Introduction
Hepatocellular carcinoma (HCC) is the sixth most
commonly diagnosed malignancy and the third leading cause of
cancer-related mortality, with an estimated 748,000 new cases and
696,000 deaths worldwide in 2008 (1). At present, patients with
intermediate-advanced HCC are eligible for palliative treatments
including transcatheter arterial chemoembolization (TACE) and the
oral multikinase inhibitor, sorafenib (2,3).
However, treatment benefits are still limited and the development
of more effective pharmacological agents is expected (4).
Sinomenium acutum Rehd. et Wils. (Fam.
Menispermaceae), a Chinese medicinal herb, has been
traditionally used for the treatment of various diseases, such as
rheumatism, fevers and neuralgia for centuries (5). Sinomenine
(7,8-didehydro-4-hydroxy-3,7-dimethoxy-17-methylmorphinane-6-one)
is the most abundant active component isolated from this herb and
has previously been demonstrated to exert anti-inflammatory,
anti-rheumatic, immunosuppressive and analgesic effects (5–10).
Sinomenine hydrochloride (SH), a hydrochloride chemical form of
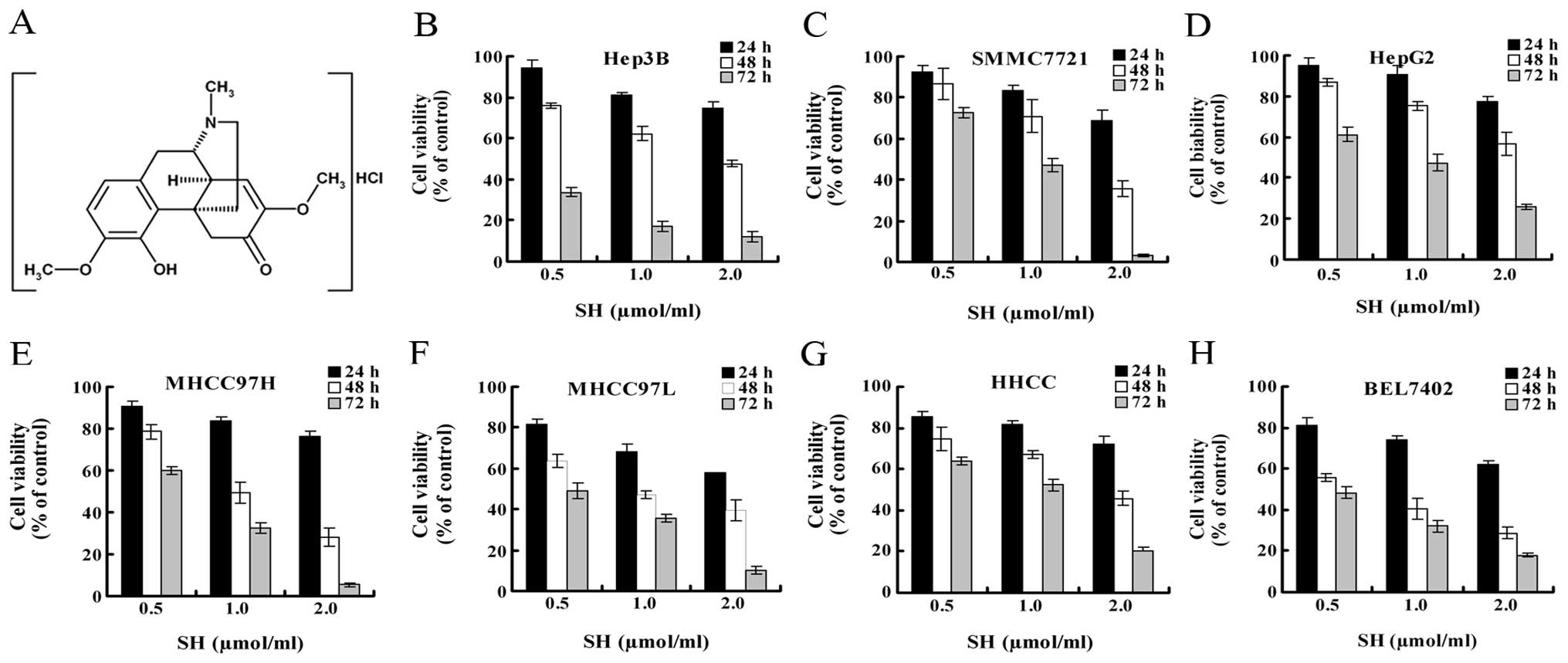
sinomenine (Fig. 1A), has been
successfully used in clinical practice in China for treating
rheumatism and neuralgia with minimal side-effects (11). Recently, a number of studies have
reported the anti-neoplastic potential of SH in vitro
against a variety of human tumor cells, including leukemia
(12), synovial sarcoma (13) and cancers of lung (14) and prostate (15).
As is currently known, cancer initiation and
development are attributed to the disruption of the normal balance
between cell proliferation and cell death (16). Cell proliferation is regulated by
the cell cycle, which is governed by a number of cyclin-dependent
kinases (CDKs) and their pivotal inhibitor, p21/WAF1/Cip1 (p21)
(17). Apoptosis is a key mode of
programmed cell death and its distinctive morphological changes
include membrane blebbing, cell shrinkage, chromatin condensation,
DNA cleavage and apoptotic bodies (18). It has been well-established that,
in most circumstances, apoptosis is regulated and executed by the
activated cysteine-aspartic proteases (caspases). Two major
signaling pathways initiate and propagate caspase activation
(19,20): the extrinsic (death receptor)
pathway originates from the recognition between death receptors
(Fas and tumor necrosis factor-receptor 1) and their ligands and
results in the activation of caspase-8 and -10 (20). The intrinsic (mitochondrial)
pathway is triggered by the disruption of the mitochondrial
transmembrane potential (Δψm), permeabilization of the
outer membrane, release of apoptogenic proteins such as cytochrome
c (Cyt c) from the mitochondrial intermembrane space
into the cytoplasm and subsequent activation of caspase-9 (21). Mitochondrial apoptosis is regulated
by anti-apoptotic Bcl-2 and pro-apoptotic Bax which localize in the
outer mitochondrial membrane (22). The extrinsic and intrinsic pathways
converge at the activation of caspase-3, a key executioner of
apoptosis (23).
The anticancer effects of various therapeutic drugs
involve cell cycle arrest and apoptosis induction (24,25).
SH has been demonstrated to inhibit cell proliferation and induce
apoptosis in a variety of human tumor cells (12–15).
However, the potential anticancer effect of SH in HCC and the
underlying molecular mechanisms remain to be investigated. In this
study, we report that SH inhibits HCC growth in vitro and
in vivo by promoting p21-associated cell cycle arrest and
inducing caspase-dependent apoptosis.
Materials and methods
Chemicals and reagents
Primary antibodies against poly(ADP-ribose)
polymerase (PARP), caspase-3, -8, -9, -10 and Omi/HtrA2 were
purchased from Cell Signaling Technology, Inc. Primary antibodies
against proliferating cell nuclear antigen (PCNA), survivin, Cyt
c, Bcl-2, Bax and p21 were obtained from Santa Cruz
Biotechnology, Inc. Matrigel was obtained from BD Biosciences. The
general caspase inhibitor, Z-VAD-FMK, was purchased from R&D
Systems. SH (98% purity), provided by the Hunan Zhengqing
Pharmaceutical Group Co. Ltd., was directly dissolved in DMEM
containing 10% fetal bovine serum for in vitro studies and
formulated in physiological saline for in vivo studies.
Cell culture
The human HCC cell lines, Hep3B, SMMC7721, HepG2,
HHCC, Bel7402, MHCC97-H and MHCC97-L, were obtained from the
Shanghai Institutes for Biological Sciences (SIBS). Cells were
grown in DMEM supplemented with 10% fetal bovine serum and
maintained in an incubator with a humidified atmosphere containing
5% CO2 at 37°C.
