Introduction
The Rho GTPases, such as Cdc42, Rac and Rho, can
impact cell morphology and migration by regulating the actin
cytoskeleton (1). The Rho GTPases
can also regulate proliferation and activate signaling pathways,
including the JNK/MAPK and JNK/SAPK signaling pathways (2–6). The
Rho GTPases are required for binding to certain proteins (1). The p21-activated kinase (Pak) family
of serine/threonine kinases are major target proteins of the Rho
GTPases (7). Six mammalian Pak
proteins have been identified in this family and have been
classified into 2 groups: group 1 Paks (Pak1–Pak3) and group 2 Paks
(Pak4–Pak6) (8). Pak4 was first
identified as an effector of Cdc42 in mouse embryonic fibroblast
cell lines (9). The overexpression
of Pak4 has been observed in a number of cancer cell lines
(10). Pak4 has also been shown to
be overexpressed in many types of cancer, such as esophageal
squamous cell carcinoma (11) and
mouse colon tumors (12). However,
to our knowledge, little is known about the individual role of Pak4
in Hep-2 laryngeal carcinoma cells.
Laryngeal squamous cell carcinoma (LSCC) constitutes
almost 2 to 3% of all malignant tumors, with a total of 159,000 new
cases of carcinoma per year, more commonly affecting males
(13). In China, diagnostic and
therapeutic modalities for laryngocarcinoma have improved over the
past few years. However, the incidence of LSCC is gradually rising,
particularly in Northeastern China (14).
In the present study, we investigated the effects of
Pak4 on Hep-2 laryngeal carcinoma cells by regulating its
expression in vitro and in vivo. The mechanisms of
the effect of Pak4 on laryngeal carcinoma cells were also
investigated.
Materials and methods
Cell lines and cell culture
Hep-2 laryngeal carcinoma cells were obtained from
the American Type Culture Collection (ATCC; Bethesda, MD) and
maintained in RPMI-1640 medium (Life Technologies, Inc.,
Gaithersburg, MD) supplemented with 10% (v/v) fetal bovine serum
(FBS) and antibiotics (100 units/ml of penicillin and 100 mg/ml of
streptomycin) at 37°C in a 5% (v/v) CO2 incubator.
siRNA against Pak4 and transfection
Pak4 small interfering RNA (siRNA) oligonucleotides
were purchased from Dharmacon, Inc. (Lafayette, CO). Cells were
seeded onto 60-mm plates for 24 h and transfected with siRNA or
control siRNA for 48 h using Lipofectamine 2000 (Invitrogen Life
Technologies, Carlsbad, CA) according to the manufacturer’s
instructions.
RT-PCR
Total RNA was isolated using an RNeasy Mini kit
(BioMed, Beijing, China). cDNA was reverse transcribed with 1
μg of total RNA, using the Takara Reverse Transcription kit
(Takara Dalian, Dalian, China). cDNA was amplified using the
following primers: the sequences of the forward and reverse primers
for Pak4 were 5′-GACATCAAGAGCGACTCGATCC-3′ and
5′-ATCACCATTATCCCCAGCGAC-3′, respectively. GAPDH was used as
the internal control. The sequences of the forward and reverse
primers for GAPDH were 5′-AGAAGGCTGGGGCTCATTTG-3′ and
5′-AGGGGCCATCCACAGTCTTC-3′, respectively. PCR was performed for 35
cycles under the following conditions: annealing at 56°C (15 sec),
extension at 72°C (30 sec) and denaturing at 94°C (30 sec) using a
Takara thermal cycler.
Western blot analysis
Total cell extracts were obtained using lysis buffer
containing 20 mM HEPES (pH 7.4), 150 mM NaCl, 1 mM
MgCl2, 1 mM EDTA, 10% glycerol, 1% Triton X-100, 1
μg/ml leupeptin and 1 μg/ml aprotinin. Equal amounts
(90 μg) of cell lysates were separated by 10%
SDS-polyacrylamide gel electrophoresis, transferred onto
polyvinylidene difluoride membranes and incubated with the
following specific antibodies: Pak4 antibody (Abcam PLC, Cambridge,
UK) was used to identify transfection efficiency. β-actin (Santa
Cruz Biotechnology, Inc., Santa Cruz, CA) was used as the internal
control. The reaction was followed by probing with
peroxidase-coupled secondary antibodies including anti-rabbit IgG,
or anti-mouse IgG antibodies at dilutions ranging from 1:1,000 to
1:2,000 (Amersham Biosciences, Needham, MA) and binding results
were visualized by enhanced chemiluminescence (Amersham Pharmacia
Biotech, Piscataway, NJ).
Colony formation assay
For the colony formation assay, cells (200 cells per
well) were seeded in 24-well tissue culture plates. Plates were
incubated for 3 weeks in a humidified incubator at 37°C. Three
weeks after seeding, colonies were stained with 0.05% crystal
violet containing 50% methanol and counted. The colonies were
counted in 4 to 5 random fields from each of the duplicate samples
by using a microscope at ×100 magnification.
Measurement of caspase-3 and -9
activities
Caspase activities were measured by colorimetric
assay kits (KeyGen Biotech. Co., Ltd., Nanjing, China) according to
the manufacturer’s instructions. After harvesting, cells were
washed in ice-cold PBS and lysed; proteins were extracted and
stored at −80°C. Cell lysates (20 μl) were then added to a
buffer containing a p-nitroaniline (pNA)-conjugated substrate for
caspase-3 (Ac-DEVD-pNA), or -9 (LEHD-pNA) to a total of 100
μl reaction volume. Incubation was carried out at room
temperature for caspase-3 and -9 followed by shaking at 500 rpm for
1 min and then incubation at room temperature for 2 h. The
concentration of the released pNA in each well was measured using a
plate-reading luminometer (Thermo Fisher Scientific, Beijing,
China). Data were from 3 independent experiments.
Cell cycle analysis
The Hep-2 laryngeal carcinoma cells
(3×105/well) were plated and incubated overnight. The
control and treated cells were trypsinized, collected in PBS and
fixed on ice with 1% paraformaldehyde, followed by 70% cold
ethanol. Following treatment with 10 μg/ml RNase, the cells
were stained with 50 μg/ml propidium iodide (PI; KeyGen
Biotech. Co., Ltd.) for 15 min at room temperature for cell cycle
analysis. The stained cells were analyzed by flow cytometry. Data
analysis was performed with CellQuest software (BD Biosciences,
Rockville, MD).
