Introduction
Renal cell carcinoma (RCC) is the most common type
of kidney cancer in adults and its incidence has increased
consistently for the past 20 years (1). The main curative treatment for RCC is
surgery, but the tumor often has recurrence or the patients have
metastatic disease after surgery (2). Therefore, the identification of
effective and specific novel targets for early diagnosis and
treatment are necessary. Insights into the biology of clear-cell
renal cell carcinoma (ccRCC) have identified multiple pathways
associated with the pathogenesis and progression of this cancer,
leading to the development of a number of agents targeting these
signaling pathways, which include the tyrosine kinase inhibitors
sorafenib, sunitinib and pazopanib, the monoclonal antibody
bevacizumab, and the mTOR inhibitors temsirolimus and everolimus
(3).
Aurora kinases are a family of conserved mitotic
regulators consisting of Aurora kinase A, B and C in humans
(4). The activities of Aurora
kinases depends on autophosphorylation of Thr288 in the activation
loop of Aurora A and phosphorylation Thr232 in Aurora B. Aurora
kinase A and B have been well characterized to be important players
in mitosis (5). They are
overexpressed in tumors such as prostate cancer, esophageal
squamous cell carcinoma, and head and neck squamous cell carcinoma,
and are shown to be positively correlated with chromosomal
instability and clinical aggressiveness in the malignancies
(6). Along with cyclins and
cyclin-dependent kinases, Aurora kinases have been reported to link
to G2/M transition of cell cycle (7). Moreover, several reports have
demonstrated that Aurora kinases interact with many important
cellular proteins related to cell cycle and cell division,
including p53 and cdc25 (8).
However, few have been reported concerning the correlation of
Aurora kinases with cell cycle regulation and proliferation in
ccRCC.
We aimed to explore the mechanisms by which Aurora
kinases exerted its effects on ccRCC by downregulation the
expression of the Aurora kinases with miRNAs and VX680
(small-molecule pan Aurora kinases inhibitor). We observed that
silencing the expression of Aurora kinases induced the inhibition
of the proliferation and metastasis, and lead to the G2/M phase
arrest in ccRCC cells. Furthermore, we confirmed the anti-tumor
activity of inhibition of Aurora kinases in HF assay and in a
xenograft model. We observed that silencing Aurora kinases with
miRNAs or treating the cells with VX680 could inhibit the
phosphorylation of ERK, therefore decrease the expression of
cdc25c, cyclinB/cdc2 and upregulate the expression and p-cdc2
(Tyr15). These changes led to inhibition of proliferation,
metastasis and the G2/M arrest in ccRCC; moreover both Aurora
kinases might be important targets for regulation of the tumor
growth.
Therefore, inhibition of Aurora kinases might
contribute to blocking the ccRCC progression. Aurora kinases appear
to be potential therapeutic targets in the management of the renal
cell carcinoma.
Materials and methods
Cell culture
Caki-1 and SN12C cells were kindly provided by
B.T.T. (National Cancer Center, Singapore). The cells were
maintained in DMEM medium (Invitrogen) supplemented with 10% fetal
bovine serum (FBS; Invitrogen), 100 IU/ml penicillin and 100
μg/ml streptomycin (Invitrogen) in a humidified incubator
containing 5% CO2 at 37°C.
Generation of the stable knockdown Aurora
A and Aurora B in the SN12C and Caki-1 cell lines
SN12C and Caki-1 cells were seeded at a density of
1×105 cells per well in 6-well plates. Following
overnight incu bation, the cells reached 50% confluence. They were
then transfected with either the AurA miRNA, AurB miRNA or control
miRNA vector (BLOCK-iT™ Pol II miR RNAi Expression Vector kit with
EmGFP, purchased from Invitrogen) using Lipofectamine 2000
(Invitrogen), according to the manufacturer’s recommended protocol.
In brief, the normal cellular medium was replaced with serum- and
antibody-free DMEM medium. The miRNAs and Lipofectamine mixture
were added directly to the six plates without removal of the
culture medium. The medium was mixed by gentle agitation. The
plates were incubated for an additional 6 h and then changed to
normal medium. The initial selection for transfected cells was
performed by growth in fresh medium containing 8 μg/ml
Blasticidin S HCl (Invitrogen). Selective pressure was maintained
by growth in a medium containing 8 μg/ml Blasticidin S HCl.
After two weeks of growth in selective media, the clone with green
fluorescence was selected and continually cultured in the normal
medium. Cells were harvested for western blot analysis the
expression of Aurora kinases. Stable transfected cells with AurA
miRNA, AurB miRNA were designated Caki-1/AurA1, Caki-1/AurA2,
Caki-1/AurB1, Caki-1/AurB2, SN12C/AurA, SN12C/AurB. Stable
transfected cells with control miRNA were designated Caki-1/C,
SN12C/C.
Analysis of cell proliferation and
viability
Cells were seeded on 96-well plates in DMEM medium
supplemented with 10% fetal bovine serum. Cells were treated with
DMSO or VX680 for 96 h and then cell viability was measured with
the Cell Counting Kit-8 (CCK-8; Dojindo Laboratories, Kumanmoto,
Japan).
Cells (stably transfected with control miRNA, AurA
miRNA, AurB miRNA, respectively) were seeded on 96-well plates in
DMEM medium supplemented with 10% FBS and the proliferation of the
cells was monitored by CCK-8 assay at 24, 48, 72 and 96 h.
Cell cycle analysis
Cells (stably transfected with control miRNA, AurA
miRNA, AurB miRNA, respectively) were cultured in DMEM medium
supplemented with 10% FBS for 96 h or cells were incubated with
either VX680 or DMSO (control) for 96 h. Those cells were then
collected and analyzed using a cellular DNA flow cytometric
analysis kit (Roche). Briefly, cells were collected after treatment
and stained with propidium iodide. Cell cycle profiles were
determined by flow cytometric analysis.
In vitro invasion assay
Invasion was determined using a variation of the
Boyden chamber assay, as described (9). Briefly, cells were trypsinized and
counted; next, 1×106 cells (SN12C cells treated with
DMSO or 0.48 μmol/l VX680 (0.2 IC50); SN12C cells
were stably transfected with control miRNA, AurA miRNA, AurB miRNA,
respectively) suspended in 200 μl of DMEM containing 0.1%
BSA. The cells were seeded into the upper compartment (Costar)
coated polycarbonate filter with a pore size of 8.0 μm in a
24-well plate. Each polycarbonate filter had been coated with 10
μl of 0.5% Matrigel before the addition of cells. DMEM
medium (600 μl) containing 10% FBS was added to the lower
compartment as a chemo attractant. After 14 h of incubation at 37°C
in 5% CO2, 90% relative humidity, the cells on the
underside of the chamber were fixed to the membrane using methanol
for 10 min. Filters were stained with HE stain at room temperature.
Cells in the upper compartment were removed using a cotton swab,
leaving only the cells on the underside of the filter, representing
those cells that had successfully invaded across the
Matrigel-coated filter. The chambers were then photographed to
compare the amount of invasive cells on the underside of the
membrane. The five visual fields were photographed in every
membranes, and manual counting of nuclear-stained cells. All
samples were run in triplicate.
The hollow fibre assay (HF assay)
The cells were harvested by a standard
trypsinisation procedure and resuspended at the desired cell
density (1×106 cells/ml). The cell suspension was
flushed into the hollow fibres, thereafter they were heat-sealed
and cut at 1-cm intervals. The fibres were incubated in DMEM medium
in 6-well plates 24 h prior to surgical implantation in 6- to
8-week-old female pure strain Balb/c-nu/nu mice (Vital River
Laboratory Animal Technology Co. Ltd). All animals were housed in
controlled environment at 25°C on a 12 h light, 12 h dark cycle.
Mice were maintained in accordance with the National Institute of
Health Guide for the Care and Use of Laboratory. Fibres were
implanted s.c. in the back of the mice (two fibres per mouse, one
for SN12C cells, one for Caki-1 cells). Separate in vitro
control fibres were also prepared and were incubated in DMEM medium
during the experiment (10 days). The mice were treated on day 3
with VX680 at 80 mg/kg by i.p. or by 50% PEG (6 mice per group).
