Introduction
Fibrosarcoma is one of the high-grade malignant soft
tissue sarcomas that commonly occurs in persons middle-aged and
older. Fibrosarcoma involves deep soft tissues of the extremities
and trunk. Although the prognosis of these patients has improved
due to the development of surgical treatment and adjuvant
chemotherapies, these therapies are not fully effective. The
probability of local recurrence is related to the completeness of
excision, with recurrence rates of 12–79% (1–3).
Fibrosarcoma metastasizes to the lungs and bone. Metastasis occurs
in 9–63% of patients and the 5-year survival rate of these patients
is 39–54% (2,3). To improve treatment outcomes, novel
antitumor therapies are urgently required.
Bisphosphonates (BPs) are effective inhibitors of
bone resorption and have been used in the treatment of metabolic
bone diseases (4).
Nitrogen-containing BPs (N-BPs), the so-called second- and
third-generation BPs, induce apoptosis in osteoclasts by inhibiting
protein prenylation in small G proteins through inhibition of
farnesyl pyrophosphate synthase in the mevalonate pathway (5). It has been reported that
third-generation BPs, such as zoledronic acid (ZOL), the most
potent N-BPs clinically available, may not only reduce bone loss,
but may also exert direct antitumor effects against various
malignant cells (6,7). We also reported that the effects of
ZOL against osteosarcoma cells and fibrosarcoma cells (8–11).
However, ZOL is rapidly cleared from the circulation within 1–2 h
(6). Furthermore, following
infusion of the standard dose of ZOL, peak plasma levels were only
1–2 μM (12). It is
therefore likely that peripheral tumors are exposed to a low
concentration of ZOL for only a few hours, and that the effects of
ZOL alone may be insufficient in vivo. Therefore,
combination therapy consisting of BPs along with other adjuvant
therapy seemed to be required for treatment of soft tissue
tumors.
There have been a number of reports regarding the
combined effects of third-generation BPs with antitumor agents in
various cancer cell lines (10,13,14)
and we have also reported that ZOL synergistically augments the
effects of antitumor agents in fibrosarcoma cell lines (8). Another well-established treatment
modality for the local treatment of malignant tumors is
radiotherapy. There have been several recent reports regarding the
combined effects of third-generation BPs with radiation in various
cancer cell lines (15,16). However, there have been no previous
reports regarding the combined effects of ZOL with ionizing
radiation (IR) in fibrosarcoma cells. Furthermore, there have been
no reports concerning the detailed mechanisms underlying the
combined effects even in other cancer cells. Therefore, this study
was performed to clarify the combined effects of BPs and IR. It has
been reported that cells in the G2 and M phases are more
radiosensitive than those in other phases of the cell cycle
(17,18), so the effects of ZOL on the cell
cycle were investigated in the present study. In addition, IR can
also directly induce DNA damage causing double-strand breaks (DSB)
and single-strand breaks (19–21),
and it also activates specific prosurvival signaling, including the
MAPK pathway, PBK-Akt pathway and NF-κB activation (21). It has also been reported that IR
induces cell death by generating reactive oxygen species (ROS).
Based on previous reports, we examined the inhibitory effects of
ZOL on these signal pathways and the effects of cotreatment with
ZOL and IR on ROS generation. Furthermore, taking clinical
application into consideration, we evaluated the differences in
antitumor effects according to dosage method and clarified one of
the mechanisms of action in fibrosarcoma cells.
Materials and methods
Reagents
ZOL [1-hydroxy-2-(1H-imidazole-l-yl)
ethylidene-bisphosphonic acid] was obtained from Novartis Pharma AG
(Basel, Switzerland). Akt inhibitor IV, Akt inhibitor VIII
(Carbiochem, San Diego, CA), U0126 (Cell Signaling Technology,
Beverly, MA), N-acetylcysteine (Nacalai Tesque Inc., Kyoto, Japan),
the caspase inhibitors zVAD-fmk, zDEVD-fmk, zIETD-fmk, zLEHD-fmk
and zAEVD-fmk (R&D Systems, Minneapolis, MN) were purchased.
Akt inhibitor IV, VIII, U0126, caspase inhibitors and
N-acetylcysteine were dissolved in dimethyl sulfoxide (DMSO). An
equivalent amount of DMSO was used as a control. The maximum volume
(%) of DMSO in the assays was 0.1%.
X-ray irradiation
Cultured cells were irradiated with 0–8.0 Gy X-rays
(Softex M-150WE; Softex Co. Ltd., Tokyo, Japan). The irradiation
conditions selected were a distance of 1 cm from the focus to the
specimen and an irradiation rate of 0.5 Gy/min in air.
Cell lines and cell culture
The human fibrosarcoma cell line HT1080 was used.
Cells were cultured in RPMI-1640 medium (Nacalai Tesque Inc.)
supplemented with 10% fetal calf serum and 1% antibiotics.
Cell viability assay
Proliferation of the cell line was determined using
the methylthiazol-diphenyl-tetrazolium (MTT) assay, as described
previously (22). HT1080 cells
were cultivated in flat-bottomed 96-well plates (Greiner
Labortechnik, Frickenhausen, Germany) at 2×103 cells per
well and incubated for 24 h, followed by incubation with various
concentrations/doses of ZOL and/or radiation for a further 72 h.
The mean of six data values for each treatment were calculated. The
linear relationship between the degree of proliferation and cell
number was evaluated within the range of the experiment.
Half-maximal inhibitory concentrations (IC50) were
determined using the non-linear regression program CalcuSyn
(Biosoft, Cambridge, UK).
Cell cycle analysis
To analyze alterations in the cell cycle, nuclear
staining with propidium iodide (Sigma-Aldrich, Tokyo, Japan) was
analyzed using a FACSCalibur flow cytometer (Becton-Dickinson,
Franklin Lakes, NJ) as described previously (8). DNA histograms were created using Cell
Quest software for Apple Macintosh (Becton-Dickinson). The ModFit
LT V2.0 software (Verity Software, Topsham, ME) was used to analyze
the data.
Detection of apoptosis
To analyze apoptosis, hypodiploid DNA (sub-G1)
populations were assayed using a FACSCalibur flow cytometer as
described previously (8).