MTT assay for cell viability
We conducted
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assays as recommended by the manufacturer. Briefly, exponentially
growing cells were trypsinized, seeded at an initial density of
8×103 per well in 96-well plates and incubated in
standard growth medium for 24 h prior to being subjected to
different treatments for the indicated periods of time. Cells were
subsequently incubated in medium containing MTT (0.5 mg/ml) for an
additional 4 h. The supernatant was removed and 150 μl DMSO
were added to each well to dissolve the intracellular formazan
compound. The absorbance was measured with an ELISA reader
(Bio-Rad, Hercules, CA, USA) at 490 nm. The relative cell viability
(%) was calculated as the percentage between SH-treated cells and
the untreated controls.
Cell cycle distribution analysis
Cells were grown in 6-cm dishes at a density of
0.5×106 cells/ml and treated with the desired
concentrations of SH in complete medium for 48 h. Cells were then
collected and fixed in 70% ethanol at −20°C overnight. After being
suspended in 0.5 ml of propidium iodide (PI) staining solution (50
μg/ml PI, 100 μg/ml RNase and 0.2% Triton X-100 in
PBS) for 30 min at room temperature in the dark, cell cycle
distribution was determined by a FACSCalibur flow cytometer
(Becton-Dickinson).
Western blot analysis
Proteins were prepared as described previously
(26). After protein
quantification, equal amounts of protein (∼30–60 μg) were
electrophoretically separated by SDS-PAGE with 12% Tris-Glycine
gels and transferred onto nitrocellulose membranes. After being
blocked with 5% non-fat dry milk in TBST buffer (weight/volume) for
1 h, the membranes were incubated with the primary antibodies at
4°C overnight, followed by incubation with a suitable horseradish
peroxidase-conjugated secondary antibody at room temperature for 1
h. Immunoreactive bands on the membranes were developed using an
electrogenerated chemiluminescence (ECL) detection system followed
by exposure to X-ray film.
Hoechst staining
Hoechst staining was performed using an
Apoptosis-Hoechst Staining kit (Beyotime), according to the
manufacturer’s instruction. Exponentially growing cells were seeded
in 6-multiwell plates at a density of 5×105 cells per
well, maintained in culture overnight and exposed to SH at 2
μmol/ml for 48 h. Cells were fixed with 4% paraformaldehyde,
stained with Hoechst 33528 and then visualized under a fluorescence
microscope (Olympus).
DNA fragmentation assay and quantitative
analysis for apoptosis
Cells were plated on 6-cm dishes at a density of
0.5×106 cells/ml, grown for 24 h and treated with
increasing concentrations of SH for 48 h. Cells were then collected
and centrifuged. For the detection of DNA ladders, intracellular
DNA was extracted using an Apoptosis DNA Ladder Detection kit
(Nanjing KeyGen Biotech. Co. Ltd.) according to the manufacturer’s
instructions. DNA samples were dissolved in TE buffer (pH 8.0),
separated by electrophoresis on a 1.5% agarose gel with ethidium
bromide (0.5 μg/ml) and then photographed using a molecular
imager (Bio-Rad). Quantitative analysis of apoptosis by flow
cytometry was assessed using an Annexin V-FITC Apoptosis Assay kit
(Joincare Biosciences, Zhuhai, China), as described in the
manufacturer’s recommendations. At least 10,000 cells were analyzed
for each sample.
Mitochondrial membrane potential
measurement
We examined Δψm using a Mitochondrial
Membrane Potential Assay kit with JC-1 (Beyotime) as described in
the manufacturer’s instructions. Cells at a density of
0.5×106 cells/ml were seeded on 6-cm dishes, grown for
24 h, treated with different concentrations of SH for 48 h and
stained with JC-1, followed by flow cytometry for the quantitative
analysis of Δψm. At least 10,000 events per sample were
recorded.
Preparation of cytosolic
fractionation
Briefly, cells were lysed with permeabilization
buffer (250 mmol/l sucrose, 20 mmol/l HEPES/KOH (pH 7.4), 1 mmol/l
EGTA, 1 mmol/l EDTA, 1 mmol/l DTT, 0.1 mmol/l PMSF, 1 μg/ml
chymostatin, 1 μg/ml leupeptin, 1 μg/ml antiparin and
1 μg/ml pepstatin A). The lysates were centrifuged at 500 ×
g for 10 min (4°C). The supernatants were further centrifuged at
13,000 × g for 30 min (4°C) and the supernatants containing
cytosolic fraction were collected.
Caspase-3 activity assay
The activity of caspase-3 was assessed using a
Caspase Colorimetric Assay kit (R&D Systems), following the
instructions provided by the manufacturer. Briefly, cells were
washed with ice-cold PBS and lysed in a lysis buffer. After being
centrifuged at 10,000 × g for 10 min (4°C), cell lysates were
incubated with 50 μl of reaction buffer and 5 μl of
caspase-3 colorimetric substrate (DEVD-p-nitroaniline) for 2 h at
37°C. The optical density of the color reaction was
spectrophotometrically measured at 405 nm.
Tumor xenograft study
Four-week-old male BALB/c athymic nude mice,
weighing 18–23.5 g, were obtained from Shanghai SLAC Laboratory
Animal Co. Ltd. (Shanghai, China) and housed in sterile laminar
flow rooms with 12-h light and dark cycles at a temperature range
of 19–23°C and a humidity of 40–60% in the Laboratory Animal Centre
of Xi’an Jiaotong University. All experimental procedures were
conducted in accordance with the institutional guidelines for
conduct and animal welfare. Mice were subcutaneously injected with
5×106 of human HCC Hep3B cells suspended in 100
μl of mixture containing serum-free DMEM and matrigel (5:1)
in the right hind legs. Thirty mice were randomly divided into the
following 4 groups: control group (n=6), SH 50 mg/kg group (n=8),
SH 100 mg/kg group (n=8) and SH 150 mg/kg group (n=8). After 24 h,
mice in the control group were intraperitoneally (i.p.) injected
with 0.1 ml of physiologic saline per 10 g body weight daily and
mice in other 3 groups were i.p. injected with 50, 100 and 150
mg/kg doses of SH in 0.1 ml of saline daily for 24 days. Mice and
their average food consumption were weighed daily to determine the
effects of SH on their general health. Tumor size was also measured
daily with vernier caliper and tumor volume was calculated as
0.5236 × length × width × width and expressed as mm3. At
the end of the experiment, mice were sacrificed and tumors were
immediately removed, weighed and then fixed in 4%
paraformaldehyde.
Immunohistochemistry staining
Paraformaldehyde-fixed tissues were embedded in
paraffin blocks and cut into 4-μm sections. Haematoxylin and
eosin (H&E) staining was conducted according to conventional
procedures. Immunohistochemistry staining was carried out using a
Dako Autostainer (Dako, Carpinteria, CA, USA) as described
previously (27). Briefly, the
sections were dewaxed, rehydrated, subjected to antigen retrieval
by pressure cooking for 5 min and incubated with serum for 15 min
to block endogenous enzyme and non-specific antigens. The sections
were incubated with appropriately diluted primary antibodies at 4°C
overnight and then with DakoCytomation EnVision-HRP reagent for 30
min, followed by incubation with diaminobenzidine (DAB) in a dark
room. After counterstaining with hematoxylin, the sections were
then dehydrated. The relative protein expression was evaluated by
the average percentage of positive cells (number of positive cells
×100/total number of cells) in 5 different random microscopic
fields (×400) in each tumor sample.
In situ apoptosis detection by TUNEL
staining
Terminal deoxynucleotidyl transferase-mediated
dUTP-biotin nick end-labeling (TUNEL) assay was performed using an
In Situ Cell Death Detection kit, POD (Roche Diagnostics)
according to the protocol supplied by the manufacturer. The average
apoptotic index of each tumor sample was calculated as described
above.
Statistical analysis
SPSS 16.0 was used to analyze statistical data. Data
are expressed as the means ± SE. Statistical comparisons were
performed using one-way ANOVA or the Student’s t-test. P<0.05
was considered to indicate a statistically significant
difference.