The mice were sacrificed at day 10 and the fibres were excised from
the mice. Excess host tissue was removed and the cells were
retrieved from the fibres (both in vitro and in vivo)
for analysis of growth (the ‘stable end-point’ modified MTT assay)
and cell cycle distribution (flow cytometric analysis to measure
cell cycle distribution of the cell population), as described
previously (10). For each
measurement 10,000 cells were counted. The total number of cells in
these fractions in cell cycle analysis was set at 100% (11).
Tumorigenicity of the cells with
silencing Aurora kinases in the xenograft model
Athymic nude mice (Balb/c-nu/nu female,
6–8-week-old) were purchased from Vital River Laboratory Animal
Technology Co. Ltd. Each cell line [SN12C cells stably transfected
with control miRNA (SN12C/C), AurA miRNA (SN12C/AurA) and AurB
miRNA (SN12C/AurB), respectively] was typsined, and washed twice
with PBS. The 5×106 cells suspended in 200 μl
0.9% NaCl were injected s.c. into the left flank of Balb/c-nu/nu
mice, five mice per group. Tumor dimensions were measured twice
every week and the volume calculated as length × width × depth ×
0.5. Percentage of relative tumor volume calculated as the tumor
volume on each day divided by the volume at the time of the first
measurement. Mice were euthanized after the injection on the 22nd
day. Tumors were removed, cleaned from adjacent tissues and
weighed.
Tumor implantation and growth in a ccRCC
xenograft model
Six-week-old female BALB/c-nu/nu nude mice (Vital
River Laboratory Animal Technology Co. Ltd) had five million SN12C
cells subcutaneously implanted in the left flank. When tumors had
grown to an average volume of 100 to 150 mm3,
tumor-bearing mice were separated into two groups of 7 animals. One
group received i.p. injections of 50% PEG300 as a vehicle control;
one group received i.p. injections of VX680 at 80 mg/kg every day.
Tumor size was measured 2–3 times per week and tumor volume was
calculated as length × width × height × 0.5. Percentage of relative
tumor growth calculated as the tumor volume on each day divided by
the volume at the time of the first measurement. The tumor growth
ratio is presented as mean ± SD. Growth curves were plotted to show
the mean relative tumor volume (RTV) within each experimental group
at the indicated time points. Mice were euthanized at the end of
the treatment period (on day 30). Tumors were removed, cleaned from
adjacent tissues, weighed and fixed in 4% polyformaldehyde,
paraffin-embedded, and then 4-μm-thick sections were
prepared. All sections were stained with H&E and were used for
subsequent immunohistochemical analysis. Parts of all sections were
stored at −80°C for western blot analysis.
Cell lysate and western blot
analysis
Lysates (cells treatment with VX680 or DMSO) were
prepared by washing cells with PBS and then following the methods
previously described (12).
Portion of four randomly selected tumors from each group were
homogenized for lysate preparation as previously described
(12). For western blot analysis,
samples transferred to a nitrocellulose membrane by semi-wet
electrophoresis (Invitrogen) were incubated with primary antibody
(rabbit anti-Aurora A, rabbit anti-Aurora B, rabbit
anti-phosphorylated Aurora A (Thr288), mouse anti-phosphorylated
Aurora B (Thr232), rabbit anti-cdc25c, rabbit anti-phosphorylated
p-cdc2 (Tyr15), mouse anti-cdc2, mouse anti-cyclin B, rabbit
anti-phosphorylated p-ERK, rabbit anti-ERK, rabbit anti-PCNA)
overnight at 4°C, detected with horseradish peroxidase-conjugated
anti-rabbit or anti-mouse IgG (Santa Cruz), and developed using an
ECL Western blot detection and analysis system (Applygen
Technologies Inc., Beijing, China). Membranes were tested for equal
loading by probing for actin.
Immunohistochemistry
Immunohistochemical staining was done on 4-μm
formalin-fixed, paraffin-embedded tissue sections. Endogenous
peroxidase activity was blocked with 3% hydrogen peroxide. Antigen
retrieval was carried out in citrate buffer (10 mmol/l, pH 6.0) for
15 min at 100°C in a microwave oven. The slides were incubated with
a primary rabbit rabbit anti-PCNA, overnight at 4°C. Sections were
then incubated with secondary anti-rabbit IgG (Santa Cruz) for 30
min. After washing with 1X TTBS, sections were incubated with
Vectastain ABC reagent (Santa Cruz). The immune complex was
visualized using DAB substrate solution (Santa Cruz). For the
quantitation of PCNA, see the description in Huang et al
(13).
Statistical analysis
All values are expressed as mean ± SD. Values were
compared using Student’s t-test. P<0.05 was considered
significant.
Results
Downregulation of Aurora kinases by
miRNAs or VX680 induces the inhibition of the proliferation in
SN12C and Caki-1 cells in vitro
To assess the activities of Aurora kinases in the
growth of ccRCC cells, we employed the miRNAs targeting either
Aurora A or B to knockdown the expression of Aurora kinases in both
SN12C cells and Caki-1 cells. The basal and active level of the
Aurora A or B was detected by western blot analysis (Fig. 1A). In both SN12C and Caki-1 cells,
silencing of Aurora kinases caused significant reduction on basal
and active levels of Aurora kinases A and B.
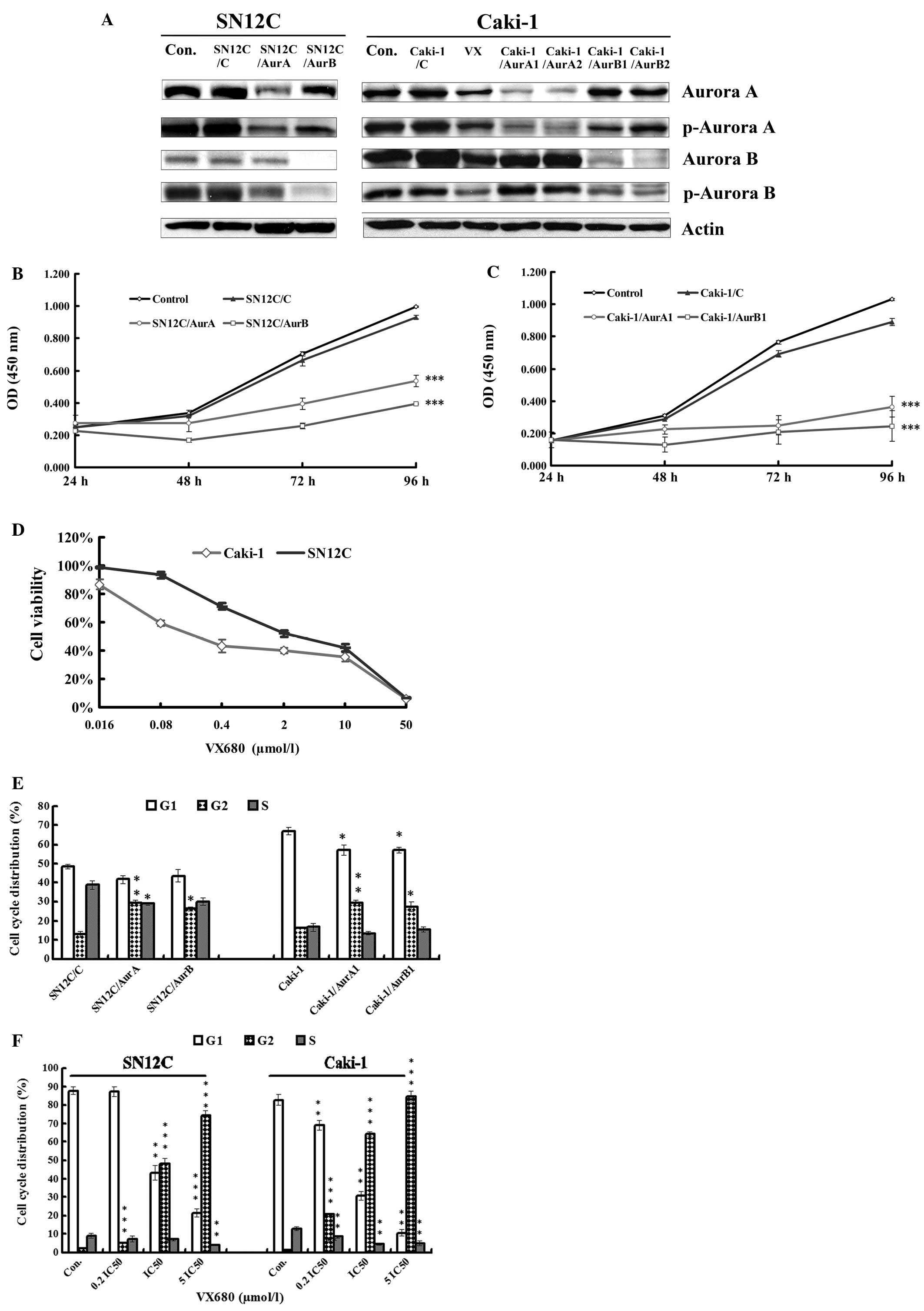 | Figure 1Antiproliferation and cell cycle
arrest caused by inhibition of Aurora kinases. (A) The expression
of Aurora A, Aurora B, p-Aurora A and p-Aurora B in whole-cell
lysates in SN12C and Caki-1 cells silencing of Aurora A and Aurora