Western blot analysis
Western blot analysis was performed as described
previously (8) using antibodies to
the following molecules: extracellular signal-regulated kinase
(ERK1/2), phosphorylated ERK1/2 (p-ERK1/2), Akt, phosphorylated Akt
(p-Akt), glyceraldehyde 3 phosphate dehydrogenase (GAPDH),
caspase-3, -9, -10, cleaved caspase -3, -9 and phosphorylated Bad
(p-Bad) (Cell Signaling Technology), Rapl A, cyclin B1 and cdc2
(Santa Cruz Biotechnology, Santa Cruz, CA), caspase-8
(Becton-Dickinson) and Bad (Assasy Designs Stressgen, Ann. Arbor,
MI). The membranes were washed thoroughly and incubated for 1 h
with horseradish peroxidase-conjugated anti-mouse or anti-rabbit
IgG (Santa Cruz Biotechnology). Enhanced chemiluminescence
(Amersham Biosciences, Tokyo, Japan) was used for detection.
Optimized detection was achieved using the Chemi Doc™
XRS+ imaging system and Quantity One analysis software
(Bio-Rad, Hercules, CA).
Transient transfection and luciferase
assay
To determine promoter activity, we used a
single-luciferase reporter assay system. HT1080 cells were plated
in 24-well plates and incubated at 37°C. At 70–80% confluence, the
cells were washed and incubated with medium containing no serum or
antibiotics for 6 h. The cells were then transfected with the NF-κB
reporter vector pGL4.32 (Promega, Madison, WI) using Lipofectamine
2000 (Invitrogen, Carlsbad, CA) reagent according to the
manufacturer’s protocol. Twenty-four hours after transfection, the
cells were treated with medium containing 1.2 μM ZOL, and 24
h after the start of treatment the cells were irradiated at 3 Gy.
One hour after irradiation, HT1080 cells were collected and lysed
for luciferase assay using the ONE-G1o™ luciferase assay system
(Promega). The light intensity was measured using a MicroLumat Plus
LB96V (Berthold Technologies, Bad Wildbad, Germany). PSV-(3 plasmid
(Promega) was used as an internal control. All luciferase assays
were carried out in triplicate.
Determination of intracellular ROS
Intracellular ROS levels were measured using
2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) (Nacalai Tesque
Inc.) as a probe. Briefly, cells were loaded with DCFH-DA by
incubation in complete medium containing 20 μM DCFH-DA for
20 min in the dark at 37°C, 5% CO2. The cells were
rinsed with PBS, resuspended by trypsinization and analyzed by flow
cytometry. The results were analyzed with Cell Quest software. Data
are expressed as (number of positive cells) × (mean of the
fluorescence intensity).
Statistical analysis
Data are expressed as means ± SD of triplicate
experiments. Statistical evaluation of the data was performed using
Student’s t-test for simple comparisons between groups and
treatments. In all analyses, P<0.05 was taken to indicate
statistical significance.
Results
Co-treatment with ZOL and IR shows
enhanced inhibitory effects on fibrosarcoma cell growth
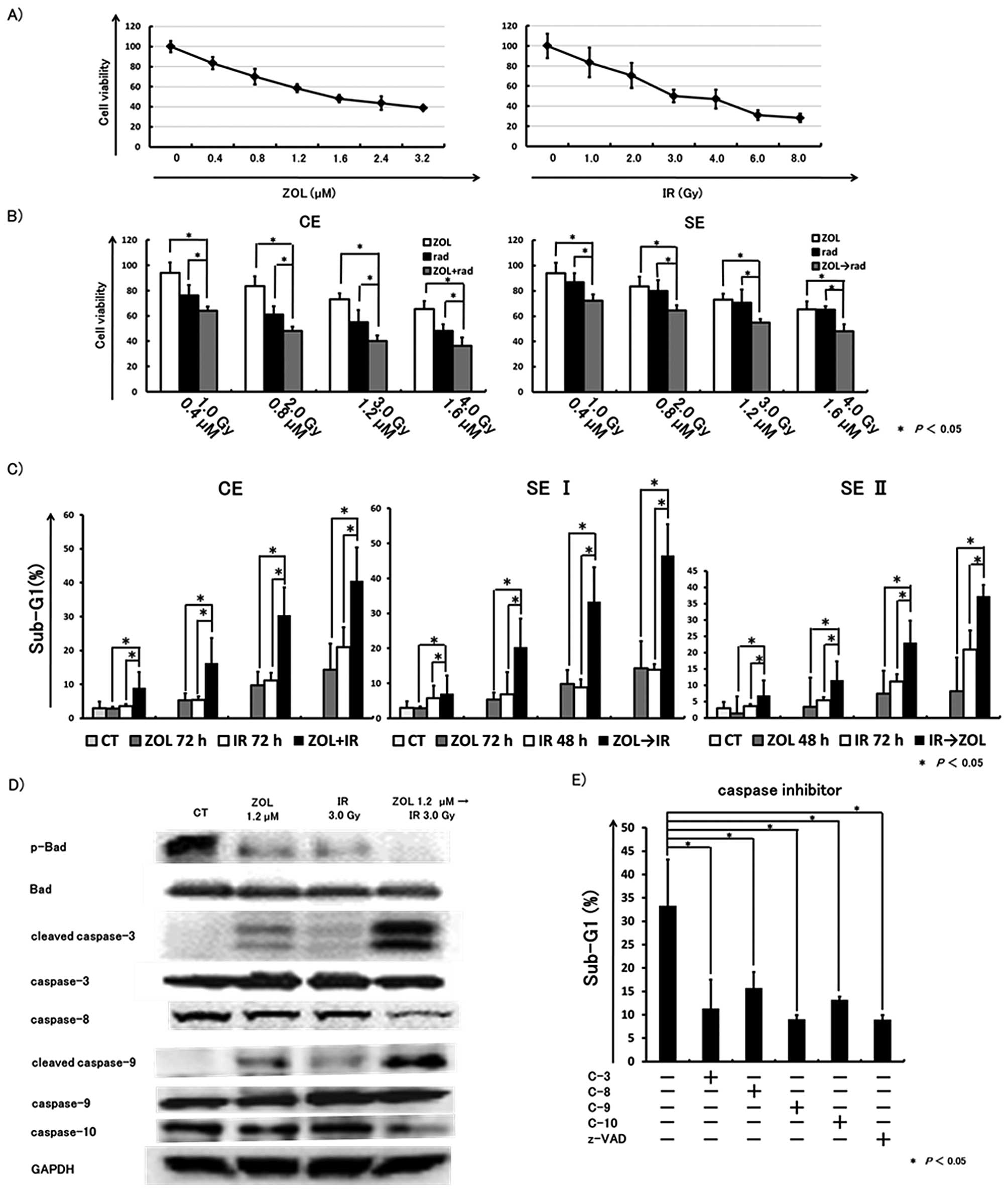
The rate of growth inhibition was evaluated by MTT
assay. The individual IC50 values for ZOL and radiation
after 72 h of exposure were 1.77 μM and 4.08 Gy,
respectively (Fig. 1A). Based on
the IC50 values, we investigated the combined effects of
ZOL at concentrations lower than the IC50 with radiation
at a lower dose than the IC50.
