Introduction
Accumulating evidence supports the notion that most
human tumors develop in an environment of chronic inflammatory
conditions (1). Consistent with
this view several epidemiological studies have connected chronic
inflammation with increased risk of cancer development (2). Recent studies have associated DNA
damage triggered by inflammation with increased transformation
potential of pre-malignant cells within tissues with no indications
of underlying infection (3)
implying that regardless of the way inflammation is generated its
components (inflammatory cells and mediators) ultimately establish
an environment that promotes tumor growth and progression.
Hallmarks of cancer-related inflammation include the
presence of leukocyte infiltrating cells, cytokines, and peptide
mediators of cellular immunity at the sites of tumors (4–6). The
balance of pro- (IL-1, IL-6, TNF-α) and anti-inflammatory cytokines
(IL-10, IL-16) and their interdependent interactions within the
neoplastic tissue are of pivotal importance in determining the
progression and the outcome of the inflammatory reactions (7). TNF-α is a major pro-inflammatory
cytokine detected in malignant cells in human breast, ovarian,
prostate and bladder tumors as well as in leukemia and lymphomas
(8). Since TNF-α is a potent
inducer of cancer cell survival and contributes to chronic
inflammation and cancer development anti-TNF strategies are used in
cancer therapy (9). IL-10 is
produced by immune and malignant cells and facilitates cancer cells
to escape immune surveillance (10). IL-10 exerts anti-inflammatory
effects by inhibiting the production of pro-inflammatory cytokines
thus acting as an inhibitor of tumor progression within the tumor
microenvironment (7).
A subset of cytokines linked to cancer and in
particular to metastasis are the chemotactic cytokines chemokines
(11). Deregulation of chemokine
networks or inappropriate activation can contribute to inflammatory
diseases and malignant transformations (12,13).
Signalling via CXCR4 chemokine receptor regulates processes such as
cell proliferation, trafficking of immune cells, migration,
adhesion and angiogenesis, as part of normal physiology (14). However, since most of these events
are also required for carcinogenesis, this receptor is the most
commonly found in cancer cells (15) and has been shown to play important
role in cancer progression as it is involved in the metastatic
spread of many human tumors, including breast cancer (13). Its unique ligand, CXCL12, is also
expressed within the tumor microenvironment as well as at distant
metastatic sites and the CXCR4-CXCL12 axis is essential in the
control of invasion and metastasis (14,16).
The cytokine/chemokine network within the tumor
microenvironment triggers signalling pathways that induce the
expression of genes involved in tumor cell growth and invasiveness,
angiogenesis, metastasis and production of more cytokines that
affect the existent network and modulate malignant progression.
Signal transducer and activator of transcription 3 (STAT3), along
with nuclear factor-kappa B (NF-κB) and hypoxia inducible factor 1
alpha (HIF-1α) are the main regulatory transcription factors
coordinating tumor initiation and progression in tissues where
cancer-related inflammation occurs (5,17).
Hypoxia is a common feature of inflammatory solid tumors that
alters the gene expression profile of genes involved in the
regulation of metabolism, angiogenesis, tissue remodelling,
proliferation and apoptosis (18).
HIF-1α promotes tumor cell growth and metastasis by upregulating
among other genes the expression of CXCR4 in ovarian and breast
cancer cells (19). On the other
hand, the p53 tumor suppressor mediates both apoptotic cell death
and anti-angiogenic effects by repressing HIF-1α and CXCR4
(20) in hypoxic tumors and
mutations inhibiting its pro-apoptotic competence allow
inflammation and NF-κB to exert their tumor-promoting effects
(21).
The HIF-1α and p53 transcription factors interfere
with each other’s protein stability and transcriptional activity in
hypoxic conditions (22–24). Previous work in our laboratory has
indicated that the p300/CBP associated factor (PCAF) is a crucial
coordinator of the action of both p53 and HIF-1α in hypoxic cancer
cells (22). PCAF, p300 and
members of the p160 family of the nuclear hormone receptor (NR)
co-activators, such as the steroid receptor co-activator 1 (SRC-1),
bear intrinsic histone acetyl transferase (HAT) activity thereby
modulating directly or indirectly the transcriptional activity of
both HIF-1α and p53 (22,25,26).
On the contrary, members of the NAD+-dependent
deacetylase (HDAC) family of sirtuins, such as SIRT-1, antagonise
HAT activity and target p53 and HIF-1α for deacetylation in
mammalian cells (27). Both SRC-1
and SIRT-1 affect several phenotypic aspects of mammary tumors
interfering with the p53 and HIF-1α pathways (27,28).
Herein, we present evidence endorsing the view that SRC-1 and
SIRT-1 are involved in the regulation of gene expression of the
potent metastatic chemokine receptor CXCR4 in hypoxic breast cancer
cells by modulating the transcriptional activity of both p53 and
HIF-1α.
Materials and methods
Cell lines, cell culture and
constructs
The human breast carcinoma cell lines MCF-7
(p53+/+) and MDA-MB-231 (p53+/−) (obtained
from ECACC) were routinely maintained in Dulbecco’s modified
Eagle’s medium (Sigma-Aldrich, UK), supplemented with 10% v/v
foetal bovine serum (Gibco, UK) and 1% penicillin/streptomycin 10
U/ml (Lonza, USA) at 37°C in a humidified atmosphere containing 5%
CO2. Cells were treated with 10 μM etoposide
(Sigma-Aldrich) and 250 μM desferrioxamine (DSFX)
(Sigma-Aldrich) for 16 h. Transient transfections were carried out
using the Polyfect transfection reagent (Qiagen, Sussex, UK),
according to the manufacturer’s instructions. Constructs used for
exogenous expression included HA-SRC-1 (29) Flag-SIRT-1 (Addgene, Cambridge, MA),
pcDNA3 (22), and β-galactosidase
(22). Human CXCR4 luciferase
reporter containing 5 consensus HREs was constructed by amplifying
the - 1619 to −258 region (counted from the translation initiation
site) and inserting it in the pGL3 promoter luciferase vector
(Promega, USA).
Quantitative RT-PCR
Quantitative reverse transcription PCR was performed
as previously described (22).