B analyzed by western blot analysis. Con, untreated control
samples; VX, samples treated with VX680 0.1 μmol/l (∼0.2
IC50) for 96 h. (B) The growth curve of proliferation of
SN12C cells transfected with control miRNA (SN12C/C), AurA miRNA
(SN12C/AurA) and AurB miRNA (SN12C/AurB). Error bars represented
standard deviation. ***P<0.001 indicates
statistically significant divergence from the non-target control
cells (SN12C/C). (C) The growth curve of proliferation of Caki-1
cells transfected with control miRNA (Caki-1/C), AurA miRNA
(Caki-1/AurA1) and AurB miRNA (Caki-1/AurB1). Error bars
represented standard deviation. ***P<0.001 indicates
statistically significant divergence from the non-target control
cells (Caki-1/C). (D) Effect of VX680 on the viability of SN12C and
Caki-1 cell lines. Cells were treated with VX680 for 96 h and cell
viability was determined by a CCK-8 assay. Error bars represented
standard deviation. (E) miRNAs mediated Aurora kinase
silencing-induced cell cycle arrest in G2/M. SN12C and Caki-1 cells
transfected with control miRNA, AurA miRNA and AurB miRNA were
cultured for 96 h, stained with Annexin V-FITC and propidium iodide
(PI) and analyzed by flow cytometry. Error bars represent standard
deviation. *P<0.05, **P<0.01 indicates
statistically significant divergence from the non-target control
cells. (F) VX680 induced cell cycle arrest in G2/M. SN12C and
Caki-1 cells were incubated with VX680 for 96 h, stained with
Annexin V-FITC and PI and analyzed by flow cytometry. Error bars
represent standard deviation. **P<0.01,
***P<0.001 indicates statistically significant
divergence from the control cells. |
We then detected the potential effects of
miRNA-mediated silencing of Aurora kinases on the growth of Caki-1
and SN12C cells in vitro by CCK-8 assay. Our results showed
that cell growth significantly slowed down after silencing of
Aurora kinases by miRNAs targeting Aurora A and Aurora B compared
with that in control in both cell lines (Fig. 1B and C).
In order to confirm the active role of Aurora
kinases on proliferation in ccRCC cell lines, we conducted the
CCK-8 assay to detect the antiproliferation effect of a
small-molecule pan Aurora kinases inhibitor, VX680, which has
inhibition constants (Ki) of 0.6, 18 and 46 nM for Aurora A, B and
C, respectively (14). SN12C and
Caki-1 cells were cultured in mediums with different concentrations
of VX680 for 96 h. It was found that VX680 obviously decreased the
growth of cells in a dose-dependent manner (Fig. 1D) in both cell lines. Half
inhibition concentration (IC50) was 2.41±0.41
μmol/l and 0.478±0.11 μmol/l in SN12C and Caki-1
cells, respectively, the antiproliferation effect of VX680 was
similar as that of silencing of Aurora kinases by miRNAs,
indicating Aurora A and Aurora B exerted activity on proliferation
in both SN12C and Caki-1 cells.
miRNAs targeting Aurora kinases or VX680
lead to the G2/M arrest in SN12C and Caki-1 cells in vitro
Since we had observed the antiproliferation effect
induced by downregulation of Aurora kinases, we speculated that the
antitumor activity was due to the cell cycle arrest. Several
reports have showed that Aurora kinases are critically involved in
cell cycle regulation and hence in proliferation in some types of
tumors (15). Therefore, we tested
whether deregulation of Aurora kinases induced by knocking down
Aurora kinases or VX680 might have effect on the cell cycle profile
in SN12C and Caki-1 cells. We found that inhibition of either
Aurora A or B by miRNAs also induced cell accumulation in G2/M
phase in both of the two cell lines (Fig. 1E), whereas a decrease in G1 phase
in Caki-1 cells. When the cells were treated with increased
concentrations of VX680 for 96 h, we observed that VX680 led to
significant G2/M arrest and reduction of G1 (Fig. 1F) in a dose-dependent manner. Cells
accumulated almost completely in G2/M in both cells treated with
5IC50 VX680 (2.4 and 12.0 μmol/l for Caki-1 and
SN12C cells, respectively) for 96 h. These results were consistent
with the prediction that the Aurora kinases were crucial for cell
cycle progression in ccRCC, suggesting that inhibition of Aurora
kinases could arrest the ccRCC cells in G2/M phase and the effect
of Aurora kinases on proliferation was partly due to the activity
on the cell cycle in ccRCC cells.
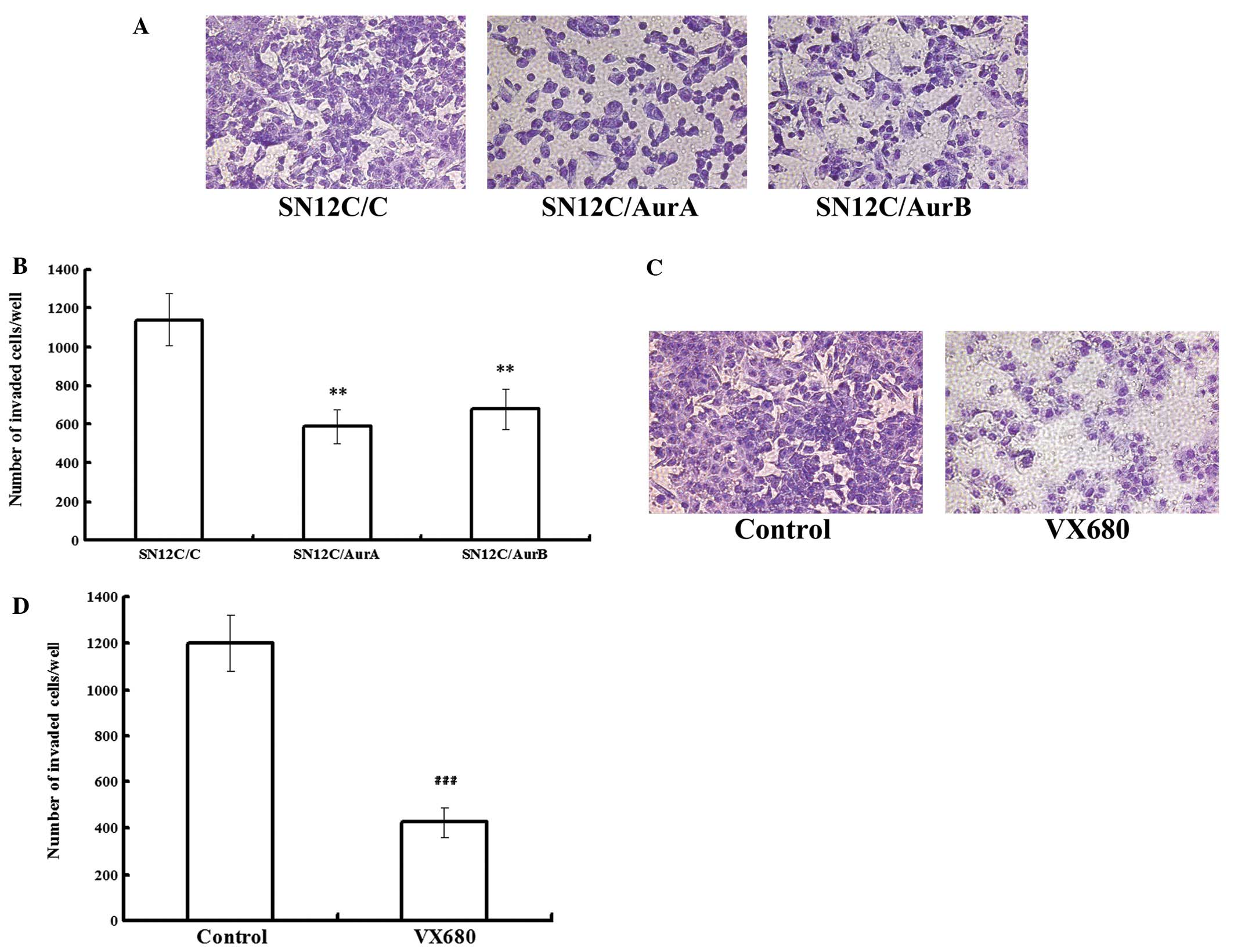
miRNAs targeting Aurora kinases or VX680
inhibit the metastasis in SN12C cells in vitro
In order to determine whether Aurora kinases were
involved in mediating the invasiveness of ccRCC cells, we conducted
transwell assays to examine the role of Aurora kinases in invasion
of SN12C cell lines. Equal numbers of cells (1×106/ml)
by treatment of either silencing of Aurora kinases or being treated
with either DMSO or 0.48 μmol/l VX680 (0.2 IC50)
were allowed to invade through a membrane coated with Matrigel,
toward a chemo attractant (10% FBS) for 14 h. The invaded cells
were fixed, stained and counted. In SN12C cells, invasion was
decreased by 48.30 and 40.58% following Aurora A and Aurora B
depletion, while the vector control cells invaded in similar
numbers to the parental SN12C cells (Fig. 2A and B), indicating that the Aurora
A and B knockdown cells were considerably less invasive than the
vector control cells. VX680 treated cells exhibited a decrease in
invasion of up to 64.59%, compared with SN12C control (Fig. 2C and D). Our data showed that
downregulation of Aurora kinases inhibited the metastasis in SN12C
cells.