Results
SH inhibits growth of human HCC
cells
The effect of SH on the growth of HCC cells were
evaluated using MTT assay. We employed 7 HCC cell lines
representing different degrees of differentiation and from diverse
genetic backgrounds to investigate the extent at which SH inhibits
the growth of HCC cells. In all the HCC cell lines, treatment with
SH at 0.5, 1 and 2 μmol/ml for 24, 48 and 72 h significantly
inhibited cell growth in a concentration- and time-dependent manner
(Fig. 1B–H).
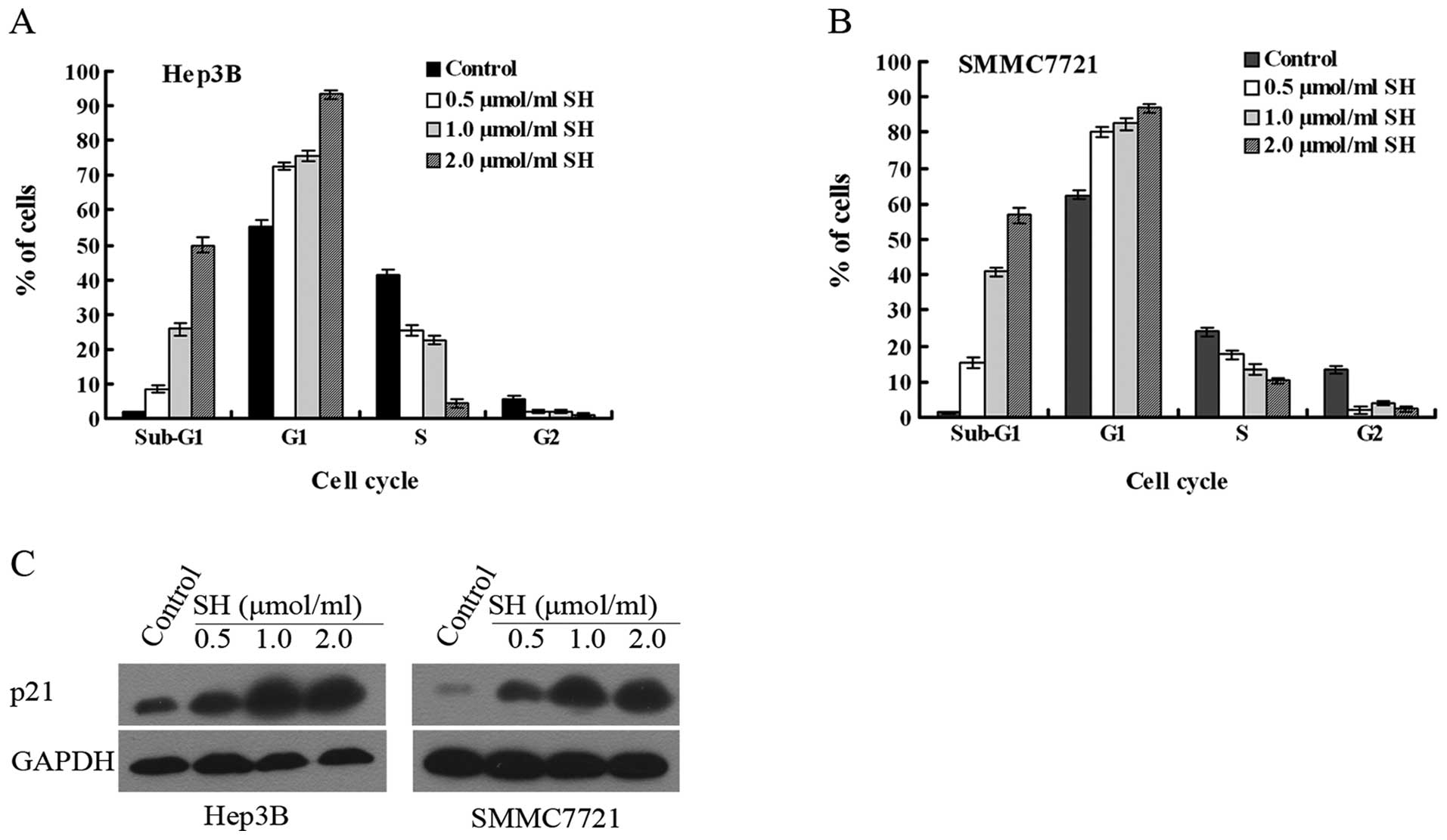
SH induces cell cycle arrest and
upregulates p21 protein expression in human Hep3B and SMMC7721 HCC
cells
We hypothesized that the SH-induced cell growth
inhibition may be associated with the modulation of cell cycle
progression. To determine this possibility, following SH treatment,
the cells were stained with PI and then subjected to flow cytometry
to analyze cell cycle distribution. In the Hep3B and SMMC7721
cells, it was shown that an accumulation of cells in the G1 phase,
accompanied by a decrease in the number of cells in the S phase and
an increase in the number of cells in the sub-G1 phase, was caused
by SH in a concentration-dependent manner following 48 h of
treatment (Fig. 2A). To elucidate
the mechanism of the cell cycle arrest induced by SH, we further
investigated the effect of SH on the cell cycle regulatory protein,
p21. Our results revealed that the protein level of p21 was
significantly elevated following 48 h of SH treatment in a
concentration-dependent manner (Fig.
2B).
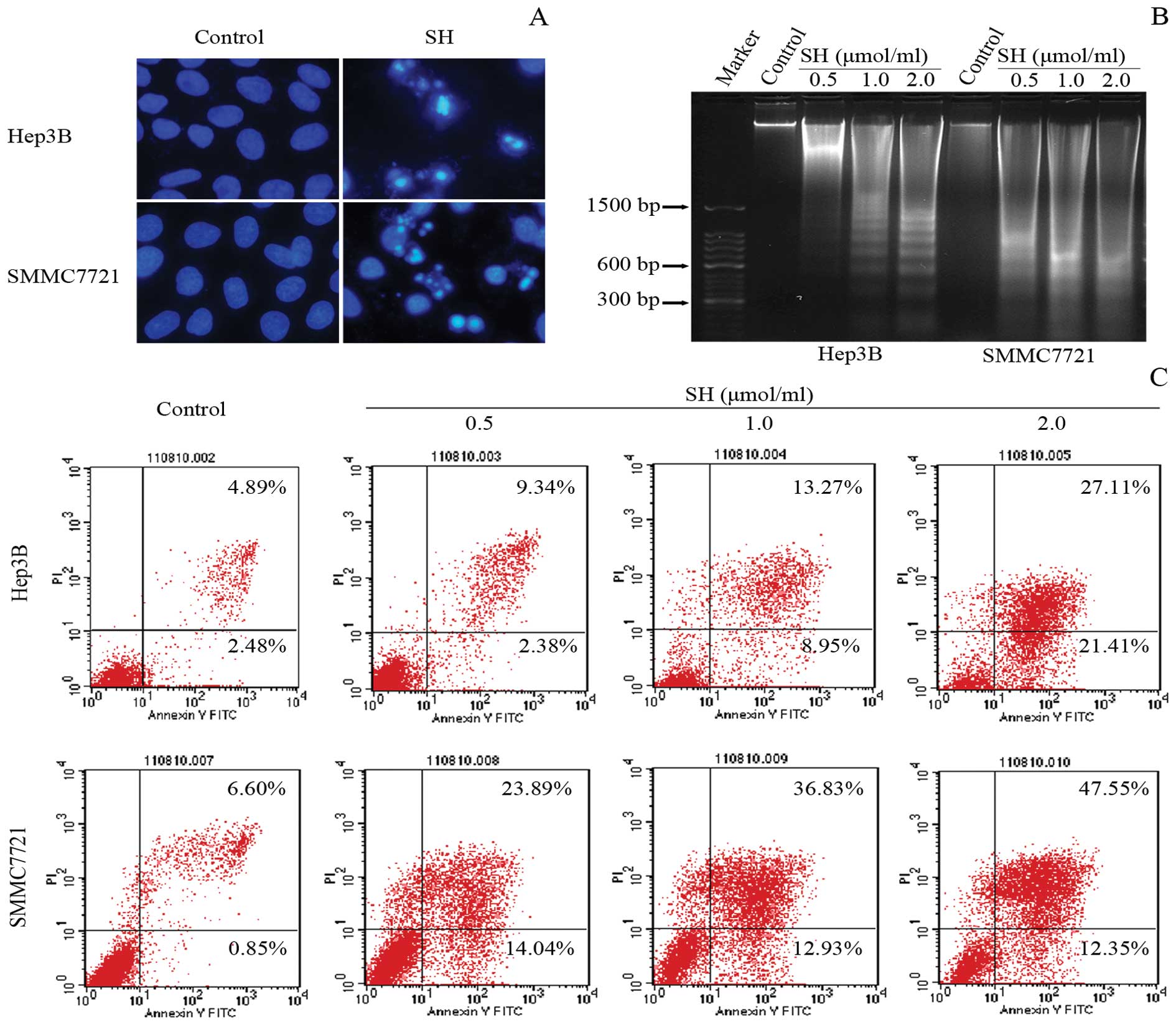
SH induces apoptosis in Hep3B and
SMMC7721 cells
We then assessed whether SH causes apoptotic cell
death in Hep3B and SMMC7721 cells. As shown in Fig. 3A, significant morphological changes
associated with apoptosis, e.g., formation of condensed and
fragmented nuclei, were induced by SH. The formation of a DNA
ladder pattern, a biochemical characteristic of apoptosis
indicating internucleosomal DNA fragmentation, was markedly
detected in a concentration-dependent manner (Fig. 3B). Correspondingly, the increase in
Annexin V-positive cells, another feature of apoptosis, was also
observed in the SH-treated cells. As shown in Fig. 3C, treatment with 0, 0.5, 1 and 2
μmol/ml of SH for 48 h induced 7.37, 11.72, 22.22 and 48.52%
of the apoptotic cell population in the Hep3B cells and 7.45,
37.93, 49.76 and 59.90% in the SMMC7721 cells, respectively.