Labeling of cells with thymidine
analogs
Actively replicating cells at the beginning of each
hour of the S phase were first labeled with the thymidine analog,
5-iodo-2′-deoxyuridine (IdU; 50 μM) (Sigma-Aldrich,
Carlsbad, CA) for 40 min, washed 3 times with PBS and then labeled
with 5-chloro-2′-deoxyuridine (CldU; 100 μM) (Sigma) for 40
min. IdU and CldU incorporated into replicating DNA were later
detected with red or green fluorescent antibodies, respectively, as
described below. Three independent replicate experiments were
performed and analyzed for the control, mock and treated cells.
Immunostaining
Slides were treated with 70% ethanol, washed in PBS,
denatured in 2.5 M HCl for 30 min and permeabilized in 0.25% Triton
X-100 for 5 min and blocked with 1% bovine serum albumin. The
slides were incubated at room temperature with the following
antibodies: i) 1:500 mouse anti-bromodeoxyuridine (detects IdU)
(Sigma); ii) 1:1,000 Alexafluor 488-conjugated anti-mouse (Sigma);
iii) 1:2,000 rat anti-bromodeoxyuridine (detects CldU) (Santa Cruz
Biotechnology, Inc.); and iv) 1:1,000 Alexafluor 633-conjugated
anti-rat antibodies (Invitrogen Life Technologies).
After counterstaining with DAPI (1 μg/ml)
(KeyGen Biotech. Co., Ltd.), photographic images were taken using
an Olympus CX71 fluorescence microscope (Olympus, Tokyo,
Japan).
Xenografted Hep-2 tumor mouse model
All in vivo experiments were approved by the
Ethics Committee of China Medical University. Human tumors were
induced by a subcutaneous (s.c.) injection of 5×106
Hep-2 cells into the dorsal flank region of nu/nu mice
(CAnN.Cg-Foxn1nu/Crl; Vital River, Beijing, China). When
tumor volumes had reached a mean volume of 50±5 mm3, the
animals were randomized into 3 groups (n=15 per group). All mice
received 2 injections, 72 h apart (days 1 and 4). Injections were
administered by mixing siRNA (20 μg) with Lipofectamine 2000
(30 μl) and PBS in a total volume of 100 μl. Tumors
were measured with calipers every 5 days and tumor volumes were
calculated (tumor volume = length × width2 × 0.52)
(15).
An additional 180 mice were used to establish
xenografts to observe survival time. For these experiments, mice
with xenograft tumors were treated as described above. Survival was
monitored until the experiments were terminated due to heavy tumor
burden.
Immunohistochemical staining
Immunohistochemical staining was performed on
4-μm sections obtained from formalin-fixed,
paraffin-embedded blocks. Endogenous peroxidase activity was
blocked with 3% hydrogen peroxide for 30 min. Antigen retrieval was
carried out in citrate buffer (10 mM, pH 6.0) for 30 min at 95°C in
a microwave oven (16). Sections
were incubated with primary antibody at 4°C overnight. Cell cycle
checkpoint-regulated proteins were probed with: anti-ataxia
telangiectasia mutated (ATM), anti-p53, anti-checkpoint kinase
(Chk)1, anti-Chk2 (Santa Cruz Biotechnology, Inc.),
anti-phospho-S345-Chk1 and anti-phospho-T68-Chk2 antibodies (Cell
Signaling Technology, Danvers, MA). Cell cycle-regulated proteins
were probed with: anti-Cyclin A (Santa Cruz Biotechnology, Inc.)
and anti-cyclin-dependent kinase 2 (CDK2) antibodies (Abcam PLC).
The sections were then incubated with a biotinylated secondary
antibody and exposed to a streptavidin complex (HRP). Positive
reactions were visualized with 3,3′-diaminobenzidine
tetrahydrochloride (DAB; Sigma), followed by counterstaining with
hematoxylin. Sections treated without primary antibodies were used
as negative controls.
Statistical analysis
Data were analyzed using GraphPad Prism 5 software.
Statistical analysis was performed using a one-tailed Student’s
t-test (unilateral and unpaired). Kaplan-Meier survival plots were
generated and comparisons between survival curves were made using
log-rank statistical analysis. P-values <0.05 were considered to
indicate statistically significant differences.
Results
mRNA and protein levels of Pak4 were
evaluated in Hep-2 laryngeal carcinoma cells
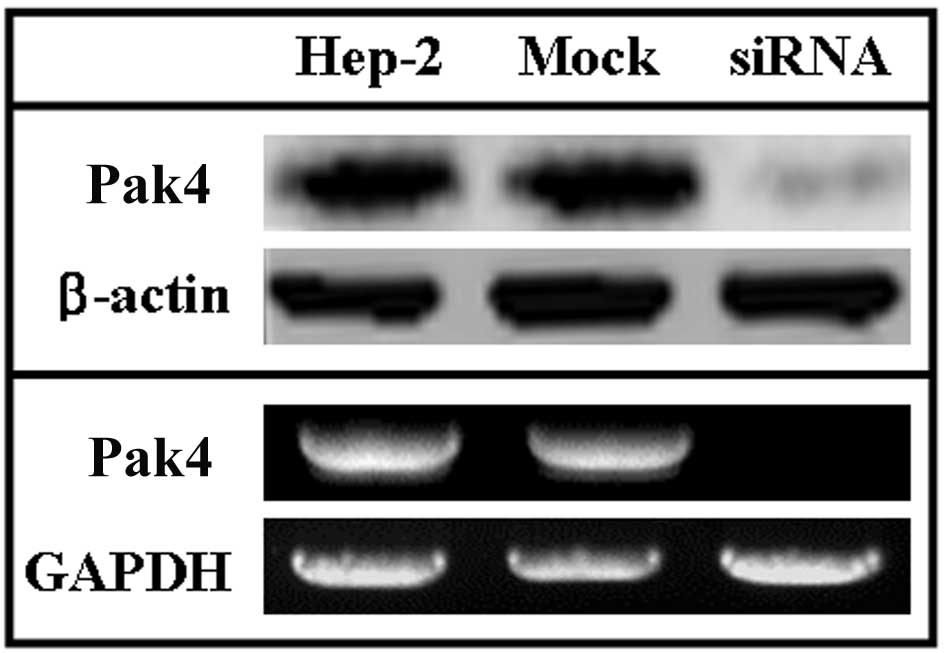
The levels of Pak4 in siRNA-treated Hep-2, untreated
and mock-treated cells were detected using western blot and RT-PCR
assays. In the western blot assays, the levels of Pak4 protein were
higher in the untreated Hep-2 and mock-treated Hep-2 cells compared
with the siRNA-treated Hep-2 cells (Fig. 1, upper panel). To determine the
correlation between the levels of Pak4 and the transcription of
Pak4, RT-PCR analysis of Pak4 was performed. In these
assays, the levels of Pak4 mRNA were higher in the untreated
Hep-2 cells compared to the treated ones and were consistent with
the levels of Pak4 detected by western blot analysis (Fig. 1, lower panel).