Inhibition of Aurora kinases by VX680
induces growth inhibition and cell cycle arrest in SN12C and Caki-1
cells tested by HF assay
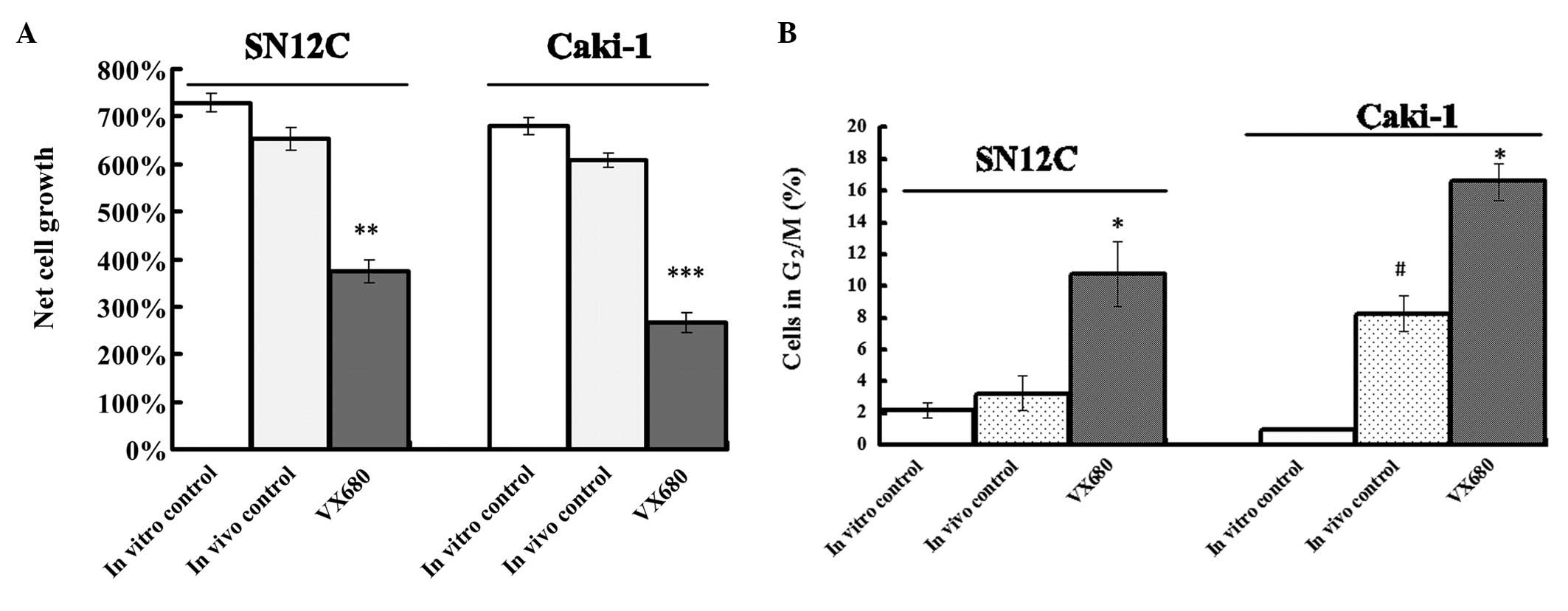
We used HF assay to detect the antitumor effect of
inhibition of Aurora kinases by VX680. The hollow fibres were quite
well tolerated by the nude mice. The VX680 treatment did not affect
the conditions of the mice beyond acceptable limits. From all
experimental groups cell suspensions were retrieved from fibres of
different animals (3 mice per group) to be assayed for cytotoxicity
of VX680. Compared to the in vivo control fibres, VX680
treatment produced a significant reduction in growth of about 32.7
and 42.6% in SN12C and Caki-1 cells, respectively (P<0.01)
(Fig. 3A). Although the cell
growth of in vivo control fibres was a little slower than in
in vitro control fibre, there were no significantly
difference in two groups (P>0.05) (Fig. 3A).
From all experimental groups cell suspensions were
retrieved from fibres of different animals (3 mice per group) to be
assayed for cell cycle phase distribution (Fig. 3B). The tendency for increased
G2/M-phase in vivo control fibres was found in SN12C and
Caki-1 cells compared with the fibres from the in vitro
control. In Caki-1 cells, G2/M phase in the in vivo control
fibres was significant different from that in the in vitro
control fibres. In VX680-treated mice, VX680 treatment resulted in
a clear G2/M-phase arrest for both cell lines (P<0.05). The
effect of VX680 on cell cycle distribution observed in HF assay was
similar as the activity of VX680 in vitro experiments.