Briefly, total RNA was extracted from MCF-7 cells using the RNeasy
kit (Qiagen), according to the manufacturer’s instructions. The RNA
was then reverse transcribed to cDNA and used for qPCR analysis
using SYBR Green fluorescent probe. Analysis was performed with the
Opticon Monitor (Bio-Rad, USA) or Realplex (Eppendorf, UK)
software. The primer sequences used for the qRT-PCR reactions are
provided in Table I.
 | Table ISummary of the primers used in ChIP,
qRT-PCR and the construction of the CXCR4-luciferase reporter. |
Table I
Summary of the primers used in ChIP,
qRT-PCR and the construction of the CXCR4-luciferase reporter.
| Primer name | Sequence
(5′→3′) | Used for |
|---|
| CXCR4 (F) |
CATGTGTCTCCCCCTTGAGT | ChIP |
| CXCR4 (R) |
TCCGCCTCTAAATTCAGACAA | ChIP |
| TNFα (F) |
CTGCCCAAGAAAGAAACCAA | ChIP |
| TNFα (R) |
AAAAAGGGAAGGCAAGAAGG | ChIP |
| CXCR4 (F) |
CAGCAGGTAGCAAAGTGACG | qRT-PCR |
| CXCR4 (R) |
ATAGTCCCCTGAGCCCATTT | qRT-PCR |
| TNFα (F) |
AGCCCATGTTGTAGCAAACC | qRT-PCR |
| TNFα (R) |
ATGAGGTACAGGCCCTCTGA | qRT-PCR |
| IL10 (F) |
TTACCTGGAGGAGGTGATGC | qRT-PCR |
| IL10 (R) |
GGCCTTGCTCTTGTTTTCAC | qRT-PCR |
| Rpl19 (F) |
ATGTATCACAGCCTGTACCTG | qRT-PCR |
| Rpl19 (R) |
TTCTTGGTCTCTTCCTCCTTG | qRT-PCR |
| CXCR4 (F) LUC |
GGTACCGTGCACAAGTGCAGAGAAGG | Construction of the
luciferase reporter |
| CXCR4 (R) LUC |
GAGCTCAAGAGGGGAGAAGGGAGGAT | Construction of the
luciferase reporter |
Immunoblotting and antibodies
Cells were harvested in high salt lysis buffer (500
mM NaCl, 50 mM Tris-HCl pH 7.5, 5 mM EDTA pH 8.0, 0.5% NP-40, 1%
Triton X-100) containing 1 μg/ml protease inhibitor cocktail
(pepstatin, aprotinin, and leupeptin) and 1 mM
phenylmethylsulphonyl fluoride (PMSF). Equal amount of protein was
loaded on polyacrylamide gels (7.5%) and resolved by SDS-PAGE.
After electroblotting and incubation with primary and secondary
antibodies, blots were developed using ECL substrate according to
the manufacturer’s instructions (Pierce, Thermo Scientific, USA).
The antibodies used were anti-HIF-1α [Calbiochem (H1a67)], anti-p53
[Santa Cruz Biotechnology (sc-126)], anti-CXCR4 [R&D (MAB
721)], anti-SRC-1 [Abcam (84)], anti-β-actin [Abcam (8227)],
anti-HA [Babco Covance (HA-11)], and anti-Flag [Sigma (M-20)].
Chromatin immunoprecipitation
Chromatin was cross-linked by addition of 1.42%
formaldehyde in the culture medium. Cross-linking was quenched by
addition of 125 mM glycine and cells were harvested in IP buffer
(150 mM NaCl, 50 mM Tris-HCl pH 7.5, 5 mM EDTA pH 8.0, 0.5% NP-40,
1% Triton X-100, 1 mM PMSF, 1 μg/ml protease inhibitor
cocktail (pepstatin, aprotinin, and leupeptin), 20 mM β-glycerol
phosphate, and 2 mM sodium orthovanadate). To shear the chromatin,
nuclear extracts were sonicated (Bioruptor sonicator), a fraction
from each lysate was kept for use as input sample and the rest of
the lysate was subjected to immunoprecipitation with 2 μg of
antibody. The reverse crosslinked DNA fragments were then amplified
in PCR reactions with specific primers (Table I) flanking the HREs within the
CXCR4 and TNF-α promoter and analyzed on agarose gel
electrophoresis.
Results
CXCR-4, TNF-α and IL-10 mRNA levels in
DSFX and etoposide-treated breast cancer cells
Solid tumors bear hypoxic fractions and are often
treated with chemotherapeutics such as the topoisomerase II
inhibitor etoposide. The therapeutic effect of these compounds
depends on their ability to trigger DNA damage and stimulate p53
mediated apoptotic response (30).
A previous report from our laboratory along with published
observations from other groups, indicate that the function of p53
differs significantly in hypoxic compared to normoxic environment
(22). Taken these observations
into account, the expression pattern of CXCR4, which is a
gene targeted by both HIF-1α (19)
and p53 (20), was followed in
MCF-7 cells under conditions where both p53 and HIF-1α were
transcriptionally active in order to delineate the molecular
mechanisms involved in the regulation of its gene expression.