Pak4 is required for tumor growth in
Hep-2 cells
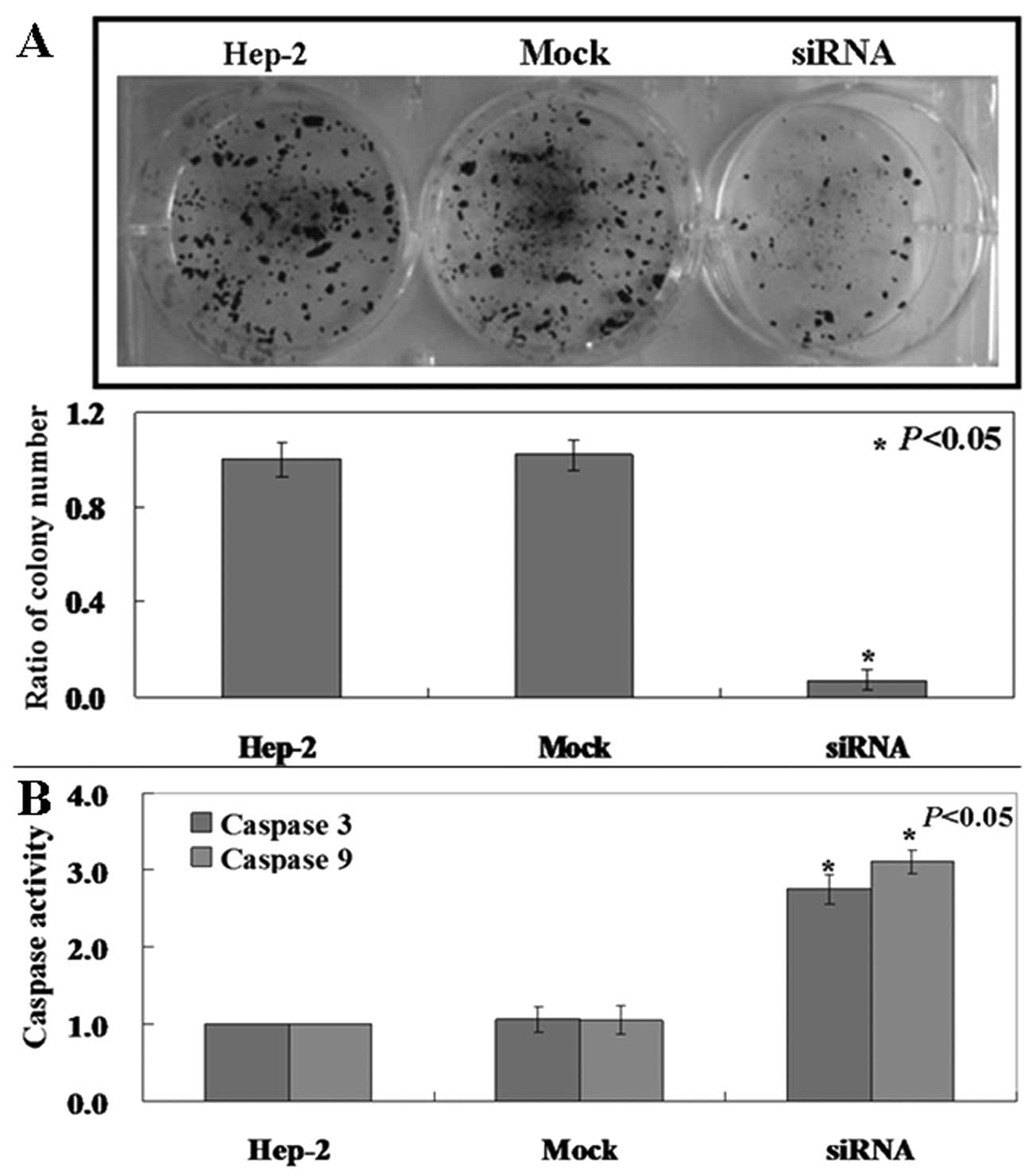
Pak4 mRNA and protein levels are high in Hep-2
cells. We determined whether its downregulation would reduce the
tumorigenicity of laryngeal carcinoma cells. Clonogenic assay
showed that the proliferation rate of siRNA-treated Hep-2 cells was
decreased compared to the untreated and mock-treated cells
(Fig. 2A, p<0.05). Caspase-3
and caspase-9 activities in the treated cells was also found to be
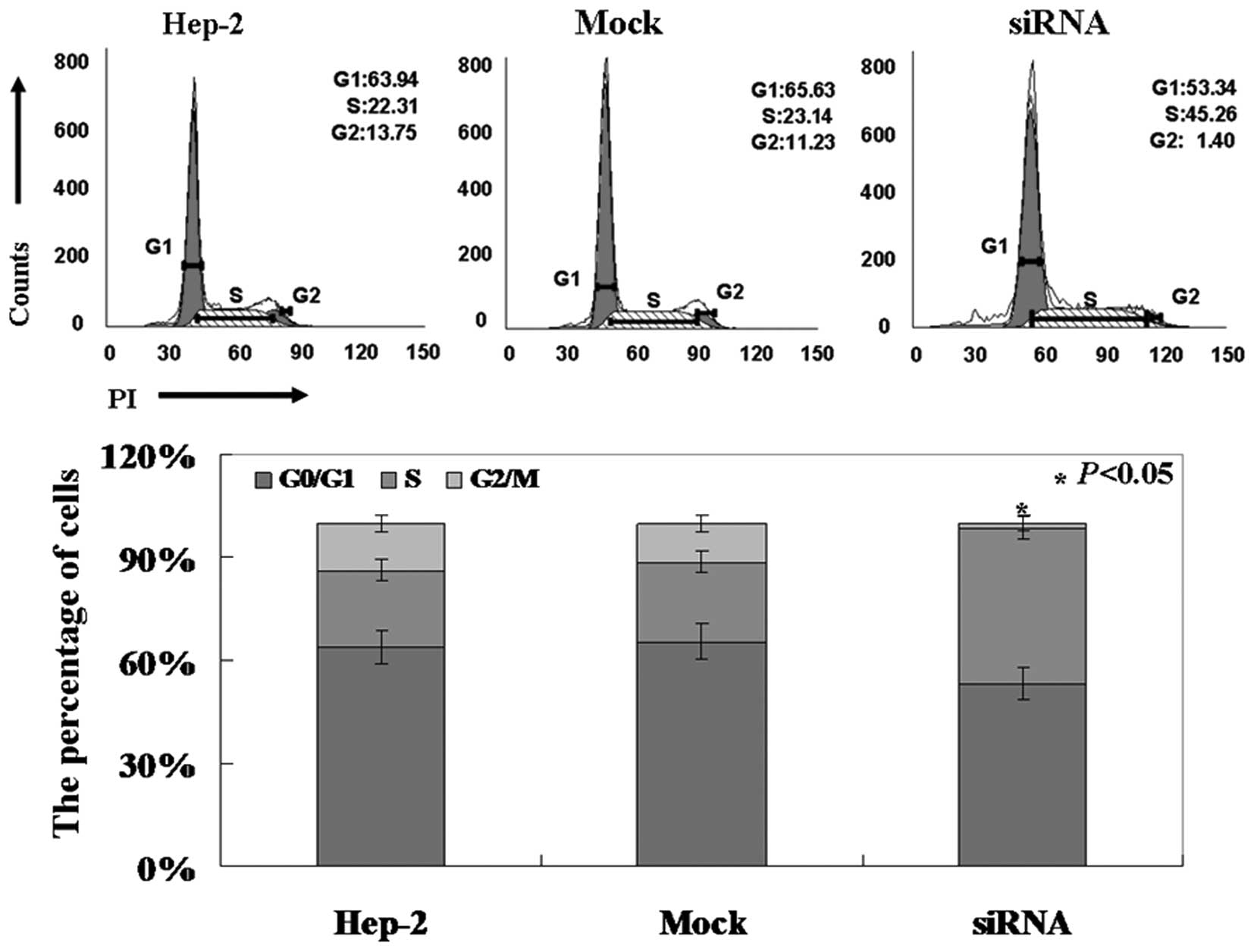
higher compared to the untreated and mock-treated cells (Fig. 2B, p<0.05). PI staining of cells
revealed that Pak4-siRNA cells were arrested in the S phase