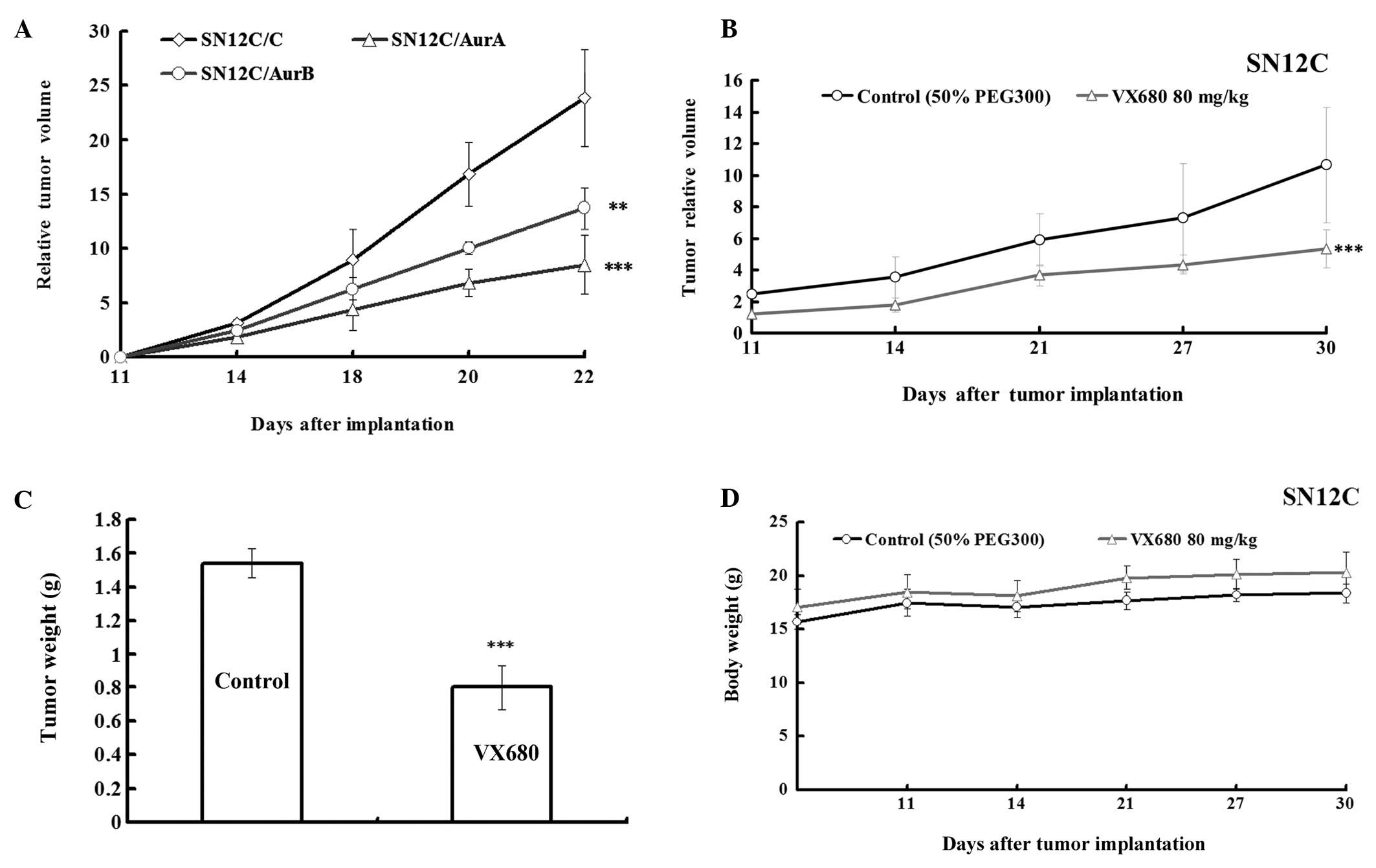
Downregulation of Aurora kinases by
silencing Aurora kinases or VX680 inhibits the growth in SN12C
xenografts
As described above, the effect of inhibition of
Aurora kinases A and B on growth of ccRCC was partly identified
in vitro and semi in vivo experiments, next we
investigated it in vivo. We explored whether the
downregulation of Aurora kinase expression in SN12C cells would
affect their ability to develop tumors in nude mice. We injected
subcutaneously SN12C cells (SN12C/C, SN12C/AurA, SN12C/AurB) into
nude mice (five nude mice per cell line) and observed the growth of
tumors. The tumors of SN12C transfected with AurA miRNA and AurB
miRNA grew slower than that in vector control (Fig. 4A). The growth rate was 35.62 and
57.44% comparing to control, respectively, according to RTV
(Table I), indicating that
silencing of Aurora kinases slowed down the growth of SN12C cells
in xenografts.
 | Table IEffects of miRNA-mediated Aurora
kinase silencing on SN12C tumor growth in athymic mice. |
Table I
Effects of miRNA-mediated Aurora
kinase silencing on SN12C tumor growth in athymic mice.
| | Body weight (g)
| Tumor size
|
|---|
| Group | No. of animals
(n) | Start | End | Volume
(mm3) | RTV | T/C (%) |
|---|
| SN12C/C | 5/5 | 17.94±0.50 | 19.57±0.77 |
2,042.04±542.80 | 23.86±4.44 | |
| SN12C/Aur A | 5/5 | 17.84±0.84 | 19.58±0.72 |
802.68±350.27*** | 8.50±2.69*** | 35.62 |
| SN12C/Aur B | 5/5 | 17.45±0.69 | 19.06±1.19 |
1,289.59±290.13** | 13.71±1.92** | 57.44 |
To further investigate the role of Aurora kinases in
growth of ccRCC in vivo, we evaluated the effect of
inhibition of Aurora kinases by VX680 on tumor growth in an
established SN12C xenograft model. As we had proved the effect of
VX680 on Caki-1 xenografts (12),
tumors in the control animals showed increased volumes and
exponential growth (Fig. 4B), the
group administrated with VX680 (80 mg/kg) suppressed tumor growth
and reduced tumor volume compared with the control with 49.9%
(P<0.001) of T/C ratio at the end of the treatment according to
RTV and a 47.9% (P<0.001) decrease according to tumor weight in
SN12C xenografts (Fig. 4B and C,
Table II). Treatment with VX680
did not alter animal body weight (Fig.
4D), peripheral blood counts, or other biological parameters
(data not shown). These results implied that the antitumor effect
of VX680-mediated Aurora kinase inhibition on the xenograft model
was not due to systemic toxicity.
| Table IIEffects of VX680 on SN12C tumors in
athymic mice. |
Table II
Effects of VX680 on SN12C tumors in
athymic mice.
| | | Body weight (g)
| Tumor size
| Tumor weight
|
|---|
| Group | Dose (mg/kg) | No. of animals
(n) | Start | End | Volume
(mm3) | RTV | T/C (%) | (g) | Inhibition (%) |
|---|
| Control | | 7/7 | 16.70±0.67 | 19.36±0.93 |
1,405.70±230.28 | 10.65±3.01 | | 1.54±0.09 | |
| VX680 | 80×23 | 7/7 | 17.12±1.62 | 20.28±1.90 |
685.09±188.66*** | 5.34±1.04*** | 49.9 | 0.8±0.13*** | 47.9 |
Inhibition of Aurora kinases by miRNAs or
VX680 influences the expression of the cell cycle regulator in
SN12C and Caki-1 cells in vitro
In order to find out whether the growth inhibition
by VX680 was caused through inhibition of the activity of Aurora
kinases in VX680-treated ccRCC cells, we examined basal and active
level of Aurora kinases in the VX680 treated SN12C and Caki-1 cell
lines by western blot. The results showed that VX680 potently
inhibited the phosphorylation of Aurora A and Aurora B in both cell
lines (Fig. 5A). Therefore, the
antiproliferation activity of VX680 was mainly due to the
inhibition of p-Aurora A and p-Aurora B in SN12C and Caki-1 cells
which was consistent with our previous results (12).