Concurrent exposure (ZOL and IR):
(CE)
HT1080 cells were cultured and incubated for 24 h,
followed by incubation with various concentrations of ZOL combined
with various doses of IR for a further 72 h. The combined treatment
induced significantly greater inhibitory effects than either used
alone.
Sequential exposure (ZOL then IR):
(SE)
After 24 h of exposure to various concentrations of
ZOL, cells were irradiated with various doses of X-rays. Each plate
was evaluated a further 48 h. The concentration/dose of
ZOL/radiation were the same as in CE. Combined treatment induced
significantly greater antitumor effects than either used alone
(Fig. 1B).
Co-treatment with ZOL and IR enhances
cytotoxic effects
The cytotoxic effects of co-treatment were evaluated
using a FACSCalibur flow cytometer. Data from three independent
experiments were collected. The combination method was as
follows.
Concurrent exposure (ZOL and IR):
(CE)
HT1080 cells were cultured in 6-well plates at
2×104 cells per well and incubated for 24 h, followed by
incubation with various concentrations of ZOL and/or IR at various
doses. After a further 24 h, cells were washed with PBS and
incubated in fresh medium for a further 48 h.
Sequential exposure (ZOL then IR): (SE
I)
After 24 h of incubation with various concentrations
of ZOL, HT1080 cells were irradiated with X-rays, then washed with
PBS and incubated in fresh medium for a further 48 h.
Sequential exposure (IR then ZOL): (SE
II)
After 24 h of incubation with various doses of IR,
HT1080 cells were incubated with various concentrations of ZOL for
24 h. The cells were then washed with PBS and incubated in fresh
medium for a further 24 h. The sub-G1 fraction in flow cytometric
analysis was increased by co-treatment with ZOL and IR, especially
in SE I treatment (Fig. 1C).
Co-treatment with ZOL and IR induces
apoptosis coupled with caspase activation and dephosphorylation of
Bad
To confirm the cytotoxic effect of ZOL combined with
IR, especially SE I, we carried out western blot analysis. After 24