(Fig. 3, p<0.05).
Hep-2 cells were obstructed in the
S/G2 transition following siRNA treatment
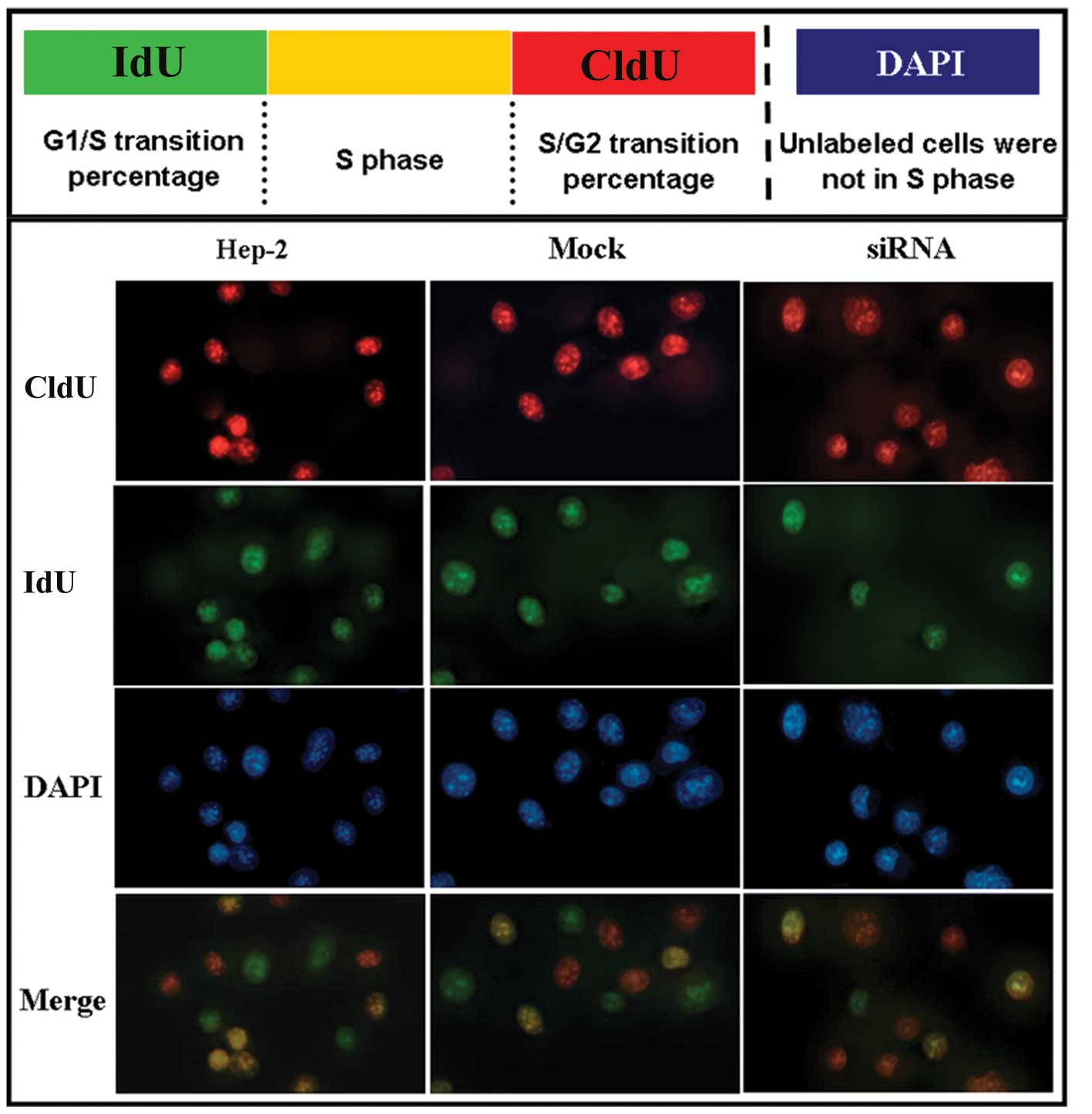
To further analyze the proliferation activity of
Hep-2 cells after siRNA treatment, a CldU/IdU double-labeling
method was applied to distinguish CldU and IdU incorporated into
cellular DNA. According to the method of Bakker et
al(17), we assessed the
G1/S and S/G2 checkpoint in Hep-2 cells
following siRNA treatment. Our labeling strategy is shown in
Fig. 4 (upper panel). In this
assay, single-labeled IdU cells (green) are cells that enter the S
phase after the first labeling period of the experiment.