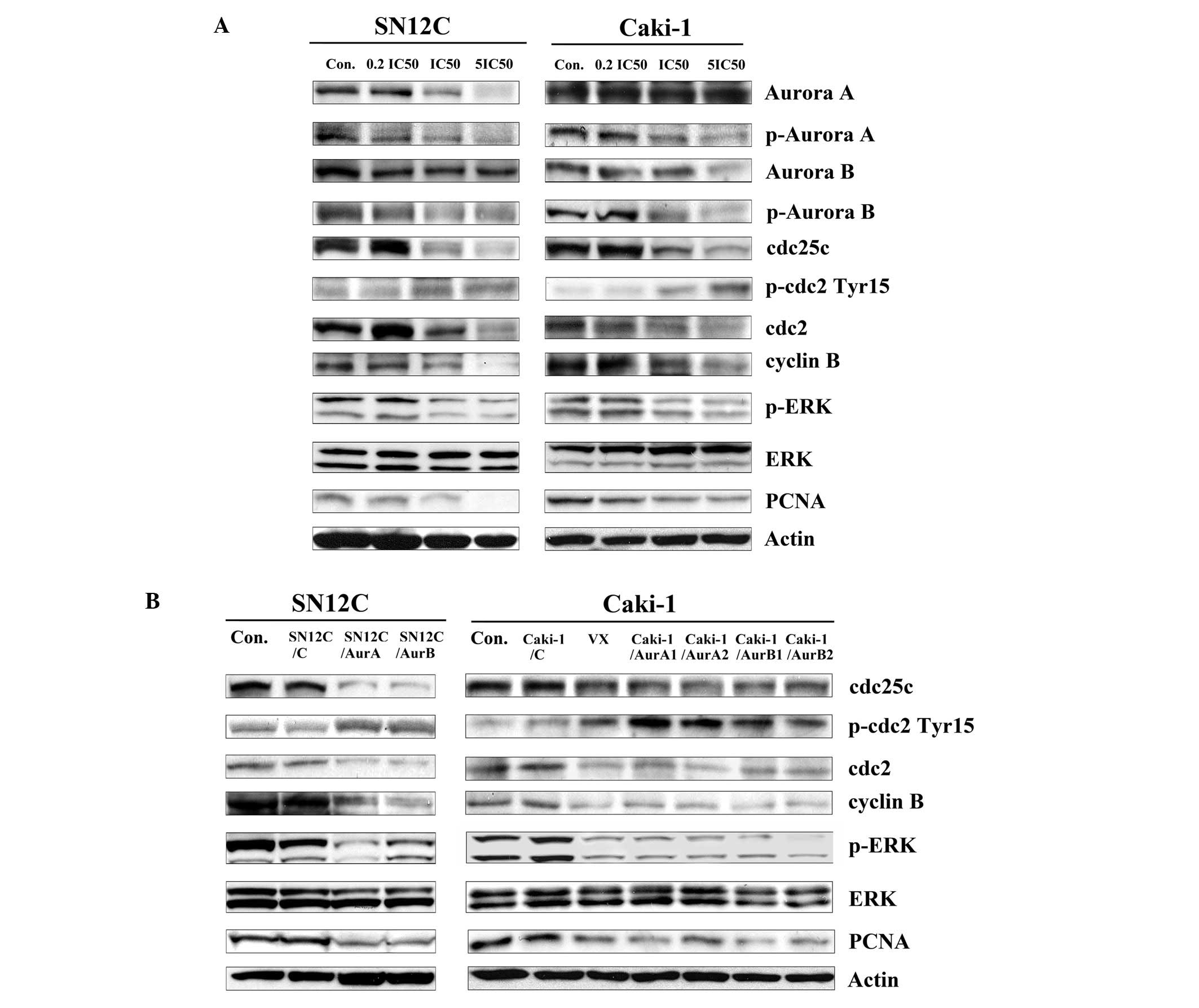 | Figure 5The effect of inhibition of Aurora
kinases on cell cycle regulators in SN12C and Caki-1 cells in
vitro. (A) SN12C and Caki-1 cells were incubated with
increasing concentrations of VX680 for 96 h. Whole-cell lysates
were subjected to western blot analysis. (B) Lysates were prepared
from SN12C, SN12C/C, SN12C/AurA and SN12C/AurB cells, Caki-1,
Caki-1/C, Caki-1/AurA1, Caki-1/AurA2, Caki-1/AurB1, Caki-1/AurB2
cells and Caki-1 cells treated with VX680 0.1 μmol/l (∼0.2
IC50) for 96 h. Con, untreated control samples; VX,
samples treated with VX680, whole-cell lysates were subjected to
western blot analysis. For western blot analysis, samples were
transferred to a nitrocellulose membrane by semi-wet
electrophoresis and incubated with indicated primary antibody
(p-Aurora A, Aurora A, p-Aurora B, Aurora B, cdc25c, p-cdc2
(Tyr15), cdc2, cyclin B, PCNA, p-ERK1/2 (Thr202/Tyr204), ERK1/2).
Actin was used as loading and transfer control. The experiment was
repeated twice and similar results were obtained. |
We further demonstrated that exposure of cells to
VX680 changed the total amounts of Aurora A or Aurora B in mitosis
to some extent, suggesting that the decreased phosphorylation of
Aurora A and Aurora B was not due only to inhibition of
phosphorylation, but also partly to Aurora kinase degradation or
downregulation.
The literature shows that Aurora kinases are
critically involved in cell cycle and thus in proliferation
(15) in some tumors. Based on the
effect of Aurora kinases on the cell cycle distribution proved by
our experiments, we next conducted experiments to examine the
mechanism of cell cycle arrest induced by inhibition of Aurora
kinases, to observe the changes of cell cycle regulator in SN12C
cells and Caki-1 cells treated with AurA miRNA, AurB miRNA and
VX680.
As reported, the cyclin B/cdc2 complex
(maturation/mitosis promoting factor) is thought to regulate
progression through the G2/M phase of the cell cycle (16). In our study, expression of cyclin B
and cdc2 was significantly and dose-dependently decreased in
treating cells with VX680 (Fig.
5A). Knockdown of AuroraA and B induced the downregulation of
cyclin B and cdc2 (Fig. 5B).
Therefore, we draw the conclusion that Aurora kinases contributed
to the regulation of cyclin B/cdc2 in SN12C and Caki-1 cells.
The activity of cdc2 is negatively regulated by the
phosphorylation of the amino acid residue threonine-14 (Thr14) and
tyrosine-15 (Tyr15). Cdc25C, a phosphatase to remove the phosphate
group from p-cdc2 (Tyr15), is the key component controlling the
entry of cells into mitosis (17,18).
Therefore, we sought to determine the phosphorylation states of
cdc2 and the expression of cdc25C in cells after inhibition of
Aurora kinases. In this study, VX680 treatment resulted in a
decrease in total cdc25c and an increase in cdc2 phosphorylation at
Tyr15 site in SN12C and Caki-1 cells (Fig. 5A). Consistent with these results,
the increased expression of p-cdc2 (Tyr15) and decreased expression
of cdc25c was found in the cell silencing of Aurora kinases
(Fig. 5B).
Members of the MAP kinase (MAPK) family have long
been known to play an important role in regulating cell growth,
differentiation, and cell death (19). One of them, the ERK pathway, is
generally considered as a survival promotion pathway (20). To investigate whether Aurora
kinases had an effect on the ERK pathway, the SN12C and Caki-1
cells were downregulated by the expression of Aurora kinase miRNAs
or VX680, and then the phosphorylation status and basal levels of
ERK was determined by western blot with specific antibodies. As
shown in Fig. 5A and B, the
phosphorylation of ERK was reduced in both cells when treated with
Aur miRNA or VX680, but there was no influence on the expression of
total ERK.
As we proved that downregulation of Aurora kinases
inhibited the growth of SN12C and Caki-1 cells, we evaluated the
level of PCNA (the proliferation marker), as shown in Fig. 5A, VX680 decreased the expression of
PCNA in a dose-dependent fashion as determined by western blot
analysis. The low level of PCNA was also observed in cell silencing
of the expression of Aurora kinases (Fig. 5B).
Downregulation of Aurora kinases by VX680
affects the cell cycle regulator in SN12C xenografts
Tumor tissues obtained from mice on day 30 after
tumor implantation were examined for expression and activity of
Aurora kinases by western blot analysis. We found that relatively
lower levels of p-Aurora A and p-Aurora B were detected in
VX680-treated tumors when compared with the control tumors
(Fig. 6A). These results were
consistent with the results from the in vitro experiments,
which showed a decrease of phosphorylation of Aurora kinases in the
VX680-treated cells (Fig. 5A). We
also found that cell proliferation within tumors, as indicated by
PCNA, was markedly reduced, as analyzed by western blot analysis
(Fig. 6A) and supported by PCNA
immunohistochemical analysis. PCNA staining in control tumors
showed 61.73±15.98% PCNA-positive cells (proliferation index),
whereas only 27.64±15.54% PCNA-positive cells were observed in the
VX680-treated group, a decrease of 55.2% (P<0.001) (Fig. 6B and C).