h of incubation with 1.2 μM ZOL, cells were irradiated at 3
Gy, washed with PBS and incubated in fresh medium for a further 48
h. Either ZOL or IR weakly affected Bad and caspases (Fig. 1D). However, ZOL and IR together
resulted in marked cleavage of caspases-3, -9 and reduced
caspases-8, -9. Moreover, co-treatment with ZOL and IR induced
dephosphorylation of Bad.
Apoptosis induced by SE I treatment is
blocked by caspase inhibitors
We also found that the pancaspase inhibitor zVAD-fmk
and caspases-3, -8,-9 and -10 inhibitors, efficiently inhibited the
sub-G1 population induced by SE I treatment. These results
indicated that the apoptosis induced by co-treatment could be
blocked by inhibition of caspases (Fig. 1E).
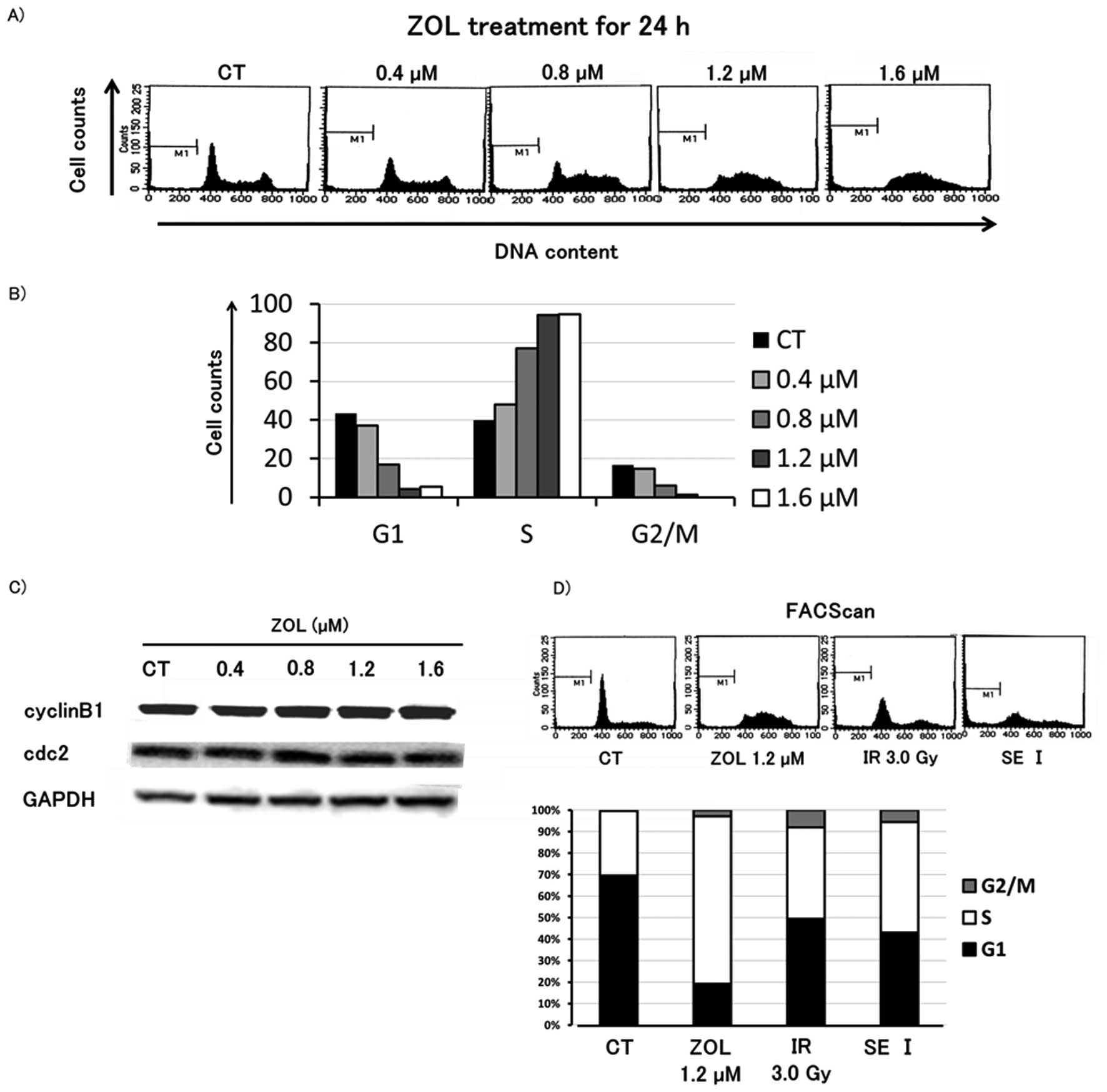
ZOL does not influence G2/M phase
cells
To evaluate the antitumor mechanism of SE I
treatment, cell cycle analysis was performed on HT1080 cells after
24 h of concurrent exposure to ZOL and IR. The results indicated an
increase in number of cells in the S phase in a dose-dependent
manner, but ZOL did not change the proportion of cells in the G2/M
phase. In addition, western blot analysis revealed that the levels
of cyclin Bl and cdc2, which are G2/M phase-related proteins, were
not altered after 24 h of exposure to ZOL. Furthermore, SE I
treatment did not change the proportion of cells in G2/M phase
compared to each single treatment (Fig. 2).
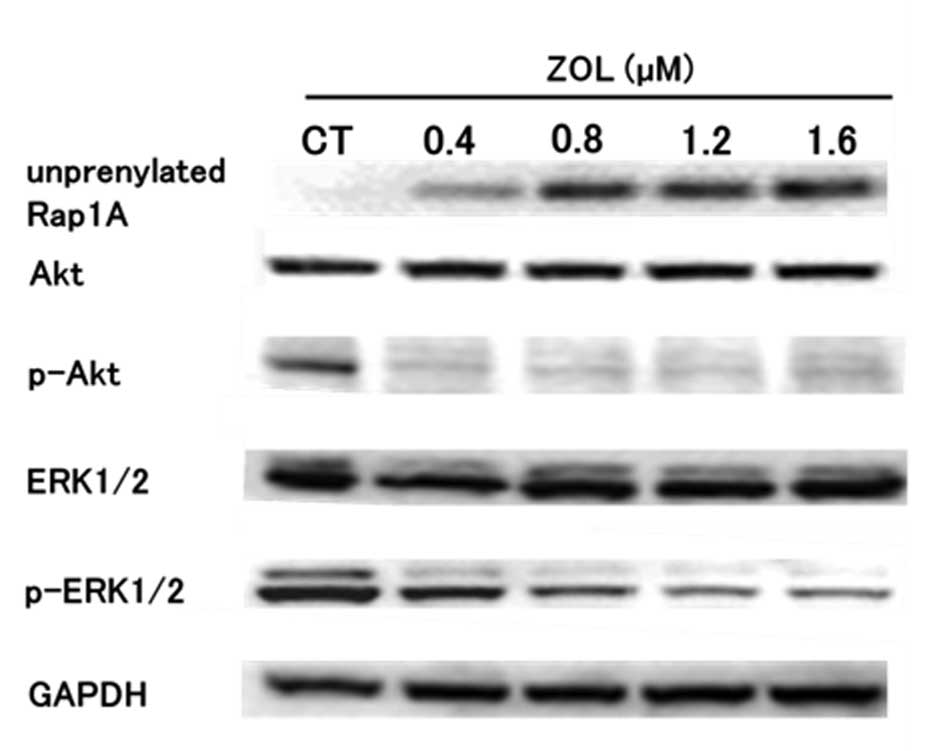
ZOL inhibits prenylation of GTP-binding
protein, and phosphorylation of Akt and ERK1/2 in HT1080 cells
To assess the involvement of the Akt pathway and
MAPK pathway in apoptosis induced by the SE I treatment, the levels
of several proteins were investigated by western blot analysis. The
levels of unprenylated Rap1A, a small G protein located upstream of
the PI3K-Akt pathway and MAPK pathway and a target of ZOL, were
increased after 24 h of ZOL treatment in HT1080 cells (Fig. 3). P-ERK1/2 levels were decreased in
a ZOL concentration-dependent manner and p-Akt levels began to
decrease markedly 24 h after ZOL treatment.