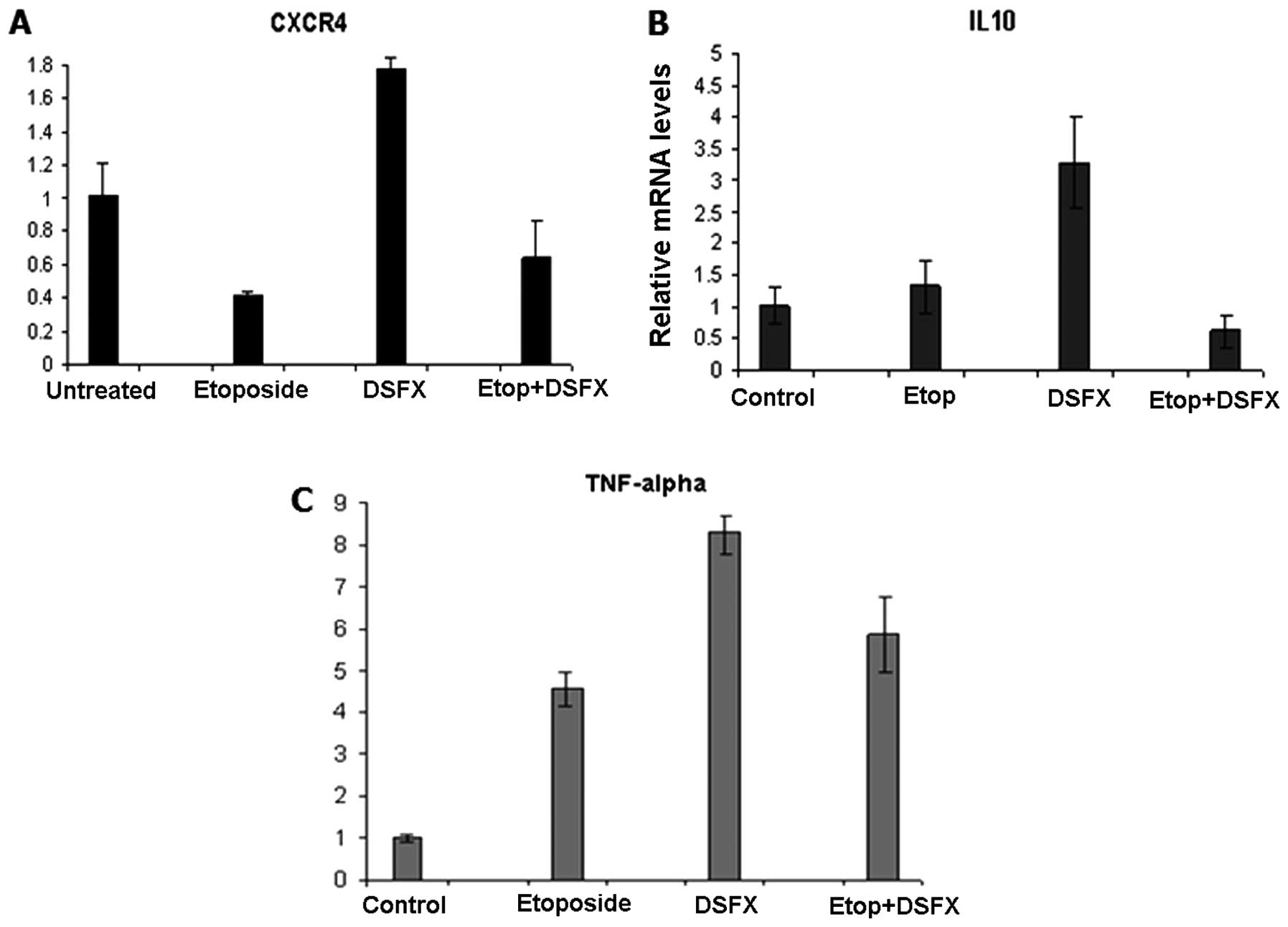
Reduced CXCR4 mRNA levels by 50% were observed in
etoposide-treated compared to untreated MCF-7 cells (Fig. 1A). In agreement with earlier
reports (19) increased by
1.7-fold CXCR4 gene expression was identified in MCF-7 cells
treated with the hypoxia mimicking agent DSFX compared to untreated
cells (Fig. 1A). Reduced
CXCR4 mRNA levels were observed in MCF-7 cells treated with
combination of etoposide and DSFX compared to untreated cells
(Fig. 1A), but these levels were
1.7-fold higher than those measured in cells treated with etoposide
alone (Fig. 1A). Given that MCF-7
cells bear wild-type p53, the reduced CXCR4 levels observed
in cells subjected to the combined treatment compared to those in
DSFX-treated cells implied a dominant p53 suppressive action over
HIF-1α (Fig. 1A).
Putative hypoxia responsive elements (HREs) were
identified within the regulatory regions of IL-10 and TNF-α
promoters (data not shown) and their mRNA levels were monitored in
a similar manner in MCF-7 cells. Induction of IL-10 and TNF-α gene
expression was detected in DSFX-treated cells (3-and 8-fold
respectively) compared to that in non-treated cells (Fig. 1B and C respectively). Combination
of etoposide and DSFX treatment repressed IL-10 expression
by ∼40% but induced TNF-α mRNA levels by 5-fold compared to
untreated cells (Fig. 1B and C).
Notably, etoposide treatment counteracted the stimulatory effect of
DSFX treatment in the case of both IL-10 and TNF-α.
HIF-1α recruitment to the regulatory
regions of the promoter of CXCR4 and TNF-α
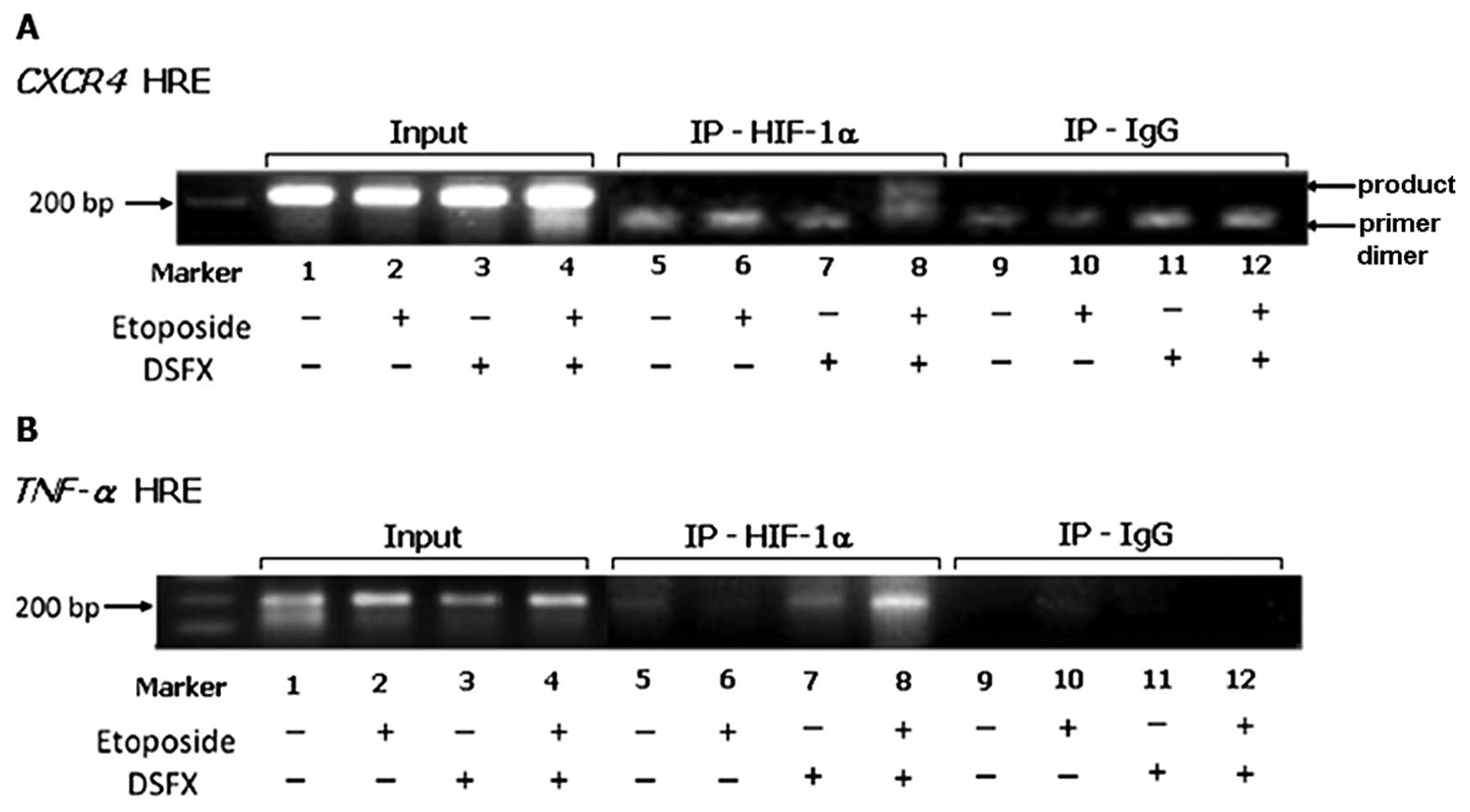
Given that etoposide-treated MCF-7 cells in hypoxia
mimicking conditions exhibited lower CXCR4 mRNA levels
compared to those observed in cells treated with DSFX only
(Fig. 1A) and taking into account
the fact that CXCR4 is known HIF-1α transcription target
(19) we were interested in
investigating the mechanisms coordinating the recruitment of HIF-1α
to the CXCR4 promoter in MCF-7 cells under diverse stress
conditions. MCF-7 cells were treated with etoposide, DSFX or
combination of both and the HIF-1α recruitment to the CXCR4
promoter was investigated employing chromatin immunoprecipitation
(ChIP) assays. It is worth noting that the HRE followed in this
study was different than that assessed by Schioppa et al
(19). Interestingly, we observed