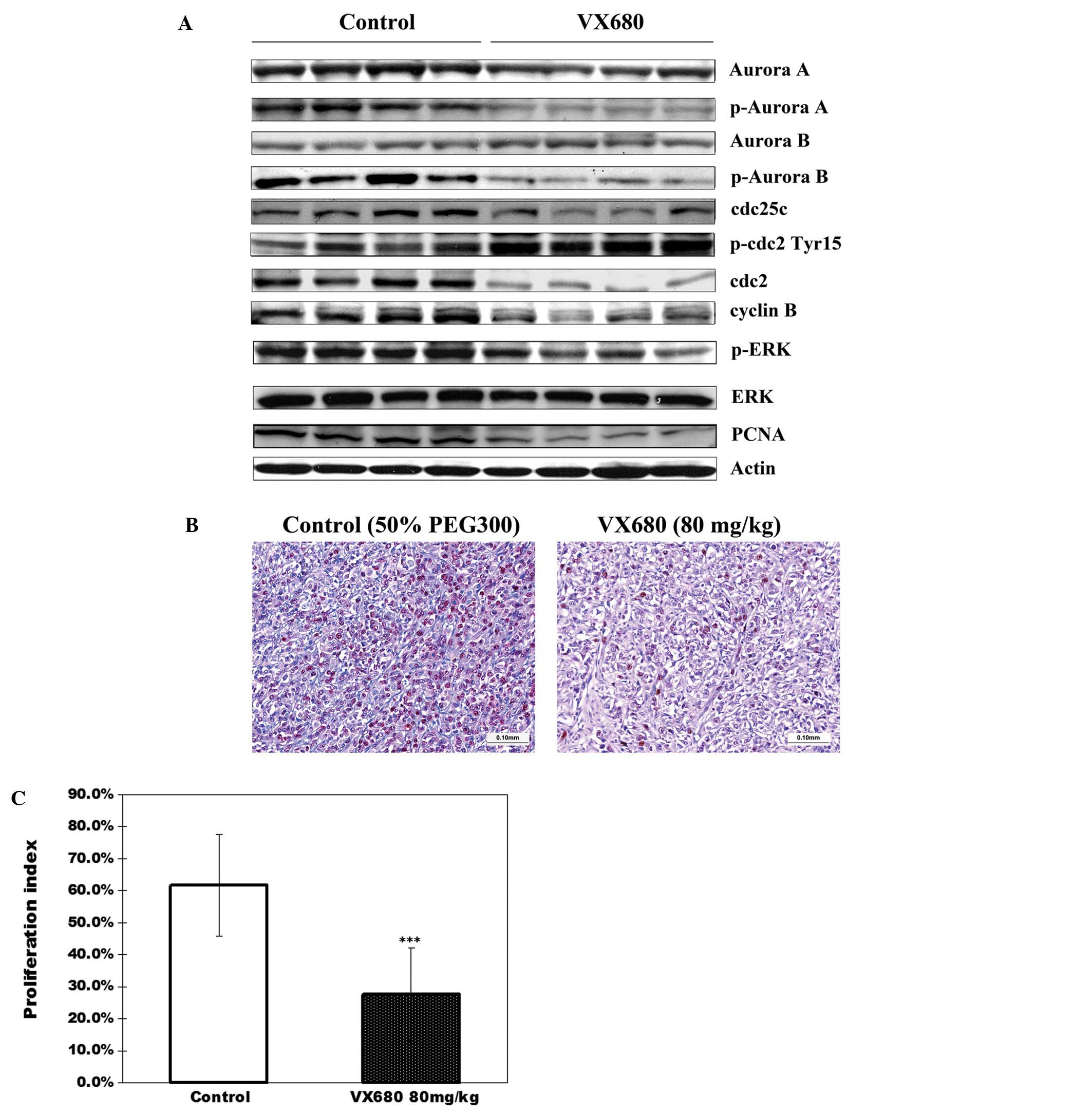 | Figure 6The effect of inhibition of Aurora
kinases by VX680 on cell cycle regulators in SN12C xenografts. (A)
A portion of four randomly selected tumors from each group (SN12C
xenograft-bearing mice treated daily with VX680 or 50% PEG300) were
homogenized for lysate preparation and analyzed by western blot for
protein expression. For western blot analysis, samples were
transferred to a nitrocellulose membrane by semi-wet
electrophoresis and incubated with primary antibody. The expression
of p-Aurora A, Aurora A, p-Aurora B, Aurora B, cdc25c, p-cdc2
(Tyr15), cdc2, cyclinB, PCNA, p-ERK1/2 (Thr202/Tyr204), ERK1/2.
Actin was used as loading and transfer control. The experiment was
repeated twice and similar results were obtained. (B)
Immunohistochemical staining for PCNA in SN12C xenograft tumors
after treatment with VX680 or 50% PEG300. (C) Quantification of a
proliferation index (PCNA) staining in SN12C xenograft tumors.
Error bars represent standard deviation. ***P<0.0001
indicates statistically significant divergence from the control
group. Inhibition of Aurora kinases by VX680 decreased the
expression of PCNA in SN12C xenograft tumors. |
Next we analyzed the alterations of cyclin B/cdc2
expression in xenograft tumors after VX680 treatment in SN12C
xenografts. We observed that cyclin B, cdc2 and cdc25c were
downregulated in the VX680-treated group relative to the control
group (Fig. 6A). Furthermore, the
protein levels of p-cdc2 obviously increased in VX680-treated mice
(Fig. 6A), whereas the active
level ERK was decreased, the basal level of ERK did not change. The
results were consistent with the results of the in vitro
experiments, further confirming that the effect of inhibition of
Aurora kinases on cell cycle regulator in ccRCC.
Discussion
Aurora A/B/C have been proved to be related with
cancer progression and to be involved in the regulation of the cell
cycle. They have been suggested as possible new anticancer targets
(21). Despite previous studies of
Aurora kinases on various tumors (22), the important roles of Aurora
kinases and their signaling pathway in ccRCC is not fully
elucidated. Our current study was an attempt to address this issue
and to clarify the potent function of Aurora kinases in human ccRCC
(23).
The functions of Aurora kinases were investigated
through dowregulating the expression of Aurora kinases in the SN12C
and Caki-1 lines. VX680 treatment or Aurora kinases silencing using
miRNAs caused inhibition of the basal and active levels of Aurora
kinases, following decrease in cell proliferation ability, leading
to arrest in G2/M phase and inhibition of metastasis in both cell
types. In HF assay, we found that the growth of SN12C and Caki-1
was inhibited and cells were arrested in G2/M phases after
treatment with VX680. Moreover, tumors injected SN12C cells with
low expression of Aurora kinases induced by miRNAs grew more slowly
than the tumors injected with SN12C control miRNA. In SN12C
xenograft model, VX680 reduced the tumor growth through blocking
the activity of Aurora kinases. Above results implied that Aurora
kinases had some effects on proliferation in ccRCC.
The mechanism of cell cycle regulation is
complicated and the cell cycle progression is tightly regulated by
the synthesis/degradation, association/dissociation, translocation
between cytoplasm and nucleius, and the
phosphorylation/dephosphorylation of several protein regulators
(24). The progression of cell
from G2 to M phase requires the coordination of various regulatory
proteins, and is initiated by formation of the cyclinB/cdc complex
through the activation of cdc25c, leading cells into mitosis
(25). Before entry to the mitotic
phase, cdc2 is kept inactive by tyrosine-15 (Tyr15) and
threonine-14 (Thr14) and dephosphorylation of the two inhibitory
subunits Tyr15 and Thr14 by cdc25c dual phosphatase will drive
cells into mitosis (26).
Therefore, disruption of any of these processes will destroy cell
cycle progression. In this study, we proved that inhibition of
Aurora kinases caused a decrease of cdc25c, leading to an
accumulation of Tyr15-phosphorylated cdc2, thus cdc2 was kept
inactive. Hence cells could not enter mitosis and arrest in G2
phase. Inhibition of Aurora kinases downregulated the expression of
cyclin B companied with the G2 phase arrest both in cells and SN12C
xenograft tumors. Taken together, those results indicated the
downregulation of cyclinB/cdc2 caused by inhibition of Aurora
kinases contributed to the G2/M accumulation in ccRCC. Thus we
concluded that Aurora kinases were the key regulators of the cell
cycle in ccRCC.
Some reports have suggested a role of ERK1/2
signaling in G2/M checkpoint control related with cdc2-Tyr15
phosphorylation following DNA damage (27) and suppression of ERK by PD98059
treatment blocked both the inhibition of cell proliferation and the
downregulation of cdc2 and cdc25c (28). The results in our present study
displayed that miRNA- or VX680-mediated Aurora kinase inhibition
could decrease the phosphorylation level of ERK in vitro and
in vivo, so we deduced that Aurora kinases might be involved
in the G2-phase arrest and cell proliferation via the induction of
ERK pathway.