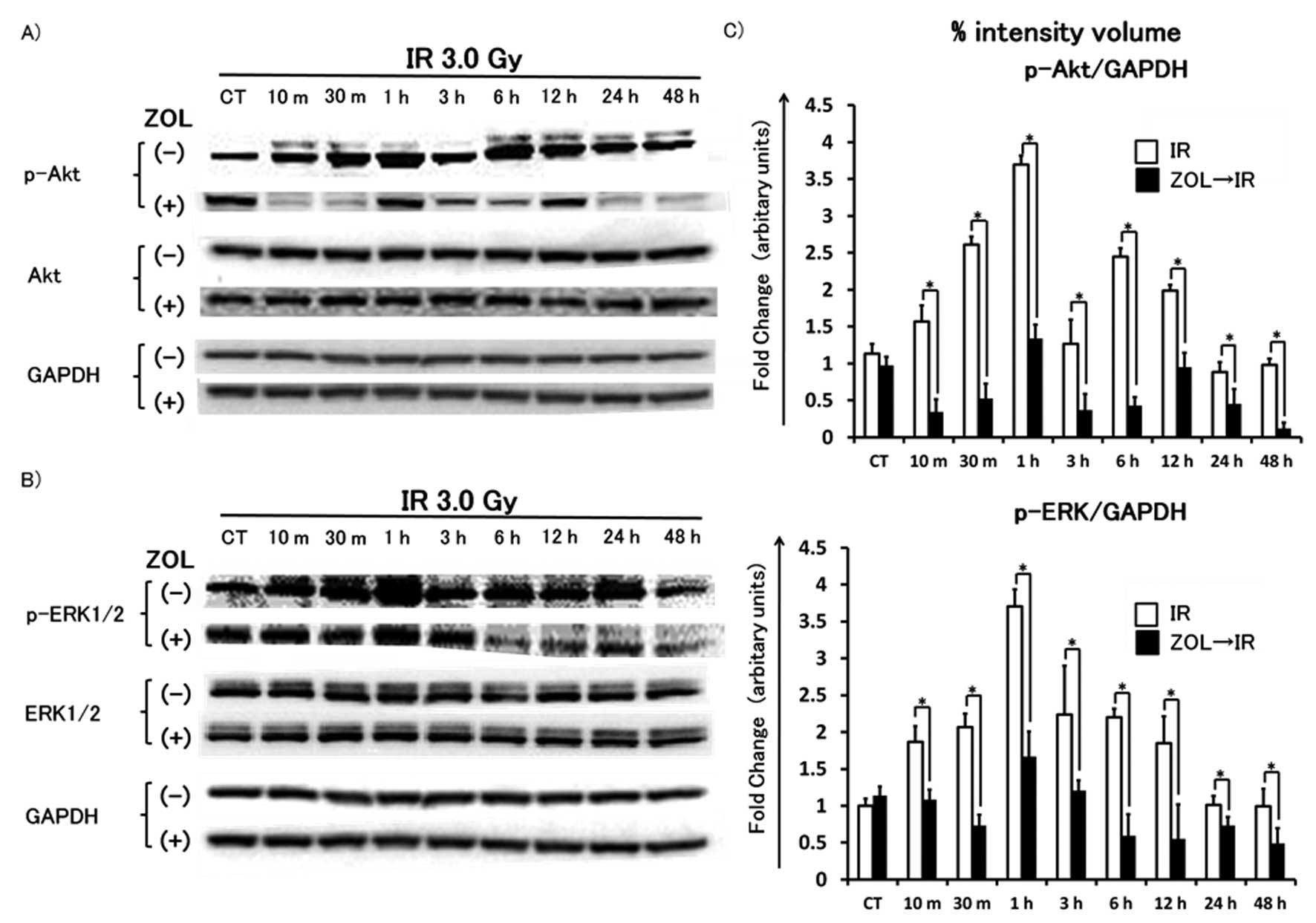
Akt and ERK1/2 phosphorylated within a
few hours after irradiation - ZOL pretreatment inhibits
phosphorylation of these proteins
After 24 h of incubation with or without 1.2
μM ZOL, HT1080 cells were washed with PBS, incubated in
fresh medium and then irradiated at a dose of 3 Gy. After 10, 30
min, 1, 3, 6, 12, 24 and 48 h of incubation, HT1080 cells were
lysed and western blot analysis was performed. Irradiation at 3 Gy
resulted in phosphorylation of Akt and ERK1/2 within 24 h,
especially at 1 h, followed by normalization within 24 h. This
phosphorylation was significantly inhibited by pretreatment with
1.2 μM ZOL compared to single radiation treatment alone
(Fig. 4).
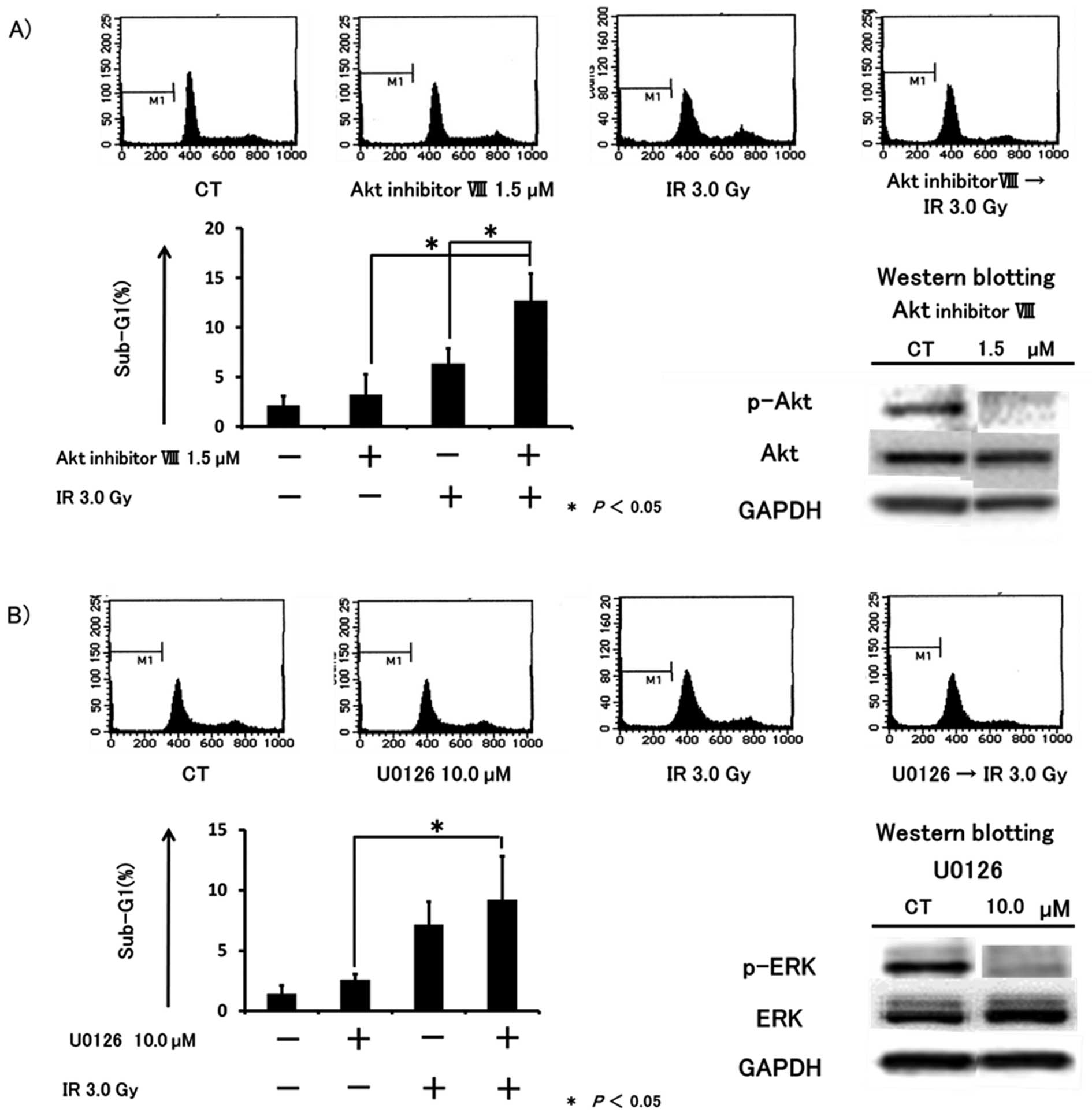
The cytotoxic effect was enhanced by
co-treatment with IR and inhibition of Akt or MEK activity
Based on previous reports (23,24)
and the present findings, we hypothesized that the cytotoxic
effects induced by SE I treatment may depend on the inhibition of
Akt and/or ERK1/2 activity by ZOL. To investigate the inhibitory
effects of ZOL on Akt and ERK1/2, HT1080 cells were exposed to Akt
inhibitor IV, Akt inhibitor VIII, a selective inhibitor of Akt with
no effect on PI3K or PDK1, or U0126, a selective MEK inhibitor.