that HIF-1α was recruited to the CXCR4 promoter only in
MCF-7 cells treated with both DSFX and etoposide (Fig. 2A, lane 8) whereas that was not the
case in MCF-7 cells treated with DSFX alone (Fig. 2A, lane 7). These findings suggested
that the HIF-1α transcriptional complex recruited to the
CXCR4 promoter in DSFX and etoposide-treated MCF-7 cells
differed from that present in these cells treated only with
DSFX.
In similar manner ChIP experiments were carried out
in MCF-7 cells treated with etoposide, DSFX or combination of both
and the HIF-1α recruitment to the putative HRE identified 6923-bp
upstream of the translation start site of the TNF-α promoter
was assessed. HIF-1α was recruited to the TNF-α promoter in
DSFX-treated MCF-7 cells (Fig. 2B,
lane 7) providing evidence that TNF-α induction in
hypoxia mimicking conditions (Fig.
1C) was a result of direct HIF-1α mediated trans-activation.
Moreover, HIF-1α was recruited to the TNF-α promoter in
MCF-7 cells treated with combination of DSFX and etoposide
(Fig. 2B, lane 8). Taken together
these results implied the existence of a selective mechanism
directing the recruitment of HIF-1α in specific subsets of its
transcriptional targets under diverse stress conditions.
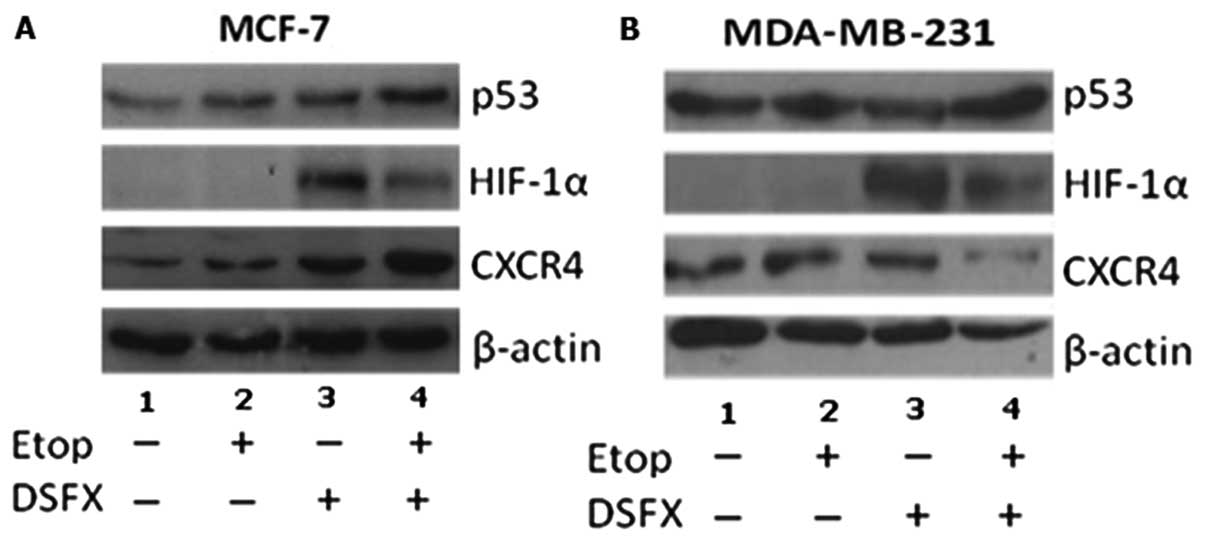
Protein levels of CXCR4 in breast cancer
cells
Western blot analysis was performed to follow the
CXCR4 protein levels under conditions of DNA-damage, hypoxia
mimicking and combination of the two stresses. Two breast cancer
cell lines differing in the p53 status (MCF-7 and MDA-MB-231) were
used for this analysis. Elevated CXCR4 protein levels were observed
under these stresses in wild-type p53 MCF-7 cells, compared to
non-treated cells (Fig. 3A,
compare CXCR4 lanes 2-4 to lane 1). Combination of DSFX and
etoposide treatment resulted in higher CXCR4 protein levels
compared to those observed in cells treated with either etoposide
or DSFX individually (Fig. 3A
compare lane 4 with lanes 2 and 3 respectively). The same analysis
was performed in MDA-MB-231 cells expressing high levels of mutated
p53G280A (31). Elevated CXCR4
protein levels were observed in etoposide and DSFX-treated
MDA-MB-231 cells (Fig. 3B compare
lanes 2 and 3 with lane 1 respectively) whereas combined etoposide
and DSFX treatment of these cells resulted in downregulation of the
CXCR4 protein levels (Fig. 3B
compare lane 4 with lane 1).