Single-labeled CldU cells (red) are cells that are left int he S
phase before the second labeling period of the experiment. Cells
labeled both with CldU and IdU (yellow) are in the S phase.
Non-labeled cells are not in the S phase. This method helped us to
identify the percentage of cells in the G1/S and
S/G2 transitions. As shown by our results, there was no
difference in the number of Hep-2 cells and mock cells in the
G1/S transition, S phase and S/G2 transition.
Non-labeled cells were considered cells in other phases (Fig. 4, lower panel). However, we found
that the number of Hep-2 cells following siRNA treatment in the
S/G2 transition was higher than that of cells in the
G1/S transition (Fig. 4,
lower panel). This indicated that Hep-2 cells were obstructed
in the S/G2 transition following siRNA treatment.
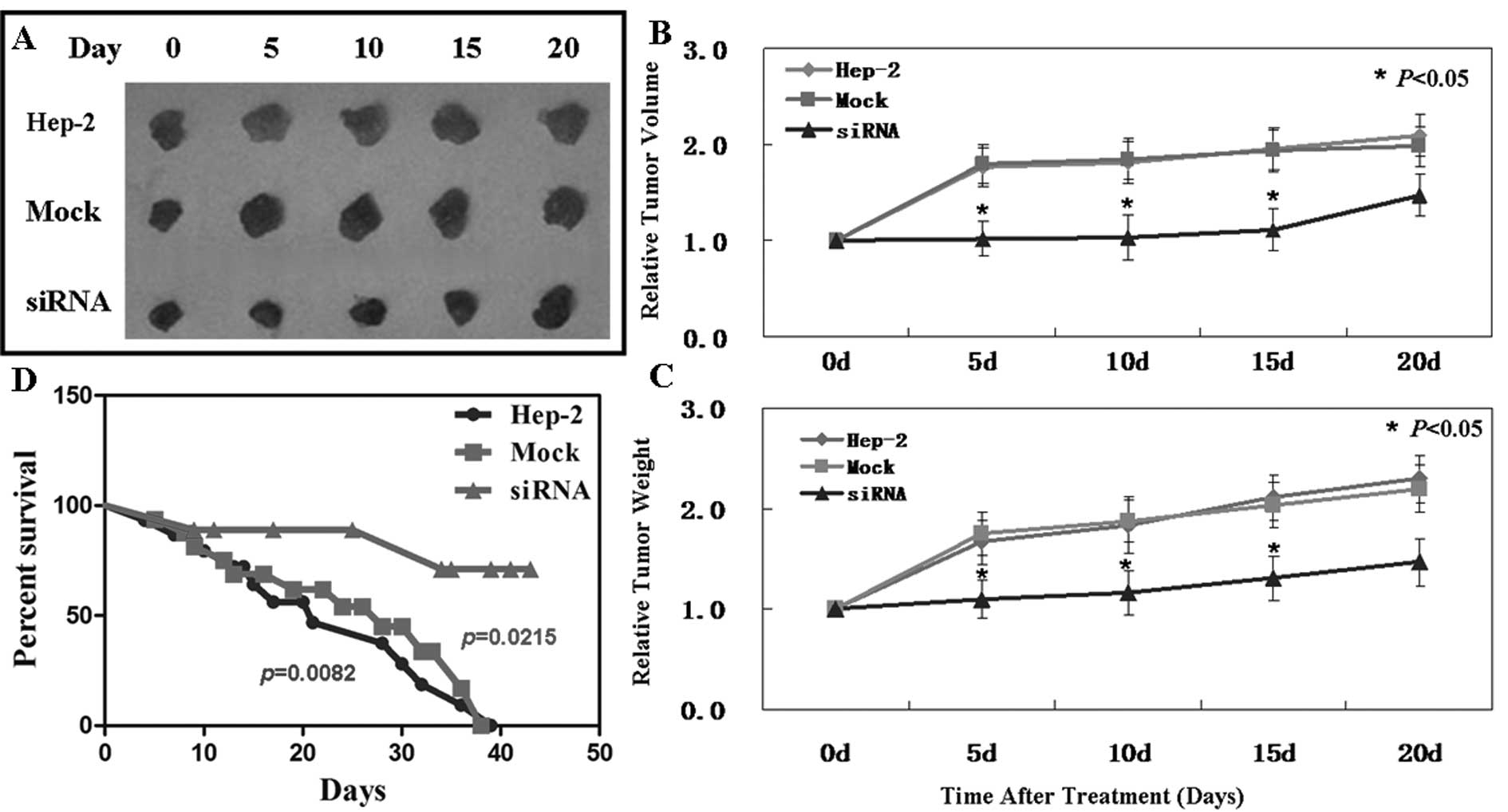
In vivo anti-tumor effect of Pak4-siRNA
on laryngeal carcinoma xenografts
The anti-tumor effect of Pak4-siRNA was analyzed in
vivo using Hep-2 laryngeal carcinoma subcutaneous xenografts. When
the established tumors reached 5–7 mm in diameter, siRNA was
injected into the tumors. Treatment with siRNA was performed by
mixing the plasmid DNA (20 μg) with Lipofectamine 2000 (30
μl) and PBS in a total volume of 100 μl per mouse and
injecting the mice intratumorally twice every 3 days. As is shown
in Fig. 5B, on day 15, the tumor
volume of the control group was 623.1±43.3 mm3, that of
the mock-treated group was 637.4±48.6 mm3 and that of
the siRNA-treated group was 324.6±32.7 mm3. These
results demonstrate the suppressive effect of Pak4-siRNA on tumor
growth. Moreover, Pak4-siRNA showed the same effects on tumor
weight (Fig. 5C). However, on day
20, tumor volume and weight showed no significant differences
between the 3 groups. Subsequently, the effectiveness of siRNA on
the xenografts gradually diminished after 15 days.