After 24 h of incubation with these reagents, p-Akt and p-ERK1/2
were detected by western blot analysis. We confirmed that p-Akt and
p-ERK1/2 were markedly inhibited by 1.25 μM Akt inhibitor IV
(data are not shown), 1.5 μM Akt inhibitor VIII (Fig. 5A), and 10.0 μM U0126
(Fig. 5B). Next, we evaluated
whether these inhibitors enhanced the cytotoxic effects of IR by
flow cytometry. After 24 h of incubation with or without Akt
inhibitor IV, VIII or U0126, cells were irradiated at a dose of 3
Gy and incubated for a further 48 h. In cultures exposed to a
combination of Akt inhibitor IV or VIII with IR, the sub-G1
population was significantly increased compared to either agent or
IR alone (Fig. 5A) (data are not
shown for Akt inhibitor IV). Sequential treatment with U0126
followed by IR tended to augment the sub-G1 population by IR, but
the results were not statistically significant (Fig. 5B). Furthermore, the cell cycle was
not altered by co-treatment with Akt inhibitor VIII or U0126 along
with IR compared to each agent or IR alone.
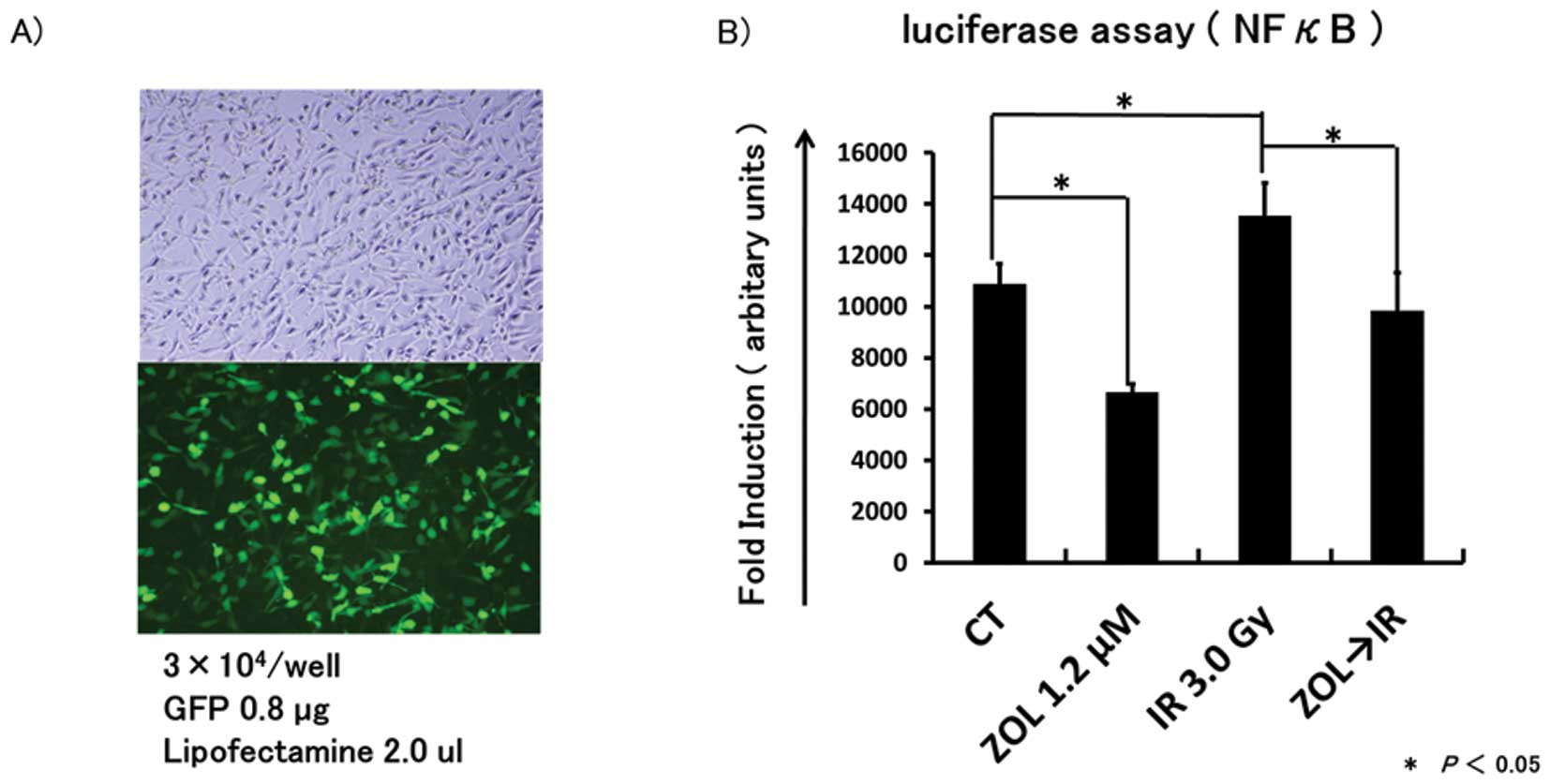
NF-κB promoter activity stimulated by IR
is inhibited by ZOL pretreatment
We investigated whether ZOL can inhibit the NF-κB
gene promoter activity, which was reported to be activated by IR
(25), using transient
transfection with the NF-κB promoter-luciferase reporter plasmid,
pGL4.32 or the empty vector PSV-β. Twenty-four hours after
transfection, HT1080 cells were treated with or without medium
containing ZOL and 24 h after the start of treatment, cells were
irradiated at a dose of 0 or 3 Gy. Then, 1 h after irradiation,
cells were collected for luciferase assay. IR stimulated NF-κB
promoter activity and ZOL inhibited the promoter activity compared
to controls (Fig. 6). Furthermore,
NF-κB promoter activity stimulated by IR was significantly
inhibited by pretreatment with ZOL.