SRC-1 and SIRT-1 affect CXCR4 gene
expression in hypoxia mimicking conditions
SRC-1 is a HIF-1 transcriptional coactivator in
normoxic and hypoxic cells (26,32)
whereas SIRT-1 deacetylase, exerts SRC-1 opposing effects in the
androgen receptor signalling pathway in prostate cancer cells
(33) and has been linked to
hypoxia-mediated transcription (34). Given the functional and physical
interplay of SRC-1 and SIRT-1 with HIF-1α (26,32,35)
together with the identification of multiple putative HREs in the
regulatory region of the CXCR4 promoter triggered our
interest to investigate the effect of these two factors on the
activity of CXCR4 promoter in hypoxic breast cancer cells.
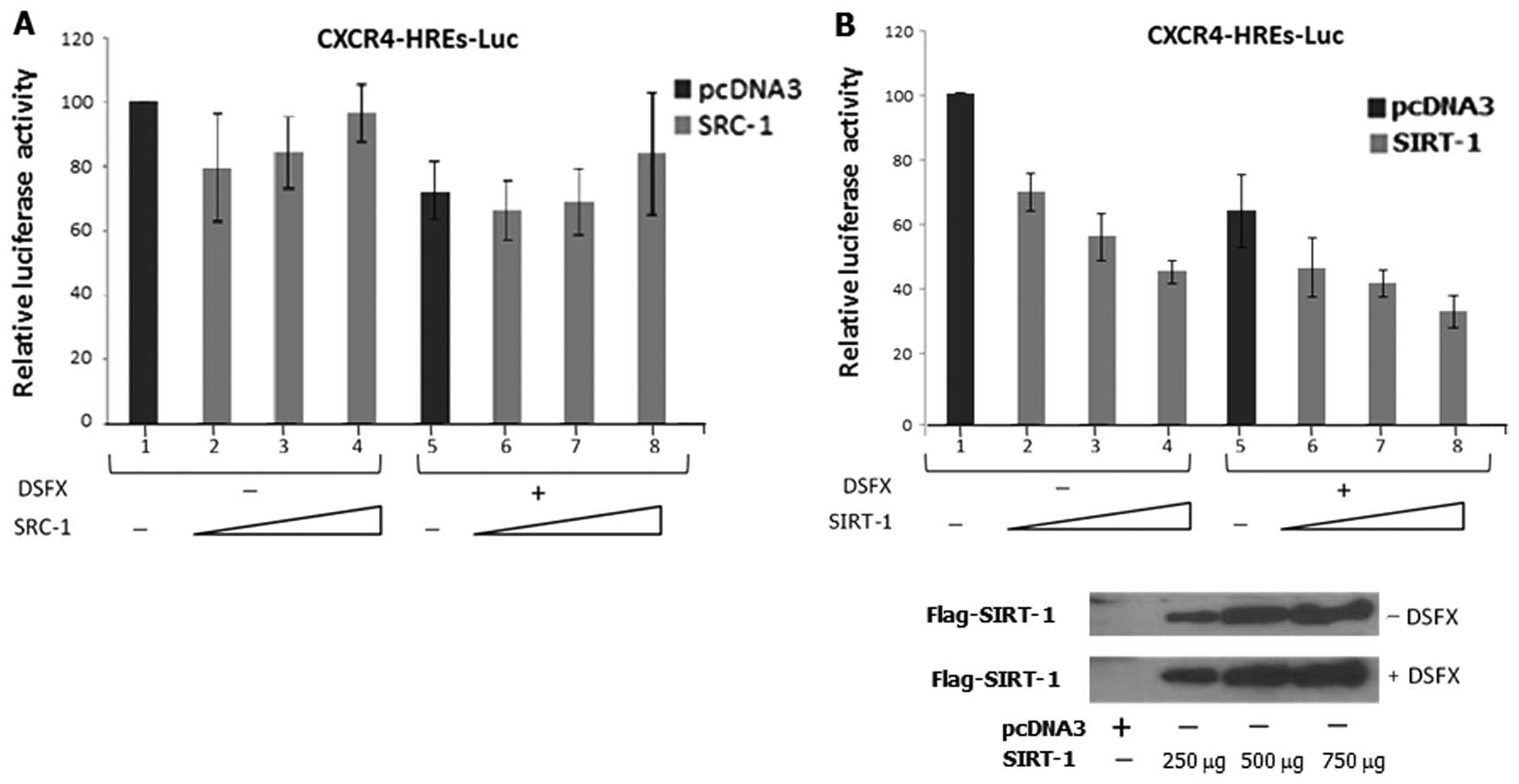
To assess the role of SRC-1 and SIRT-1 on CXCR4 promoter
activity under hypoxia mimicking conditions, we performed
luciferase reporter assays in MCF-7 cells transfected with the
CXCR4-luc reporter and exogenously expressing either SRC-1 or
SIRT-1 under normoxic or hypoxia mimicking conditions.
Initial reduction of CXCR4 luciferase
reporter activity was evident in MCF-7 normoxic cells expressing
exogenously transfected SRC-1 (Fig.
4A compare bar 1 with bar 2) which gradually increased
following increasing amounts of transfected SRC-1 (Fig. 4A compare bars 3, 4 with bar 2).