A significantly improved survival rate was observed
in the mice treated with siRNA (Fig.
5D). The mice began to die first in the siRNA-treated group on
day 9, and then in the control and mock-treated groups on days 3
and 4, respectively. At the end of this experiment, there were 10
out of 15 mice left alive in the siRNA group. However, all mice
died in the other 2 groups.
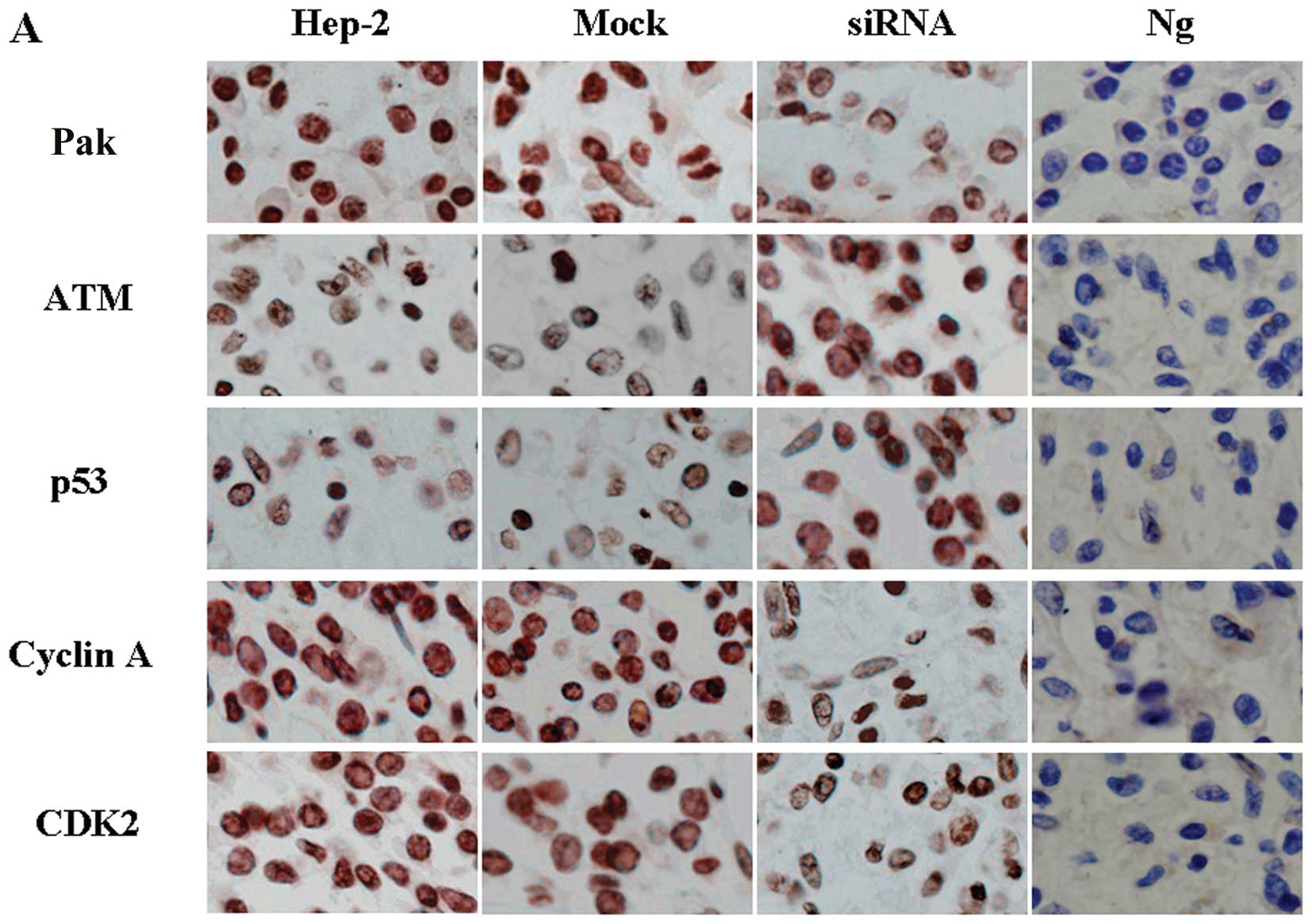
Downregulation of Pak4 causes S phase
arrest and is associated with the upregulation of p53
To identify the mechanism of the cell cycle arrest
induced by Pak4, immunohistochemical staining was performed to
detect changes in cell cycle-related proteins. The results
demonstrated that the downregulation of Pak4 induced a significant
decrease in the levels of cyclin A (Fig. 6A). Decreased levels of CDK2 were
also detected (Fig. 6A). In
further experiments, we found that p53 was activated following the
downregulation of Pak4 (Fig. 6A).
These results indicate that the cell cycle arrest is dependent on
p53 function in these cells.
Downregulation of Pak4-induced arrest at
the S/G2 transition depends on both Chk1 and Chk2
The protein kinases, ATM and Chk1/Chk2, are major
components of the mechanisms that oversee the control of DNA
replication and genomic integrity (18). In our study, we found that the
levels of Chk1 and Chk2 were not altered in the siRNA-treated group
compared with the control and mock-treated groups (Fig. 6B). However, the levels of P-Chk1
and P-Chk2 were significantly increased in the siRNA-treated group
(Fig. 6B). To examine the
hypothesis that the ATM/Chk1/Chk2-p53 pathway is activated by the
downregulation of Pak4 in the siRNA-treated group, we detected the
levels of ATM, the upstream protein of Chk1 and Chk2. We found that
ATM levels also increased (Fig.
6B). These data are consistent with the results of the cell
cycle arrest at the S/G2 transition.
Discussion
Pak4 was originally identified as a protein which
binds strongly to Cdc42 and mediates Cdc42-induced cytoskeletal
organization and cell shape (9).