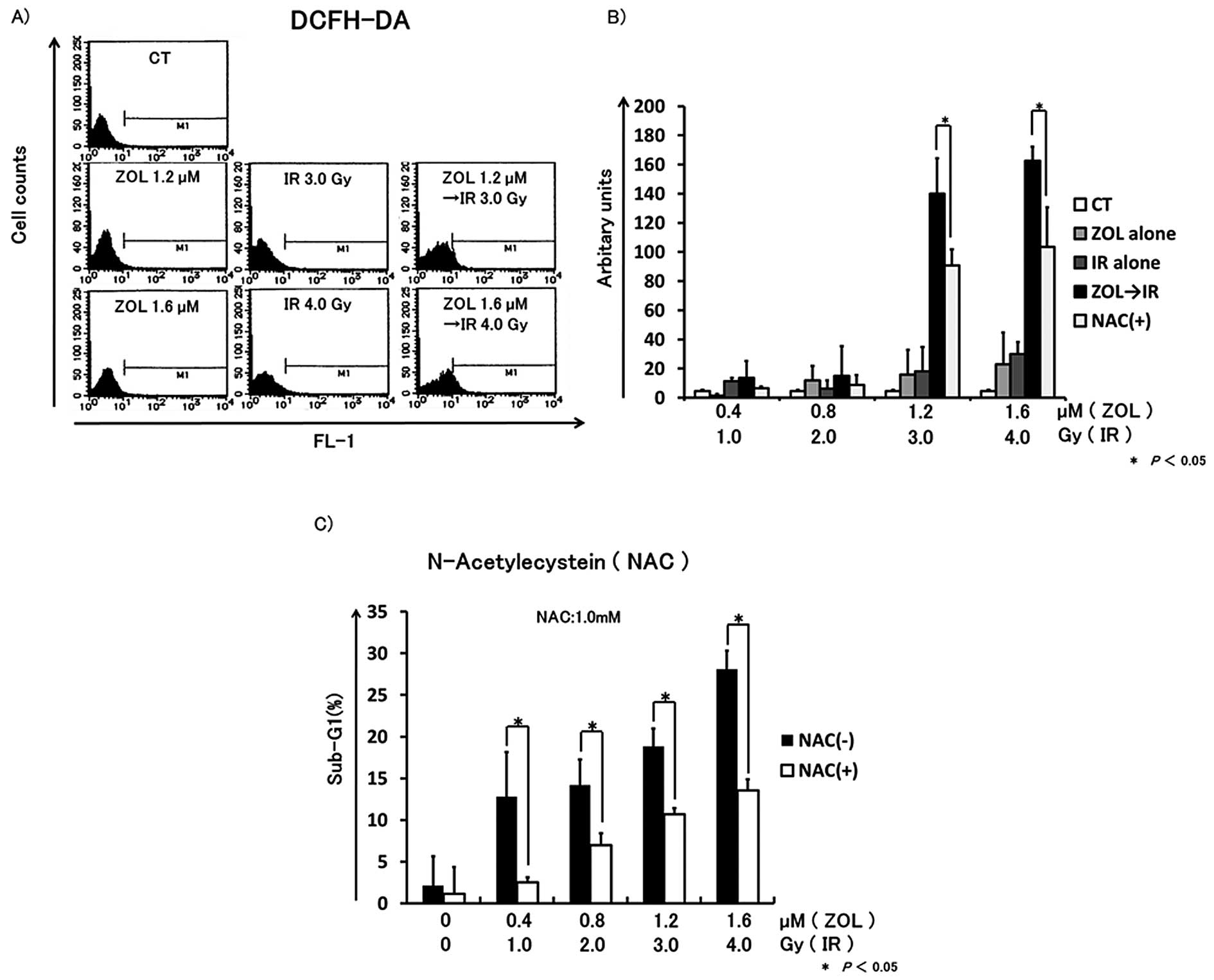
ROS generation induced by IR is enhanced
by ZOL, and the increasing cell death induced by SE I treatment is
inhibited by N-acetylcysteine (NAC)
To determine the involvement of ROS generation in
cell death induced by SE I treatment in HT1080 cells, we monitored
ROS generation by flow cytometry. After 24 h of incubation, cells
(2×104) were treated with various concentrations of ZOL
in the presence or absence of 1 mM NAC for 24 h, then exposed to
IR. Forty-eight hours after IR, cells were incubated with DCFH-DA,
and the fluorescence intensity was measured by flow cytometry. In
comparison to IR alone or ZOL alone, ROS generation was
significantly increased after SE I treatment and the ROS generation
was inhibited by NAC treatment. Furthermore, the sub-G1 population
in the HT1080 cells treated with NAC was significantly reduced
compared to the untreated control group (Fig. 7).