CXCR4 luciferase reporter activity was lower in cells
treated with the hypoxia mimicking agent DSFX compared to the
untreated cells (Fig. 4A compare
bars 1–4 with bars 5–8). Slight induction of the CXCR4
luciferase reporter was observed in cells transfected with the
highest amount of SRC-1 (Fig. 4A
compare bars 6 and 7 to bar 8) suggesting that overexpressed SRC-1
can reverse the suppressive effect of hypoxia on the activity of
the CXCR4 luciferase reporter.
In contrast to SRC-1, exogenous expression of SIRT-1
gradually reduced the CXCR4 luciferase reporter activity in
a dose-dependent manner in both normoxic (Fig. 4B, compare bars 2–4 to bar 1) and
hypoxia mimicking conditions (Fig.
4B, compare bars 6–8 to bar 5) in MCF-7 cells lending support
to the view that SIRT-1 exacerbates the repressive effect of
hypoxia on the activity of the CXCR4 luciferase
reporter.
Effect of SRC-1 and SIRT-1 on CXCR4
protein levels
To explore further the effect of SRC-1 and SIRT-1 in
the modulation of the CXCR4 expression, we assessed the CXCR4
protein levels in untreated, etoposide and DSFX-treated MCF-7 cells
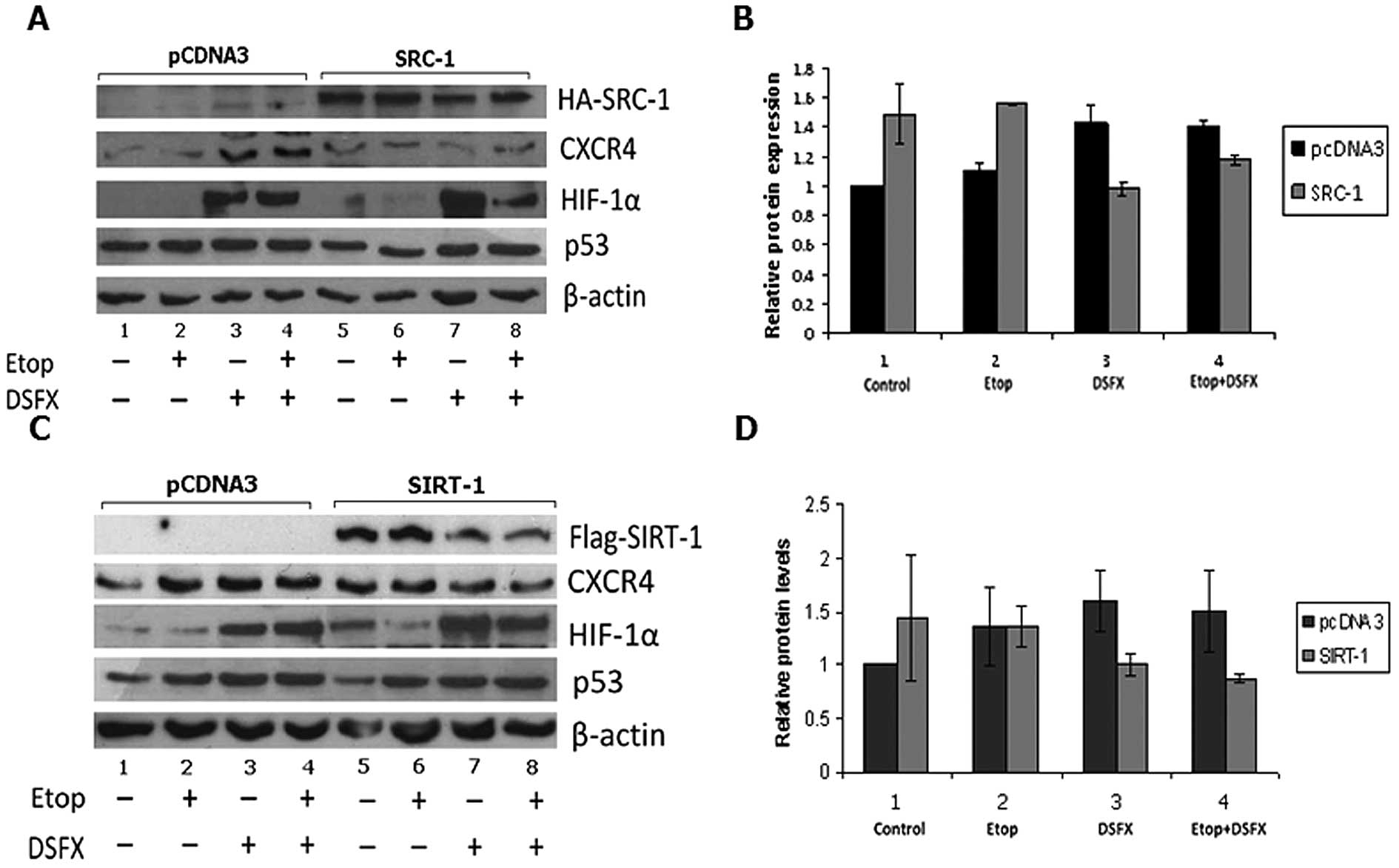
exogenously expressing either SRC-1 (Fig. 5A and B) or SIRT-1 (Fig. 5C and D). CXCR4 protein level in
each condition was compared to the level of this protein in cells
transfected with the empty vector pcDNA3. SRC-1 transfection
resulted in an average 50% increase of CXCR4 protein in untreated
normoxic cells (Fig. 5A and B,
compare black bar 1 to grey bar 1). Etoposide treatment increased
the CXCR4 protein levels by 40% in SRC-1 exogenously expressing
cells compared to pcDNA3-transfected cells (Fig. 5A and B, compare black bar 2 to grey
bar 2). In contrast, 29% decrease of CXCR4 protein levels were
observed in DSFX-treated MCF-7 cells overexpressing SRC-1 compared
to non-transfected cells (Fig. 5A and
B, compare black bar 3 with grey bar 3). Combination of
etoposide and DSFX treatment in SRC-1 overexpressing cells reduced
CXCR4 protein levels by 16% compared to cells transfected with
pcDNA3 under the same conditions (Fig.
5A and B compare black bar 4 with grey bar 4).