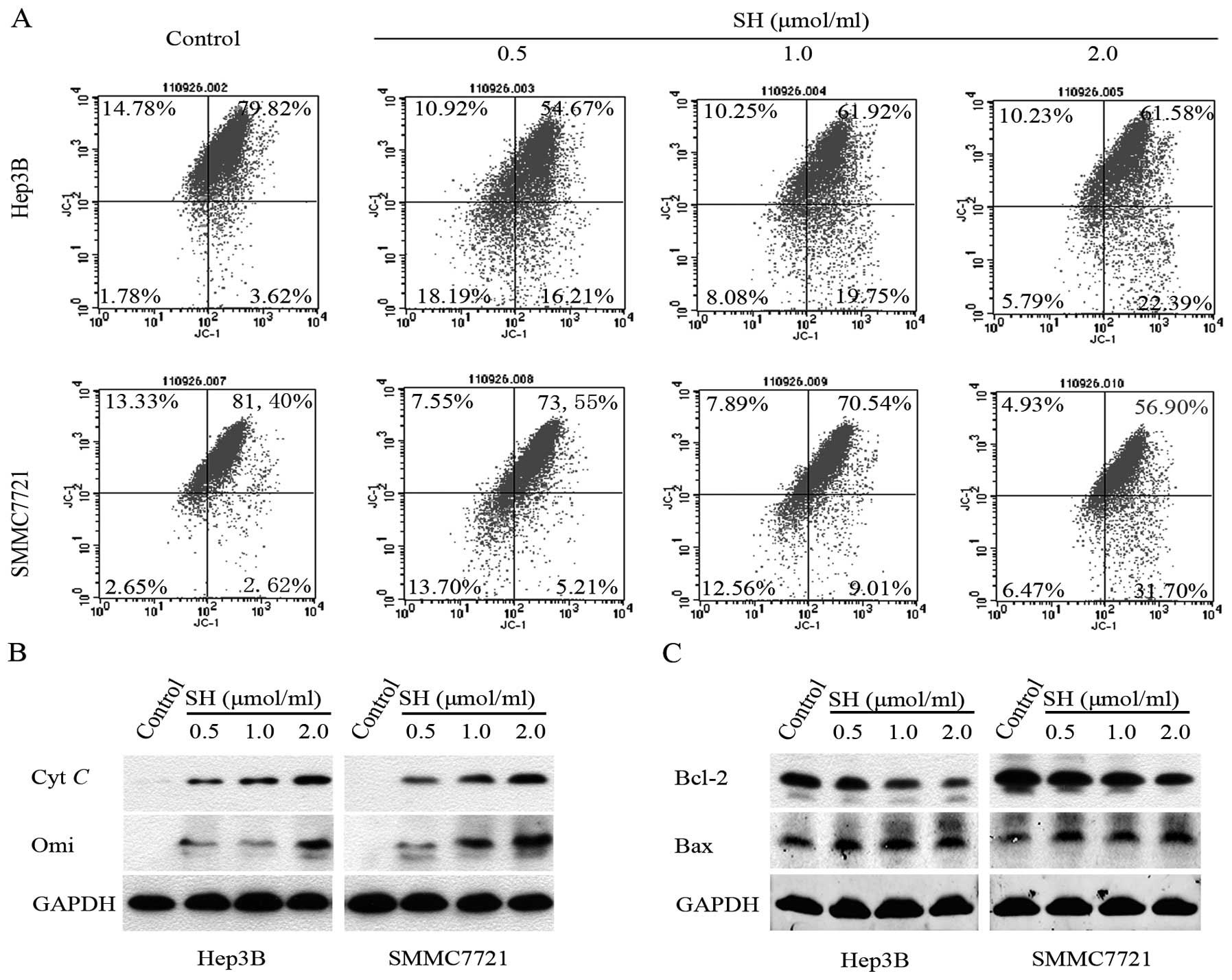
SH disrupts Δψm, promotes the
release of Cyt c and Omi/HtrA2 from the mitochondria and decreases
the Bcl-2/Bax ratio in Hep3B and SMMC7721 cells
As the mitochondria play a key role in propagating
apoptotic signaling, we examined the effect of SH on Δψm
by JC-1 staining. Quantitative analysis using flow cytometry
revealed that the percentage of cells with collapse of
Δψm increased following treatment with SH in a
concentration-dependent manner in the Hep3B and SMMC7721 cells
(Fig. 4A). Additionally, we found
that SH treatment gradually increased the levels of apoptogenic Cyt
c and Omi/HtrA2 proteins in the cytosolic fraction in a
concentration-dependent manner (Fig.
4B). As the altered Bcl-2/Bax ratio is a precursor for the
release of apoptogenic proteins, we further examined whether the
Bcl-2/Bax ratio decreased upon SH treatment in HCC cells. As shown
in Fig. 4C, SH treatment resulted
in an upregulation of Bax protein and a downregulation of Bcl-2
protein expression in a concentration-dependent manner.