It has been reported that specific blockage of ERK
pathway in colon tumor cells could inhibit the alterations in cell
cell contact and motility that is required for metastasis (29). Our results support those findings.
We observed that inhibition of Aurora kinases by miRNAs or VX680
could reduce the metastasis ability of ccRCC cells accompanied with
blocking the activity of ERK.
In conclusion, in this study, we described the
contribution of Aurora kinase signaling to cell proliferation,
metastasis and cell cycle regulation in ccRCC. Inhibition of Aurora
kinases downregulated the activity of ERK, therefore regulating the
expression of the cell cycle regulator cdc25c and cdc2.
Downregulation of Aurora kinases also caused the decrease of cyclin
B. Hence we deduced that Aurora kinases could exerted its effect on
cell cycle and proliferation through affecting the formation of
cyclinB/cdc2 via ERK signal in ccRCC.
In this study, we only investigated the function of
Aurora kinases on the cell cycle, proliferation, metastasis and the
relationship of Aurora kinases with ERK phosphorylation in
ccRCC.
Thus, although we proved that inhibition of Aurora
kinases might block the ccRCC progression, and Aurora kinases might
become therapy targets, more studies are needed on the complex
networks of the Aurora kinases pathway and their role(s) in
ccRCC.
Acknowledgements
We thank Dr Jindong Chen (Kidney
Cancer Research Laboratory, Department of Urology University of
Rochester Medical Center) for help in revising the manuscript. This
study was supported by The National Natural Science Foundation of
China (Approval number: 81102025).
References
|
1.
|
Bensalah K and Patard JJ: Kidney cancer in
2010: drugs, surgery and survival in RCC. Nat Rev Urol. 8:66–68.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Chowdhury S and Choueiri TK: Recent
advances in the systemic treatment of metastatic papillary renal
cancer. Expert Rev Anticancer Ther. 9:373–379. 2009. View Article : Google Scholar
|
|
3.
|
Chowdhury S, Matrana MR, Tsang C, et al:
Systemic therapy for metastatic non-clear-cell renal cell
carcinoma: recent progress and future directions. Hematol Oncol
Clin North Am. 25:853–869. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Keen N and Taylor S: Aurora-kinase
inhibitors as anticancer agents. Nat Rev Cancer. 4:927–936. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Tyler RK, Shpiro N, Marquez R and Eyers
PA: VX-680 inhibits Aurora A and Aurora B kinase activity in human
cells. Cell Cycle. 6:2846–2854. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Yang H, He L, Kruk P, et al: Aurora-A
induces cell survival and chemoresistance by activation of Akt
through a p53-dependent manner in ovarian cancer cells. Int J
Cancer. 119:2304–2312. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Dutertre S, Cazales M, Quaranta M, et al:
Phosphorylation of CDC25B by Aurora-A at the centrosome contributes
to the G2-M transition. J Cell Sci. 117:2523–2531. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Gizatullin F, Yao Y, Kung V, et al: The
Aurora kinase inhibitor VX-680 induces endoreduplication and
apoptosis preferentially in cells with compromised p53-dependent
postmitotic checkpoint function. Cancer Res. 66:7668–7677. 2006.
View Article : Google Scholar
|
|
9.
|
Merk BC, Owens JL, Lopes MB, et al: STAT6
expression in glioblastoma promotes invasive growth. BMC Cancer.
11:1842011. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Cloos J, Temmink O, Ceelen M, et al:
Involvement of cell cycle control in bleomycin-induced mutagen
sensitivity. Environ Mol Mutagen. 40:79–84. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Temmink OH, Prins HJ, van Gelderop E and
Peters GJ: The Hollow Fibre Assay as a model for in vivo
pharmacodynamics of fluoropyrimidines in colon cancer cells. Br J
Cancer. 96:61–66. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Li Y, Zhang ZF, Chen J, et al:
VX680/MK-0457, a potent and selective Aurora kinase inhibitor,
targets both tumor and endothelial cells in clear cell renal cell
carcinoma. Am J Transl Res. 2:296–308. 2010.PubMed/NCBI
|
|
13.
|
Huang D, Ding Y, Luo WM, et al: Inhibition
of MAPK kinase signaling pathways suppressed renal cell carcinoma
growth and angiogenesis in vivo. Cancer Res. 68:81–88. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Harrington EA, Bebbington D, Moore J, et
al: VX-680, a potent and selective small-molecule inhibitor of the
Aurora kinases, suppresses tumor growth in vivo. Nat Med.
10:262–267. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Teicher BA: Newer cytotoxic agents:
attacking cancer broadly. Clin Cancer Res. 14:1610–1617. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Suzuki T, Urano T, Miki Y, et al: Nuclear
cyclin B1 in human breast carcinoma as a potent prognostic factor.
Cancer Sci. 98:644–651. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Perdiguero E and Nebreda AR: Regulation of
Cdc25C activity during the meiotic G2/M transition. Cell Cycle.
3:733–737. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Singh SV, Herman-Antosiewicz A, Singh AV,
et al: Sulforaphane-induced G2/M phase cell cycle arrest involves
checkpoint kinase 2-mediated phosphorylation of cell division cycle
25C. J Biol Chem. 279:25813–25822. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Pearson G, Robinson F, Beers Gibson T, et
al: Mitogen-activated protein (MAP) kinase pathways: regulation and
physiological functions. Endocr Rev. 22:153–183. 2001.PubMed/NCBI
|
|
20.
|
Lee SJ, Park K, Ha SD, et al: Gleditsia
sinensis thorn extract inhibits human colon cancer cells: the role
of ERK1/2, G2/M-phase cell cycle arrest and p53 expression.
Phytother Res. 24:1870–1876. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Meraldi P, Honda R and Nigg EA: Aurora
kinases link chromosome segregation and cell division to cancer
susceptibility. Curr Opin Genet Dev. 14:29–36. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Gautschi O, Heighway J, Mack PC, et al:
Aurora kinases as anticancer drug targets. Clin Cancer Res.
14:1639–1648. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Lee EC, Frolov A, Li R, et al: Targeting
Aurora kinases for the treatment of prostate cancer. Cancer Res.
66:4996–5002. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Lee YM, Ting CM, Cheng YK, et al:
Mechanisms of 2-methoxyestradiol-induced apoptosis and G2/M
cell-cycle arrest of nasopharyngeal carcinoma cells. Cancer Lett.
268:295–307. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Margolis SS, Perry JA, Weitzel DH, et al:
A role for PP1 in the Cdc2/Cyclin B-mediated positive feedback
activation of Cdc25. Mol Biol Cell. 17:1779–1789. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Ray G, Dhar G, Van Veldhuizen PJ, et al:
Modulation of cell-cycle regulatory signaling network by
2-methoxyestradiol in prostate cancer cells is mediated through
multiple signal transduction pathways. Biochemistry. 45:3703–3713.
2006. View Article : Google Scholar
|
|
27.
|
Cheng Y, Qiu F, Ye YC, et al: Oridonin
induces G2/M arrest and apoptosis via activating ERK-p53 apoptotic
pathway and inhibiting PTK-Ras-Raf-JNK survival pathway in murine
fibrosarcoma L929 cells. Arch Biochem Biophys. 490:70–75. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Hsu YL, Kuo PL, Lin LT and Lin CC: Asiatic
acid, a triterpene, induces apoptosis and cell cycle arrest through
activation of extracellular signal-regulated kinase and p38
mitogen-activated protein kinase pathways in human breast cancer
cells. J Pharmacol Exp Ther. 313:333–344. 2005. View Article : Google Scholar
|
|
29.
|
Guruvayoorappan C and Kuttan G:
Amentoflavone inhibits experimental tumor metastasis through a
regulatory mechanism involving MMP-2, MMP-9, prolyl hydroxylase,
lysyl oxidase, VEGF, ERK-1, ERK-2, STAT-1, NM23 and cytokines in
lung tissues of C57BL/6 mice. Immunopharmacol Immunotoxicol.
30:711–727. 2008. View Article : Google Scholar
|