SIRT-1 transfection increased CXCR4 protein levels
in untreated cells but reduced the protein levels of this receptor
in cells treated with etoposide, DSFX and combination of both
compared to untransfected cells (Fig.
5C and D). SIRT-1 transfection conferred an ∼2-fold increase in
unstressed breast cancer cells compared to pcDNA3 transfected cells
(Fig. 5C and D, compare black bar
1 with grey bar 1). However in etoposide, DSFX and etoposide and
DSFX-treated cells, SIRT-1 decreased CXCR4 protein levels by 19, 34
and 43%, respectively, compared to MCF-7 cells transfected with
pcDNA3 (Fig. 5C and D, compare
black bars 2–4 to grey bars 2–4, respectively).
Discussion
Several studies converge to the conclusion that
perturbations of the inflammatory response increase the likelihood
of pathogenesis in diseases related to metabolism, auto-immunity,
and malignant transformation (5,9,36,37).
Breast cancer for example is characterized by elevated secretion of
pro-inflammatory (IL-1, TNF-α, IL-6) or anti-inflammatory cytokines
(IL-10), chemokines and chemokine receptors (CXCL8, CXCR4), and
angiogenic factors (VEGF) (38).
This network of pro-inflammatory components rivals the inhibitory
effect of the anti-inflammatory cytokines (IL-4) on the growth of
breast cancer cells (39) and
promotes cell proliferation, tumorigenesis and metastasis (9). Thus, it is clear that studying the
regulation of the expression levels and activity of cytokines and
cytokine receptors is important for understanding the pathways that
promote cancer progression.
HIF-1 and NF-κB act synergistically promoting the
expression of pro-inflammatory, pro-angiogenic and pro-survival
molecules thus converting pro-inflammatory into oncogenic signals
by transactivating gene expression of genes involved in
angiogenesis, metastasis and switch of cellular energy metabolism
from oxidative phosphorylation to glycolysis (VEGF, GLUT-1, Epo)
(8,17). On the other hand, the stress
responsive tumor suppressor protein p53 restrains neoplastic
transformation via its pro-apoptotic and anti-proliferative
function (20,21). In an attempt to provide additional
mechanistic insights into the molecular pathways that contribute to
breast cancer progression via cancer-related inflammation we
evaluated the roles of HIF-1α and p53 transcription factors in the
regulation of the expression of inflammatory cytokines.
In line with earlier studies indicating induction of
CXCR4 gene expression in hypoxic breast and ovarian cancer
cells (19) elevated CXCR4 mRNA
levels were observed in DSFX-treated MCF-7 breast cancer cells
compared to those determined in normoxic conditions (Fig. 1A). Furthermore, the inflammatory
genes IL-10 and TNF-α were upregulated in hypoxia
mimicking conditions (Fig. 1B and
C). HIF-1α and NF-κB pathways mediate upregulation of the
pro-inflammatory TNF-α levels in hypoxic macrophages
(40) but the finding that
TNF-α is upregulated in epithelial cancer cells via the same
signalling pathways provides an additional indication that
inflammation and cancer are closely linked (41).
In an effort to understand the molecular mechanisms
involved in CXCR4 and TNF-α gene expression under
diverse types of stress, the recruitment of HIF-1α to the promoter
of these genes was followed in MCF-7 cells treated with etoposide
or DSFX either individually or in combination. HIF-1α was recruited
to the CXCR4 promoter only in cells treated with combination
of etoposide and DSFX whereas in the TNF-α promoter HIF-1α
was detected in the chromatin immunocomplexes of cells treated with
both DSFX alone and combination of DSFX with etoposide. Possible
explanation for the selective recruitment of HIF-1α to the
CXCR4 or TNF-α promoter could be the different
composition of the HIF-1α transcription complexes in the two cases
targeting this transcription factor to one or the other promoter
depending on the environmental conditions, or differential HIF-1α
post-translational modifications.
To test this hypothesis the contribution of SRC-1
and SIRT-1, which are common modulators of both HIF-1α and p53
exerting opposing effects on the activity of these transcription
factors, to the regulation of CXCR4 cellular levels was
investigated. Our findings suggest that exogenous SRC-1 induced
CXCR4 protein levels in normoxic and etoposide-treated MCF-7 cells
but repressed its cellular levels in DSFX-treated MCF-7 cells
(Fig. 5A and B). Furthermore,
exogenously expressed SIRT-1 reduced CXCR4 protein levels and
CXCR4 luciferase reporter activity in DSFX-treated MCF-7
cells (Figs. 5C and D and 4B respectively). Additionally, in MCF-7
cells treated with either etoposide alone or with combination of
etoposide and DSFX, SIRT-1 overexpression reduced CXCR4 protein
levels compared to non-transfected cells in the same conditions
(Fig. 5C and D).
Collectively the data presented here provide
evidence that the functional interplay between HIF-1α and p53 and
possibly other transcription factors mediated by SRC-1 and SIRT-1
under diverse types of stress in hypoxic breast cancer cells
modulate the gene expression of the inflammatory CXCR4 chemokine
receptor.
References
|
1.
|
Balkwill F and Mantovani A: Inflammation
and cancer: back to Virchow? Lancet. 357:539–545. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
de Visser KE, Eichten A and Coussens LM:
Paradoxical roles of the immune system during cancer development.
Nat Rev Cancer. 6:24–37. 2006.
|
|
3.
|
Allavena P, Garlanda C, Borrello MG, Sica
A and Mantovani A: Pathways connecting inflammation and cancer.
Curr Opin Genet Dev. 18:3–10. 2008. View Article : Google Scholar
|
|
4.
|
Lin WW and Karin M: A cytokine-mediated
link between innate immunity, inflammation, and cancer. J Clin
Invest. 117:1175–1183. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar
|
|
6.
|
Shiao SL, Ganesan AP, Rugo HS and Coussens
LM: Immune microenvironments in solid tumours: new targets for
therapy. Genes Dev. 25:2559–2572. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Mantovani A, Sozzani S, Locati M, Allavena
P and Sica A: Macrophage polarization: tumor-associated macrophages
as a paradigm for polarized M2 mononuclear phagocytes. Trends
Immunol. 23:549–555. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Aggarwal BB, Shishodia S, Sandur SK,
Pandey MK and Sethi G: Inflammation and cancer: how hot is the
link? Biochem Pharmacol. 72:1605–1621. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Balkwill F: TNF-alpha in promotion and
progression of cancer. Cancer Metastasis Rev. 25:409–416. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Mocellin S, Marincola FM and Young HA:
Interleukin-10 and the immune response against cancer: a
counterpoint. J Leukoc Biol. 78:1043–1051. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Ali S and Lazennec G: Chemokines: novel
targets for breast cancer metastasis. Cancer Metastasis Rev.