SH induces apoptosis through
caspase-dependent pathway in human Hep3B and SMMC7721 HCC
cells
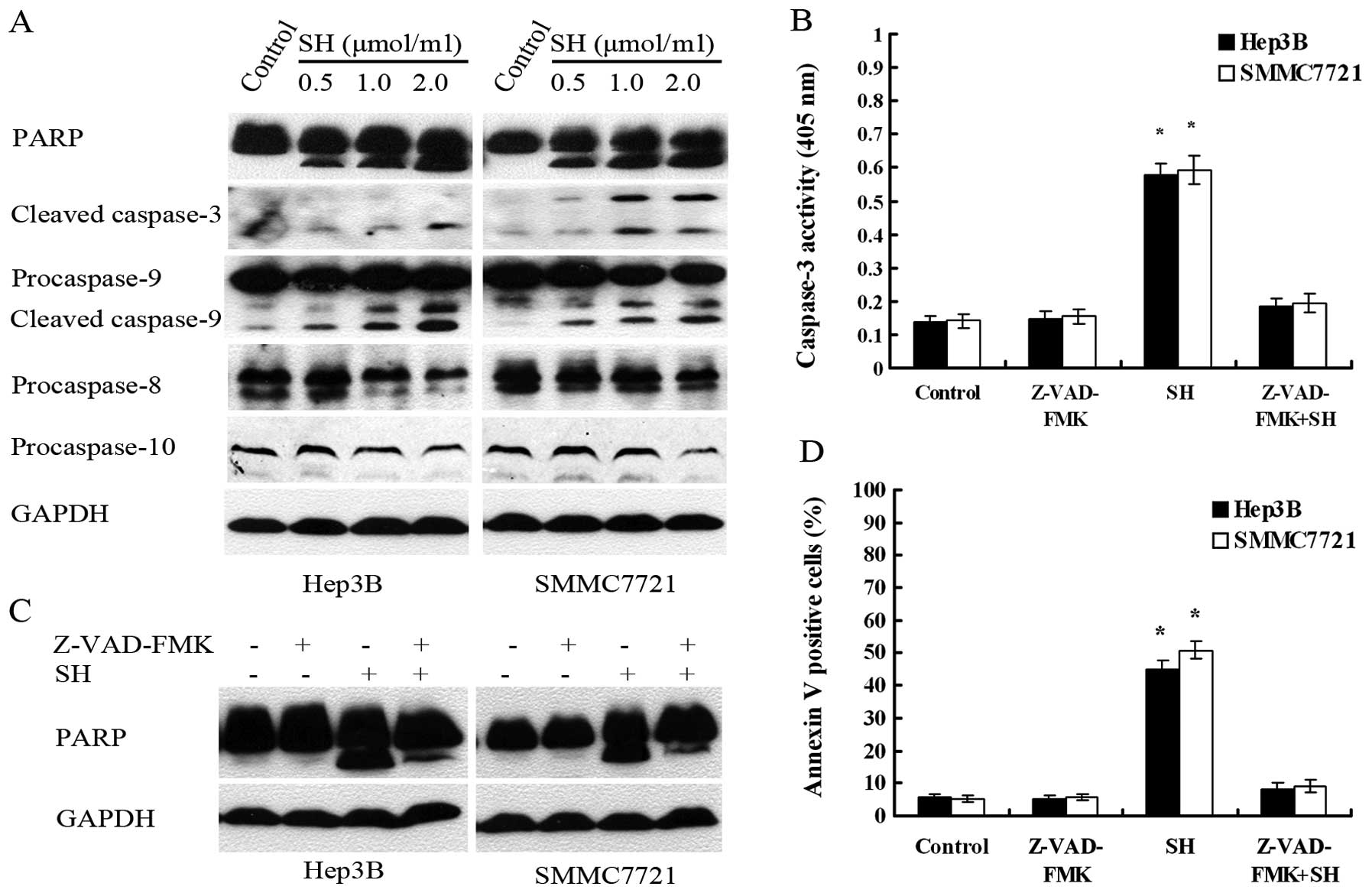
Based on the above findings, we then determined
whether the activation of a caspase cascade is involved in
SH-induced apoptosis in Hep3B and SMMC7721 cells. Western blot
analysis showed that SH treatment decreased the protein levels of
precursors of caspase-8 and -10 and increased the levels of cleaved
caspase-9, caspase-3 and PARP, a known substrate of caspase-3
(23,28), in a concentration-dependent manner
(Fig. 5A). To gain an insight into
the contribution of caspases to SH-induced apoptosis, cells were
pre-treated with a general caspase inhibitor, Z-VAD-FMK, prior to
SH treatment. We confirmed that pre- treatment with Z-VAD-FMK
efficiently inhibited SH-induced caspase activation, which was
revealed by the reduction in caspase-3 activity (Fig. 5B) and PARP cleavage (Fig. 5C). As expected, Z-VAD-FMK
efficiently suppressed SH-induced apoptosis as shown by flow
cytometry analysis (Fig. 5D).
These results suggest that the SH-induced apoptosis in HCC cells
depends on the activation of a caspase cascade.
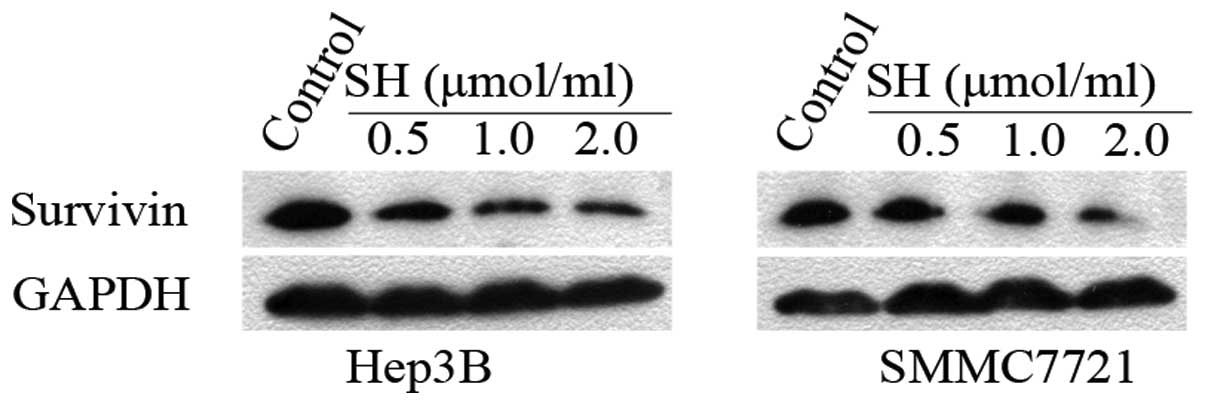
SH downregulates the expression levels of
survivin protein
As a key regulator of mitosis and apoptosis,
survivin inhibits p21 expression and caspase activation and plays
an important role in HCC tumor cell proliferation and survival
(29–32). We therefore anticipated that SH may
decrease the expression levels of survivin in Hep3B and SMMC7721
cells. As shown in Fig. 6,
compared with the untreated controls, treatment with SH for 48 h
significantly caused a concentration-dependent decrease in the
level of survivin protein expression.