Gnesutta et al found that Pak4 regulates cell growth and
survival (19). There is
increasing evidence that Pak4 is overexpressed in human tumors and
cancer cell lines (10–12). Consistent with previous studies, in
this study, we demonstrate that the serine/threonine kinase, Pak4,
is overexpressed in Hep-2 cells. In order to determine the role of
Pak4 in Hep-2 cells, we established Pak4-knockdown cell lines by
using siRNA. We also confirmed that the downregulation of Pak4
inhibits proliferation and induce S phase arrest in Hep-2 cells.
Similarly, Pak4 knockdown displayed anti-tumor activities in
laryngeal carcinoma xenografts. In further experiments, by using
CldU and IdU double staining, we found that Pak4 knockdown
obstructed S/G2 transition.
Through the course of detecting the mechanism of S
arrest, we found that the levels of cyclin A and CDK2 were
decreased. Beamish et al(20) also found that the inhibition of
cyclin A and CDK2 resulted in mitotic cell arrest with the
activation of the spindle assembly checkpoint. We confirmed that
p53, the inhibitor of cyclin A, was activated by Pak4 knockdown.
This is the cause of the inhibition of cyclin A.
Indeed, the most significant results of our study
was that Pak4 knockdown obstructed S/G2 transition. Chk2
plays important roles in the DNA damage response, the signaling of
the ATM/Chk2/p53 pathway and in cell cycle checkpoints including
the S checkpoint (21,22). The response typically leads to the
activation of p53, predominantly through ATM and Chk2 (23,24).
The outcome of p53 activation ranges from cell cycle arrest and DNA
repair to apoptosis (25,26). Consistent with previous studies, we
found that the ATM/Chk2/p53 pathway was activated by Pak4
knockdown. The expression of ATM was higher in the treated compared
to the untreated cells. Accompanied with the upregulation of ATM,
p-Chk2 was also increased. Of note, in our study, Chk1 was also
activated by Pak4 knockdown. Chk1 plays an important role in DNA
repair and is essential for the maintenance of genomic stability
(27,28). In previous studies, Ahmed et
al(29) found that the small
molecule, reactivation of p53 and induction of tumor cell apoptosis
(RITA), activated the canonical ATM/ATR DNA damage response pathway
that leads to the activation of Chk1 and Chk2 phosphorylation. In
our study, we also confirmed that the phosphoryltion of Chk1 and
Chk2 was increased accompanied with the activation of ATM.
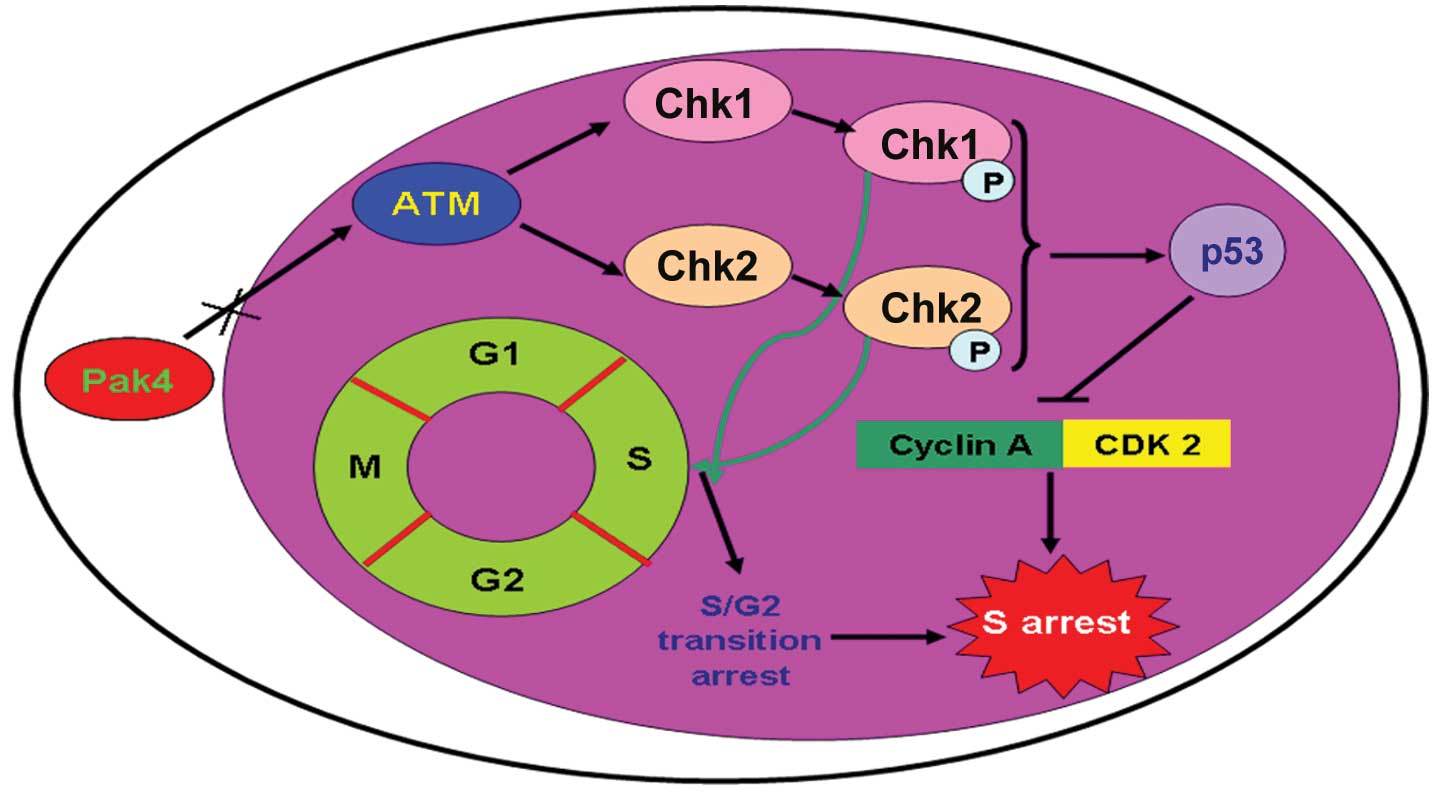
In summary, we confirmed the anti-tumor effects of
the downregulation of Pak4 on Hep-2 laryngeal carcinoma cells in
all our experiments. We found that the downregulation of Pak4
activated the ATM/Chk1/2/p53 signaling pathway in laryngeal
carcinoma cells. After the signaling pathway was activated,
apoptosis was induced by activated caspase-3 and caspase-9. We also
found that activation of the ATM/Chk1/2/p53 pathway promoted
S/G2 transition arrest (Fig. 7). The data presented in our study
maybe provide a novel insight into laryngeal carcinoma
treatment.
