–348. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Seki A, Coppinger JA, Jang CY, Yates JR
and Fang G: Bora and the kinase Aurora A cooperatively activate the
kinase Plk1 and control mitotic entry. Science. 20:1655–1658. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Choi HJ, Fukui M and Zhu BT: Role of
cyclin B1/Cdc2 up-regulation in the development of mitotic
prometaphase arrest in human breast cancer cells treated with
nocodazole. PLoS One. 6:e243122011. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Wang ZX, Xue D, Liu ZL, Lu BB, Bian HB,
Pan X and Yin YM: Overexpression of polo-like kinase 1 and its
clinical significance in human non-small cell lung cancer. Int J
Biochem Cell Biol. 44:200–210. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Lens SM, Voest EE and Medema RH: Shared
and separate functions of polo-like kinases and aurora kinases in
cancer. Nat Rev Cancer. 10:825–841. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Chang DC, Xu N and Luo KQ: Degradation of
cyclin B is required for the onset of anaphase in mammalian cells.
J Biol Chem. 278:37865–37873. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Jorgensen TJ, Tian H, Joseph IB, Menon K
and Frost D: Chemosensitization and radiosensitization of human
lung and colon cancers by antimitotic agent, ABT-751, in athymic
murine xenograft models of subcutaneous tumor growth. Cancer
Chemother Pharmacol. 59:725–732. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Hsiao YC, Hsieh YS, Kuo WH, Chiou HL, Yang
SF, Chiang WL and Chu SC: The tumor-growth inhibitory activity of
flavanone and 2’-OH flavanone in vitro and in vivo through
induction of cell cycle arrest and suppression of cyclins and CDKs.
J Biomed Sci. 14:107–119. 2007.PubMed/NCBI
|
|
25.
|
Shyu KG, Huang ST, Kuo HS, Cheng WP and
Lin YL: Antitumor activity of a novel bis-aziridinylnaphthoquinone
(AZ4) mediating cell cycle arrest and apoptosis in non-small cell
lung cancer cell line NCI-H4601. Acta Pharmacol Sin. 28:559–566.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Lee YM, Ting CM, Cheng YK, Fan TP, Wong
RN, Lung ML and Mak NK: Mechanisms of 2-methoxyestradiol-induced
apoptosis and G2/M cell-cycle arrest of nasopharyngeal carcinoma
cells. Cancer Lett. 268:295–307. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Zhang HY, Gu YY, Li ZG, Jia YH, Yuan L, Li
SY, An GS, Ni JH and Jia HT: Exposure of human lung cancer cells to
8-chloro-adenosine induces G2/M arrest and mitotic catastrophe.
Neoplasia. 6:802–812. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Chen YL, Lin SZ, Chang JY, Cheng YL, Tsai
NM and Chen SP: In vitro and in vivo studies of a novel potential
anticancer agent of isochaihulactone on human lung cancer A549
cells. Biochem Pharmacol. 72:308–319. 2006. View Article : Google Scholar : PubMed/NCBI
|