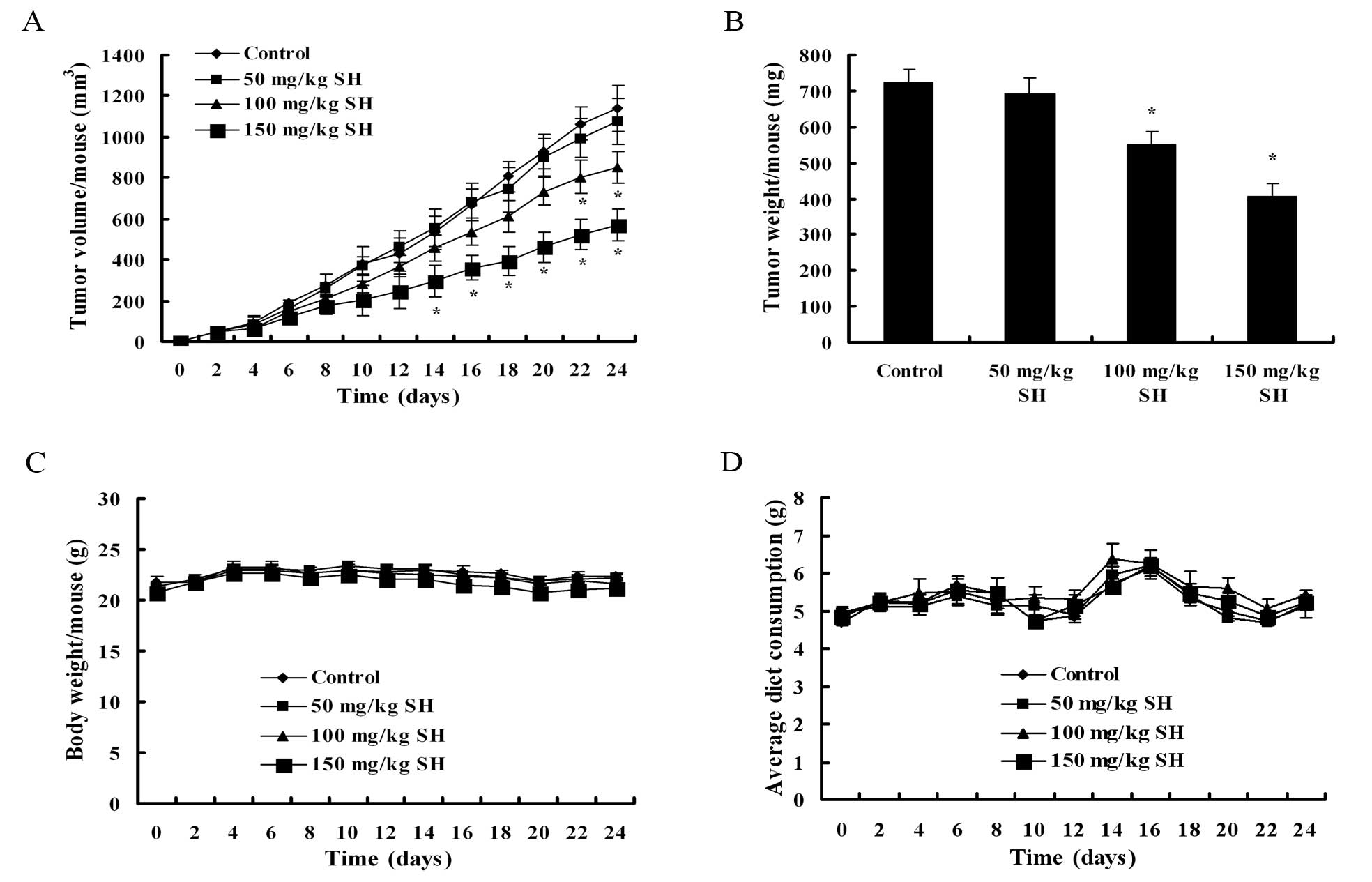
SH attenuates growth of human HCC
subcutaneous xenografts in athymic nude mice
To further evaluate the antitumor activity of SH, we
conducted an in vivo study with an athymic nude mice
xenograft model by subcutaneous inoculation of human Hep3B HCC
cells. All animals survived throughout the experiment. Fig. 7A shows that SH at the doses of 50,
100 and 150 mg/kg for 24 days caused a 5.51, 25.10 (P<0.05) and
50.01% (P<0.05) inhibition of tumor volume, respectively, as
compared with the controls. Similarly, the tumor weight per mouse
in the 50, 100 and 150 mg/kg SH-treated groups was 4.56, 23.89
(P<0.05) and 43.79% (P<0.05) less, respectively, than that in
the control group treated with the vehicle (Fig. 7B). We did not find any gross sign
of toxicity following SH administration, demonstrated by no obvious
changes in body weight and dietary consumption throughout the study
(Fig. 7C and D).
SH inhibits cell proliferation, induces
apoptosis and modulates the expression levels of p21, cleaved
caspase-3 and survivin proteins in Hep3B xenografts
The in vivo anti-proliferative effect of SH
treatment on HCC xenografts was investigated by PCNA
immunostaining. Qualitative analysis showed that the number of
PCNA-positive cells decreased from 82.2% in the control group to
78.5, 57.7 (P<0.05) and 38.9% (P<0.05) in the 50, 100 and 150
mg/kg SH-treated groups, respectively (Fig. 8A–C). TUNEL staining was performed
to assess the in vivo apoptosis induction by SH
administration. The number of apoptotic cells increased from 3.2%
in the control group to 4.1, 14.7 and 32.1% (P<0.05) in the 50,
100 and 150 mg/kg SH-treated groups, respectively (Fig. 8D–F). Correspondingly, SH treatment
at the doses of 100 and 150 mg/kg increased the expression levels
of p21 and cleaved caspase-3 proteins, as compared with the control
and 50 mg/kg SH-treated groups (Fig.
8G–L). Additionally, as expected, the decreased number of
survivin-stained cells was also observed in the tumor samples from
the SH-treated groups (Fig. 8M–O).
Overall, these data demonstrate the potential molecular mechanisms
by which SH exerts its in vivo anti-neoplastic effect on
HCC.
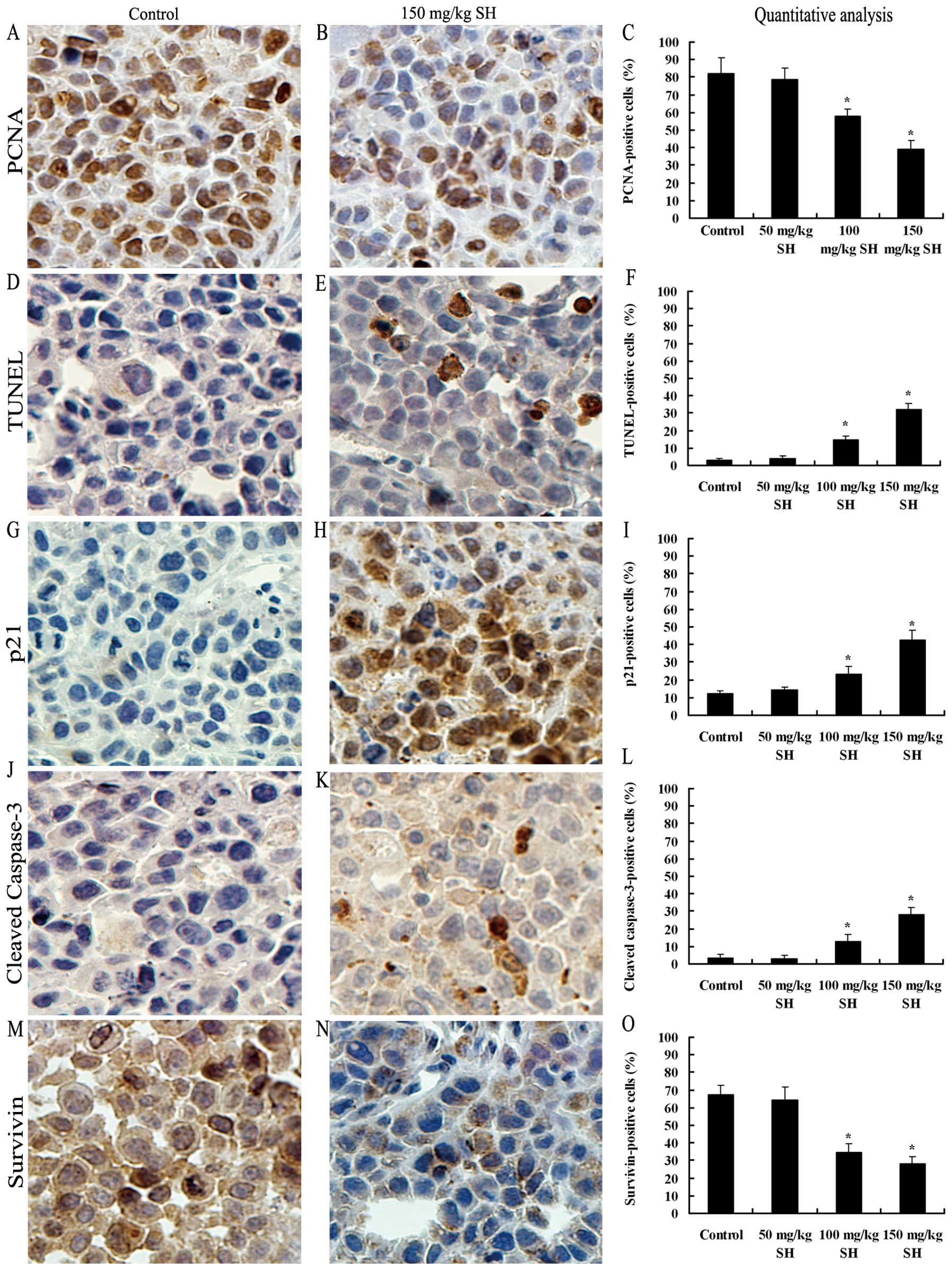 | Figure 8.SH inhibits cell proliferation,
induces apoptosis and modulates the protein expression of p21,
cleaved caspase-3 and survivin in Hep3B tumor xenografts. Tumors
were cut into sections and immunohistochemistry staining was
performed. Representative images of PCNA, TUNEL, p21, cleaved
caspase-3 and survivin staining at ×400 magnification from the
control and 150 mg/kg SH-treate4d groups are shown in (A and B), (D
and E), (G and H), (J and K) and (M and N), respectively. The
average percentage of (C) PCNA-positive cells, (F) TUNEL-positive
cells, (I) p21-positive cells, (L) caspase-3-positive cells and (O)
survivin-positive cells was counted at ×400 magnification in 5
randomly selected areas in each tumor sample. Data are expressed as
the means ± SE of mice in each group. *P<0.05
compared with the control group. Bars, SEs; SH, sinomenine
hydrochloride; HCC, hepatocellular carcinoma. |
Discussion
Available evidence suggests that some
anti-inflammatory drugs, such as aspirin act as cancer preventive
and therapeutic agents (33,34).