26:401–420. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Balkwill F: Chemokine biology in cancer.
Semin Immunol. 15:49–55. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Allavena P, Germano G, Marchesi F and
Mantovani A: Chemokines in cancer-related inflammation. Exp Cell
Res. 317:664–673. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Luker KE and Luker GD: Functions of CXCL12
and CXCR4 in breast cancer. Cancer Lett. 238:30–41. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Balkwill F: The significance of cancer
cell expression of the chemokine receptor CXCR4. Semin Cancer Biol.
14:171–179. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Uygur B and Wu WS: SLUG promotes prostate
cancer cell migration and invasion via CXCR4/CXCL12 axis. Mol
Cancer. 10:1392011. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Balkwill F: Cancer and the chemokine
network. Nat Rev Cancer. 4:540–550. 2004. View Article : Google Scholar
|
|
18.
|
Denko NC, Fontana LA, Hudson KM, et al:
Investigating hypoxic tumor physiology through gene expression
patterns. Oncogene. 22:5907–5914. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Schioppa T, Uranchimeg B, Saccani A, et
al: Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp
Med. 198:1391–1402. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Mehta SA, Christopherson KW,
Bhat-Nakshatri P, et al: Negative regulation of chemokine receptor
CXCR4 by tumor suppressor p53 in breast cancer cells: implications
of p53 mutation or isoform expression on breast cancer cell
invasion. Oncogene. 26:3329–3337. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Webster GA and Perkins ND: Transcriptional
cross-talk between NF-kappaB and p53. Mol Cell Biol. 19:3485–3495.
1999.PubMed/NCBI
|
|
22.
|
Xenaki G, Ontikatze T, Rajendran R, et al:
PCAF is an HIF-1alpha cofactor that regulates p53 transcriptional
activity in hypoxia. Oncogene. 27:5785–5796. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Blagosklonny MV, An WG, Romanova LY,
Trepel J, Fojo T and Neckers L: p53 inhibits hypoxia-inducible
factor-stimulated transcription. J Biol Chem. 273:11995–11998.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
An WG, Kanekal M, Simon MC, Maltepe E,
Blagosklonny MV and Neckers LM: Stabilization of wild-type p53 by
hypoxia-inducible factor 1alpha. Nature. 392:405–408. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Lee SK, Kim HJ, Kim JW and Lee JW: Steroid
receptor coactivator-1 and its family members differentially
regulate trans-activation by the tumor suppressor protein p53. Mol
Endocrinol. 13:1924–1933. 1999. View Article : Google Scholar
|
|
26.
|
Ruas JL, Poellinger L and Pereira T: Role
of CBP in regulating HIF-1-mediated activation of transcription. J
Cell Sci. 118:301–311. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Rajendran R, Garva R, Krstic-Demonacos M
and Demonacos C: Sirtuins: molecular traffic lights in the
crossroad of oxidative stress, chromatin remodeling, and
transcription. J Biomed Biotechnol. 2011:3682762011. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Xu J, Wu R-C and O’Malley BW: Normal and
cancer-related functions of the p160 steroid receptor co-activator
(SRC) family. Nat Rev Cancer. 9:615–630. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Onate SA, Tsai SY, Tsai MJ and O’Malley
BW: Sequence and characterization of a coactivator for the steroid
hormone receptor superfamily. Science. 270:1354–1357. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Demonacos C and La Thangue NB: Drug
discovery and the p53 family. Prog Cell Cycle Res. 5:375–382.
2003.PubMed/NCBI
|
|
31.
|
Keimling M and Wiesmuller L: DNA
double-strand break repair activities in mammary epithelial cells -
influence of endogenous p53 variants. Carcinogenesis. 30:1260–1268.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Carrero P, Okamoto K, Coumailleau P,
O’Brien S, Tanaka H and Poellinger L: Redox-regulated recruitment
of the transcriptional coactivators CREB-binding protein and SRC-1
to hypoxia-inducible factor 1alpha. Mol Cell Biol. 20:402–415.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Lavery DN and Bevan CL: Androgen receptor
signalling in prostate cancer: the functional consequences of
acetylation. J Biomed Biotechnol. 2011:8621252011. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Zhang Q, Tang X, Lu QY, Zhang ZF, Brown J
and Le AD: Resveratrol inhibits hypoxia-induced accumulation of
hypoxia-inducible factor-1alpha and VEGF expression in human tongue
squamous cell carcinoma and hepatoma cells. Mol Cancer Ther.
4:1465–1474. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Lim J-H, Lee Y-M, Chun Y-S, Chen J, Kim
J-E and Park J-W: Sirtuin 1 modulates cellular responses to hypoxia
by deacetylating hypoxia-inducible factor 1α. Mol Cell. 38:864–878.
2010.PubMed/NCBI
|
|
36.
|
De Nardo D and Latz E: NLRP3 inflammasomes
link inflammation and metabolic disease. Trends Immunol.
32:373–379. 2011.PubMed/NCBI
|
|
37.
|
Strober W and Fuss IJ: Proinflammatory
cytokines in the pathogenesis of inflammatory bowel diseases.
Gastroenterology. 140:1756–1767. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Mantovani A, Marchesi F, Porta C, Sica A
and Allavena P: Inflammation and cancer: breast cancer as a
prototype. Breast. 16(Suppl 2): S27–S33. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Toi M, Bicknell R and Harris AL:
Inhibition of colon and breast carcinoma cell growth by
interleukin-4. Cancer Res. 52:275–279. 1992.PubMed/NCBI
|
|
40.
|
Murdoch C and Lewis CE: Macrophage
migration and gene expression in response to tumor hypoxia. Int J
Cancer. 117:701–708. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Hussain SP and Harris CC: Inflammation and
cancer: an ancient link with novel potentials. Int J Cancer.
121:2373–2380. 2007. View Article : Google Scholar : PubMed/NCBI
|