Acknowledgements
We thank Dr Dong-ying Wu for his
valuable comments and excellent technical assistance.
References
|
1.
|
Van Aelst L and D’Souza-Schorey C: Rho
GTPases and signaling networks. Genes Dev. 11:2295–2322.
1997.PubMed/NCBI
|
|
2.
|
Bagrodia S, Dérijard B, Davis RJ and
Cerione RA: Cdc42 and PAK-mediated signaling leads to Jun kinase
and p38 mitogen-activated protein kinase activation. J Biol Chem.
270:27995–27998. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Dutartre H, Davoust J, Gorvel JP and
Chavrier P: Cytokinesis arrest and redistribution of
actin-cytoskeleton regulatory components in cells expressing the
Rho GTPase CDC42Hs. J Cell Sci. 109:367–377. 1996.PubMed/NCBI
|
|
4.
|
Coso OA, Chiariello M, Yu JC, et al: The
small GTP-binding proteins Rac1 and Cdc42 regulate the activity of
the JNK/SAPK signaling pathway. Cell. 81:1137–1146. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Minden A, Lin A, Claret FX, et al:
Selective activation of the JNK signaling cascade and c-Jun
transcriptional activity by the small GTPases Rac and Cdc42Hs.
Cell. 81:1147–1157. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Brown JL, Stowers L, Baer M, et al: Human
Ste20 homologue hPAK1 links GTPases to the JNK MAP kinase pathway.
Curr Biol. 6:598–605. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Kumar R, Gururaj AE and Barnes CJ:
p21-activated kinases in cancer. Nat Rev Cancer. 6:459–471. 2006.
View Article : Google Scholar
|
|
8.
|
Arias-Romero LE and Chernoff J: A tale of
two Paks. Biol Cell. 100:97–108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Abo A, Qu J, Cammarano MS, et al: PAK4, a
novel effector for Cdc42Hs, is implicated in the reorganization of
the actin cytoskeleton and in the formation of filopodia. EMBO J.
17:6527–6540. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Callow MG, Clairvoyant F, Zhu S, et al:
Requirement for PAK4 in the anchorage-independent growth of human
cancer cell lines. J Biol Chem. 277:550–558. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Fang MZ, Jin Z, Wang Y, et al: Promoter
hypermethylation and inactivation of
O6-methylguanine-DNA methyltransferase in esophageal
squamous cell carcinomas and its reactivation in cell lines. Int J
Oncol. 26:615–622. 2005.PubMed/NCBI
|
|
12.
|
Thompson HJ and Singh M: Rat models of
premalignant breast disease. J Mammary Gland Biol Neoplasia.
5:409–420. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Jemal A, Bray F, Center MM, et al: Global
cancer statistics. CA Cancer J Clin. 61:69–90. 2011. View Article : Google Scholar
|
|
14.
|
Che XH, Chen H, Xu ZM, et al:
14-3-3epsilon contributes to tumour suppression in laryngeal
carcinoma by affecting apoptosis and invasion. BMC Cancer.
10:3062010. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Alessandri G, Filippeschi S, Sinibaldi P,
et al: Influence of gangliosides on primary and metastatic
neoplastic growth in human and murine cells. Cancer Res.
47:4243–4247. 1987.PubMed/NCBI
|
|
16.
|
Dover R and Patel K: Improved methodology
for detecting bromodeoxyuridine in cultured cells and tissue
sections by immunocytochemistry. Histochemistry. 102:383–387. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Bakker PJ, Stap J, Tukker CJ, et al: An
indirect immunofluorescence double staining procedure for the
simultaneous flow cytometric measurement of iodo- and
chlorodeoxyuridine incorporated into DNA. Cytometry. 12:366–372.
1991. View Article : Google Scholar
|
|
18.
|
Nojima H: Protein kinases that regulate
chromosome stability and their downstream targets. Genome Dyn.
1:131–148. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Gnesutta N, Qu J and Minden A: The
serine/threonine kinase PAK4 prevents caspase activation and
protects cells from apoptosis. J Biol Chem. 276:14414–14419.
2001.PubMed/NCBI
|
|
20.
|
Beamish H, de Boer L, Giles N, et al:
Cyclin A/cdk2 regulates adenomatous polyposis coli-dependent
mitotic spindle anchoring. J Biol Chem. 284:29015–29023. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Stracker TH, Usui T and Petrini JH: Taking
the time to make important decisions: the checkpoint effector
kinases Chk1 and Chk2 and the DNA damage response. DNA Repair
(Amst). 8:1047–1054. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Löbrich M and Jeggo PA: The impact of a
negligent G2/M checkpoint on genomic instability and cancer
induction. Nat Rev Cancer. 7:861–869. 2007.PubMed/NCBI
|
|
23.
|
Karlseder J, Broccoli D, Dai Y, et al:
p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2.
Science. 283:1321–1325. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Antoni L, Sodha N, Collins I and Garrett
MD: CHK2 kinase: cancer susceptibility and cancer therapy - two
sides of the same coin? Nat Rev Cancer. 7:925–936. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Wells BS, Yoshida E and Johnston LA:
Compensatory proliferation in Drosophila imaginal discs
requires Dronc-dependent p53 activity. Curr Biol. 16:1606–1615.
2006.PubMed/NCBI
|
|
26.
|
Shrivastav M, De Haro LP and Nickoloff JA:
Regulation of DNA double-strand break repair pathway choice. Cell
Res. 18:134–147. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Niida H, Murata K, Shimada M, et al:
Cooperative functions of Chk1 and Chk2 reduce tumour susceptibility
in vivo. EMBO J. 29:3558–3570. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Paulsen RD and Cimprich KA: The ATR
pathway: fine-tuning the fork. DNA Repair (Amst). 6:953–966. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Ahmed A, Yang J, Maya-Mendoza A, et al:
Pharmacological activation of a novel p53-dependent S-phase
checkpoint involving CHK-1. Cell Death Dis. 2:e1602011. View Article : Google Scholar : PubMed/NCBI
|