As a drug with the potent anti-inflammatory and immunosuppressive
efficacy against arthritis in clinical practice, SH has been found
to exhibit the in vitro anti-neoplastic activities against a
variety of tumor cell lines (12–15).
However, to date, its effect on HCC remains unknown. In the present
study, our findings demonstrated that SH treatment caused the
growth inhibition of human HCC cell lines in culture in
vitro in a concentration- and time-dependent manner, as well as
that of xenografts in vivo in nude mice, associated with the
arrest of cell cycle progression and the induction of apoptosis.
Furthermore, the detailed molecular mechanisms may involve the
increased protein expression of p21, the activation of
mitochondrial events and a caspase cascade, as well as the
downregulation of survivin.
p21, a key member of CDK inhibitors, has been proven
to cause cell cycle arrest by universally binding to the CDK-cyclin
complexes. Consistent with this fact, we found that SH promoted
cell cycle arrest by increasing the protein expression of p21 in
HCC cells. p21 is mainly regulated by the p53 tumor suppressor
protein. However, approximately 50% of human cancers, including HCC
harbor p53 mutations (35,36). Our data suggest that the SH-induced
p21 upregulation involves the p53-independent pathway, as Hep3B
cells are deficient in functional p53 (37) and no increase in p53 protein
expression was observed in the SH-treated SMMC7721 cells in which
the p53 gene is wild (data not shown).
The disturbance in the balance between cell
proliferation and apoptosis is responsible for cancer development
(16). As a result,
chemotherapeutic agents mostly exert anticancer effects by the
induction of apoptosis (21). In
the present study, SH induced-apoptosis was confirmed by the
characteristic morphological changes, DNA ladder formation and
increase in Annexin V-labeled cells in a concentration-dependent
manner in human Hep3B and SMMC7721 HCC cells.
The transduction of an apoptotic signal into cells
may alter the permeability of the membranes of the mitochondria and
Δψm, which results in the translocation of apoptogenic
proteins, such as Cyt c and Omi/HtrA2 from the
intra-membrane space into the cytoplasm (38). Cytosolic Cyt c activates
caspase-9, which subsequently leads to the activation of caspase-3
(19). As an antagonist of
inhibitors of apoptosis (IAPs), Omi/HtrA2 can competitively
displace caspases from IAPs and abolish the caspase-inhibitory
effect of IAPs (39,40). In the present study, SH treatment
disrupted Δψm, which was measured as the fluorescence
intensity ratio of JC-1 red J-aggregates/green monomers and caused
a concomitant increase in Cyt c and Omi/HtrA2 levels in the
cytosolic fraction. Furthermore, mitochondrial apoptosis induced by
pro-apoptotic Bax can be prevented by anti-apoptotic Bcl-2 by
inhibiting the release of Cyt c(22). We found that SH treatment decreased
Bcl-2 and increased Bax, which further demonstrate the role of the
mitochondria in SH-induced apoptosis.
The cleavage of caspase-9, caspase-3 and PARP was
observed in this study. Moreover, procaspase-8 and -10 levels were
decreased following SH treatment, indicating the activation of
caspase-8 and -10. These results suggest that the extrinsic death
receptor and intrinsic mitochondrial pathways participate in
SH-induced apoptosis. It would be of interest to investigate
whether the activation of caspase-8 and -10 is responsible for the
activation of the mitochondrial pathway in SH-induced apoptosis and
to examine whether SH affects Fas-associated proteins. Our results
further demonstrate that the SH-induced apoptosis depends on
caspase activation since the general caspase inhibitor, Z-VAD-FMK,
at a concentration that blocks the activation of caspase-3 and the
cleavage of PARP, prevents SH-induced apoptosis.
As a member of the IAP family, survivin is almost
absent in normal adult tissues, but abundantly overexpressed in the
majority of human cancers including HCC, and is associated with
tumor progression, treatment failure and poor prognosis (29,30).
Survivin can inhibit the function of p21 and the activation of
caspases and is a target for certain anticancer drugs, such as
silibinin and silymarin (26,30–32,41,42).
We found that SH decreased the expression level of survivin
protein, which might contribute to the increase of p21 and
activation of caspases induced by SH.
A number of studies have shown that the
histamine-releasing properties of sinomenine are possibly
responsible for its anti-rheumatic effect (5). Of note, Yang et al recently
reported that histamine decarboxylase-knockout mice exhibit a
significantly increased rate of colon and skin carcinogenesis,
which is attributed to the increase in immature myeloid cells
(IMCs). IMCs can promote the growth of cancer xenografts and
exogenous histamine can induce the differentiation of IMCs to
inhibit tumor growth (43).
Additionally, it has been reported that SH inhibits angiogenesis
in vitro and in vivo(44). Therefore, the histamine release and
anti-angiogenic effects induced by SH may also contribute to its
inhibitory effect on the growth of HCC xenografts in nude mice in
our present study. It would of interest to investigate whether SH
induces its chemopreventive and chemotherapeutic effects by
promoting the release of histamine and suppressing angiogenesis and
to elucidate the correlation between SH-released histamine and the
signaling molecules involved in SH-induced apoptosis in this
study.
In conclusion, SH suppresses the growth of human HCC
cells in vitro and in vivo, which may be possibly
attributed to cell cycle arrest and apoptosis induction. SH
upregulates p21, downregulates the Bcl-2/Bax ratio, promotes the
release of Cyt c and Omi/HtrA2 from the mitochondria into
the cytoplasm, disrupts Δψm and induces the cleavage of
caspases. Additionally, survivin is a potential molecular target of
SH. The unique properties of SH may render it a promising candidate
in the prevention and therapy of HCC. However, multidisciplinary
approaches are required to improve its anticancer activity.
Acknowledgements
This study was supported by a grant
from the National Natural Science Foundation of China
(30771895).
References:
|
1.
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Lencioni R, Chen XP, Dagher L and Venook
AP: Treatment of intermediate/advanced hepatocellular carcinoma in
the clinic: how can outcomes be improved? Oncologist. 15:42–52.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Rahbari NN, Mehrabi A, Mollberg NM, et al:
Hepatocellular carcinoma: current management and perspectives for
the future. Ann Surg. 253:453–469. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Kishi Y, Hasegawa K, Sugawara Y and Kokudo
N: Hepatocellular carcinoma: current management and future
development-improved outcomes with surgical resection. Int J
Hepatol. 2011:7281032011. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Yamasaki H: Pharmacology of sinomenine, an
anti-rheumatic alkaloid from Sinomenium acutum. Acta Med
Okayama. 30:1–20. 1976.PubMed/NCBI
|
|
6.
|
Liu L, Riese J, Resch K and Kaever V:
Impairment of macrophage eicosanoid and nitric oxide production by
an alkaloid from Sinomenium acutum. Arzneimittelforschung.
44:1223–1226. 1994.PubMed/NCBI
|
|
7.
|
Liu L, Resch K and Kaever V: Inhibition of
lymphocyte proliferation by the anti-arthritic drug sinomenine. Int
J Immunopharmacol. 16:685–691. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Liu L, Buchner E, Beitze D, et al:
Amelioration of rat experimental arthritides by treatment with the
alkaloid sinomenine. Int J Immunopharmacol. 18:529–543. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Feng H, Yamaki K, Takano H, Inoue K,
Yanagisawa R and Yoshino S: Effect of sinomenine on
collagen-induced arthritis in mice. Autoimmunity. 40:532–539. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Cheng Y, Zhang J, Hou W, et al:
Immunoregulatory effects of sinomenine on the T-bet/GATA-3 ratio
and Th1/Th2 cytokine balance in the treatment of mesangial
proliferative nephritis. Int Immunopharmacol. 9:894–899. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Xu M, Liu L, Qi C, Deng B and Cai X:
Sinomenine versus NSAIDs for the treatment of rheumatoid arthritis:
a systematic review and meta-analysis. Planta Med. 74:1423–1429.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Tong XM, Zhang J, Shen Y, Xie JJ and Jin
J: Sinomenine enhanced aclarubicin-induced apoptosis by blocking
NF-kappa B pathway in HL-60 cells. J Med Plant Res. 5:635–643.
2011.
|
|
13.
|
Li XJ, Yue PY, Ha WY, et al: Effect of
sinomenine on gene expression of the IL-1 beta-activated human
synovial sarcoma. Life Sci. 79:665–673. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Jiang T, Zhou L, Zhang W, et al: Effects
of sinomenine on proliferation and apoptosis in human lung cancer
cell line NCI-H460 in vitro. Mol Med Rep. 3:51–56.
2010.PubMed/NCBI
|
|
15.
|
Fan J, Wang JC, Chen Y, et al: Sinomenine
induces apoptosis of prostate cancer cells by blocking activation
of NF-kappa B. African J Biotechnol. 10:3480–3487. 2011.
|
|
16.
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Graña X and Reddy EP: Cell cycle control
in mammalian cells: role of cyclins, cyclin-dependent kinases
(CDKs), growth suppressor genes and cyclin-dependent kinase
inhibitors (CKIs). Oncogene. 11:211–219. 1995.PubMed/NCBI
|
|
18.
|
Loo DT and Rillema JR: Measurement of cell
death. Methods Cell Biol. 57:251–264. 1998. View Article : Google Scholar
|
|
19.
|
Budihardjo I, Oliver H, Lutter M, Luo X
and Wang X: Biochemical pathways of caspase activation during
apoptosis. Annu Rev Cell Dev Biol. 15:269–290. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Jin Z and El-Deiry WS: Overview of cell
death signaling pathways. Cancer Biol Ther. 4:139–163.
2005.PubMed/NCBI
|
|
21.
|
Ghobrial IM, Witzig TE and Adjei AA:
Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin.
55:178–194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Adams JM and Cory S: The Bcl-2 protein
family: arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Slee EA, Adrain C and Martin SJ:
Executioner caspase-3, -6 and -7 perform distinct, non-redundant
roles during the demolition phase of apoptosis. J Biol Chem.
276:7320–7326. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Delhalle S, Duvoix A, Schnekenburger M,
Morceau F, Dicato M and Diederich M: An introduction to the
molecular mechanisms of apoptosis. Ann NY Acad Sci. 1010:1–8. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Zeng J, Sun Y, Wu K, et al:
Chemopreventive and chemotherapeutic effects of intravesical
silibinin against bladder cancer by acting on mitochondria. Mol
Cancer Ther. 10:104–116. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Lu XL, He SX, Ren MD, Wang YL, Zhang YX
and Liu EQ: Chemopreventive effect of saikosaponin-d on
diethylinitrosamine-induced hepatocarcinogenesis: Involvement of
CCAAT/enhancer binding protein β and cyclooxygenase-2. Mol Med Rep.
5:637–644. 2012.PubMed/NCBI
|
|
28.
|
Oliver FJ, de la Rubia G, Rolli V,
Ruiz-Ruiz MC, de Murcia G and Murcia JM: Importance of
poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson
from an uncleavable mutant. J Biol Chem. 273:33533–33539. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Mita AC, Mita MM, Nawrocki ST and Giles
FJ: Survivin: key regulator of mitosis and apoptosis and novel
target for cancer therapeutics. Clin Cancer Res. 14:5000–5005.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Ito T, Shiraki K, Sugimoto K, et al:
Survivin promotes cell proliferation in human hepatocellular
carcinoma. Hepatology. 31:1080–1085. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Shin S, Sung BJ, Cho YS, et al: An
anti-apoptotic protein human survivin is a direct inhibitor of
caspase-3 and -7. Biochemistry. 40:1117–1123. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Tamm I, Wang Y, Sausville E, et al:
IAP-family protein survivin inhibits caspase activity and apoptosis
induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer
Res. 58:5315–5320. 1998.PubMed/NCBI
|
|
33.
|
Neugut AI: Aspirin as adjuvant therapy for
colorectal cancer: a promising new twist for an old drug. JAMA.
302:688–689. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Jankowski JA and Limburg PJ: Aspirin
therapy for cancer: it is never too late. Br J Cancer.
105:1105–1106. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Hollstein M, Sidransky D, Vogelstein B and
Harris CC: p53 mutations in human cancers. Science. 253:49–53.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Oda T, Tsuda H, Scarpa A, Sakamoto M and
Hirohashi S: p53 gene mutation spectrum in hepatocellular
carcinoma. Cancer Res. 52:6358–6364. 1992.PubMed/NCBI
|
|
37.
|
Puisieux A, Galvin K, Troalen F, et al:
Retinoblastoma and p53 tumor suppressor genes in human hepatoma
cell lines. FASEB J. 7:1407–1413. 1993.PubMed/NCBI
|
|
38.
|
Goldsmith KC and Hogarty MD: Targeting
programmed cell death pathways with experimental therapeutics:
opportunities in high-risk neuroblastoma. Cancer Lett. 228:133–141.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Suzuki Y, Imai Y, Nakayama H, Takahashi K,
Takio K and Takahashi R: A serine protease, HtrA2, is released from
the mitochondria and interacts with XIAP, inducing cell death. Mol
Cell. 8:613–621. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Hegde R, Srinivasula SM, Zhang Z, et al:
Identification of Omi/HtrA2 as a mitochondrial apoptotic serine
protease that disrupts inhibitor of apoptosis protein-caspase
interaction. J Biol Chem. 277:432–438. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Singh RP, Tyagi A, Sharma G, Mohan S and
Agarwal R: Oral silibinin inhibits in vivo human bladder tumor
xenograft growth involving down-regulation of survivin. Clin Cancer
Res. 14:300–308. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Katiyar SK, Roy AM and Baliga MS:
Silymarin induces apoptosis primarily through a p53-dependent
pathway involving Bcl-2/Bax, cytochrome c release and caspase
activation. Mol Cancer Ther. 4:207–216. 2005.PubMed/NCBI
|
|
43.
|
Yang XD, Ai W, Asfaha S, et al: Histamine
deficiency promotes inflammation-associated carcinogenesis through
reduced myeloid maturation and accumulation of
CD11b+Ly6G+ immature myeloid cells. Nat Med.
17:87–95. 2011. View Article : Google Scholar
|
|
44.
|
Kok TW, Yue PY, Mak NK, Fan TP, Liu L and
Wong RN: The anti-angiogenic effect of sinomenine. Angiogenesis.
8:3–12. 2005. View Article : Google Scholar
|