Discussion
New treatment strategies are necessary to improve
the prognosis of soft tissue sarcoma. In the search for new
treatment strategies and antitumor agents, as well as from our
previous studies (8–11,26),
we found that ZOL was a potent enhancer of radiation-induced
apoptosis in fibrosarcoma cells. First, we compared three patterns
of administration, i.e., CE, SE I, and SE II, to determine the most
effective dosage method for clinical application. The results
indicated that SE I treatment exerted the most potent antitumor
effects, so we performed further investigation of the cytotoxic
effects of SE I. The increase in proportion of cells in sub-G1, and
the induction of caspase-3 cleavage by SE I treatment suggested
that it induced apoptosis, and we assumed that apoptosis was
induced through not only the mitochondrial apoptotic pathway but
also via the death receptor pathway because caspase-8,-9 and -10
inhibitors decreased the proportion of cells in sub-G1.
Next, we examined the mechanisms underlying the
cytotoxic effects of SE I treatment. Based on previous reports that
cells in the G2 and M phases are more radiosensitive than those in
other phases of the cell cycle (17,18),
we hypothesized that synergistic effects could be obtained if ZOL
arrested the cell cycle in G2/M phases. However, the proportion of
cells in G2/M phase was not increased by ZOL.
Based on our results, we investigated another
mechanism for the cytotoxic effects of SE I treatment. Whereas IR
is an effective treatment for malignant tumor cells by directly
causing DNA damage (27,28), radioadaptive resistance, a specific
prosurvival signaling network or radioprotective mechanisms
activated by IR, has also been reported (21,29).
The term radioadaptive response was originally used to describe a
reduced cell sensitivity to a higher challenge dose when a smaller
inducing radiation dose had been applied earlier (30). NF-κB is a transcription factor that
plays a key role in tumor radioadaptive resistance. It has been
reported that DNA binding of NF-κB was activated by IR (31,32)
and was regulated by various signaling cascades, e.g., the TNF-α
pathway, Ras-PI3K-Akt pathway, and Ras-MAPK pathway (33–36).
In addition, several genes, such as the anti-apoptotic Bel family,
cyclin Bl, cyclin D2, superoxide dismutases (SOD) that suppress ROS
generation and HER-2, were identified as effector genes of NF-κB
and these upregulated both cell proliferation and viability
(21). Furthermore, it has been
reported that blocking NF-κB activation increases the apoptotic
response and decreases growth and clonogenic survival of several
human cancer cell lines (37,38).
In the present study, we evaluated the inhibitory effects of ZOL on
radioadaptive signaling, which was unfavorable in treatment of
malignant tumor cells. We have reported previously that ZOL
inhibited proliferation and induced apoptosis in HT1080 cells by
inhibiting activation of small G protein prenylation (8). In the present study, we showed that
ZOL alone inhibited phosphorylation of Akt and ERK1/2, which are
downstream of the PI3K-Akt pathway and MAPK pathway. Furthermore,
pretreatment with ZOL also inhibited phosphorylation of Akt and
ERK1/2, which were activated by IR and combined treatment with IR
and MEK inhibitor/Akt inhibitors augmented the cytotoxic effects in
HT1080 cells. These results supported the findings of recent
studies that inhibition of the Ras-PI3K-Akt pathway or Ras-MAPK
pathway enhanced the cytotoxic effects of IR (39–41).
These results suggested that ZOL inhibited PI3K-Akt signaling or
MAPK signaling via the inhibition of small G protein prenylation as
one mechanism underlying the combined effects of ZOL and IR
treatment. In addition, ZOL markedly inhibited Akt phosphorylation.
These results suggested that ZOL may directly inhibit
phosphorylation of Akt by regulation of the inositol phospholipid
pathway, which is upstream of AKT, and not via its effects on small
GTPases, because ZOL negatively regulates lipid metabolism.
In the present study, IR induced Akt and ERK1/2
phosphorylation in HT1080 cells within 24 h (especially at 1 h)
after irradiation, similar to the findings of previous studies
(23,24). Furthermore, we showed that
pretreatment with ZOL also inhibited the activation of NF-κB in
HT1080 cells, although transcription of NF-κB was activated 1 h
after irradiation. These results suggested a mechanism by which SE
I was the most effective treatment as follows. SE I treatment
induced apoptosis synergistically by ZOL inhibition of the
phosphorylation of Akt, ERK1/2 and activation of NF-κB, which are
involved in radioadaptive signaling, and were upregulated within a
very short time. However, it has been reported that treatment of
cells with a MEK/ERK inhibitor shows either little or no effect on
IR-induced apoptosis (42,43). Therefore, the role of ERK
activation on cell radiosensitivity has not been clarified, and
further investigations are necessary.
On the other hand, the term ROS refers to a group of
molecules such as peroxides and free radicals derived from oxygen
that are highly reactive toward biomolecules. ROS are produced not
only by endogenous sources, but also by exogenous sources, such as
IR, chemicals, toxins and pollutants (44). Elevated ROS levels can create
oxidative stress in the cell and chronic exposure to this stress
can result in permanent changes in the genome (45). It is well known that the large and
sudden increase in ROS generation in cells by IR can lead to
apoptotic cell death (46).
Excessive ROS, e.g., that generated by IR, induces mitochondrial
apoptosis, because mitochondrial DNA is highly sensitive to
mutations caused by endogenous ROS (47,48).
DSB by ROS can arise when ROS-induced DNA damage interferes with
either DNA replication or transcription (49,50).
In the present study, intracellular ROS levels in HT1080 cells were
increased by SE I treatment, and the cytotoxic effects enhanced by
co-treatment with ZOL were reduced by NAC treatment. These results
suggested that ROS generation played a significant role in the
combined effects of ZOL with IR. The mechanism was thought to be as
follows. As a result of unprenylation of small GTPase by ZOL, NF-κB
was inactivated via inhibition of the PI3K-Akt pathway and MAPK
pathway downstream of small GTPases and then ROS generation was
increased by inhibition of NF-κB effector genes, e.g., MnSOD and
CuSOD. However, further studies are required as it is difficult to
clarify the precise mechanism underlying the combined effect based
only on our results.
We showed for the first time that ZOL significantly
enhances radiation-induced apoptosis in human fibrosarcoma cells.
Although the detailed mechanism has not been reported, we
demonstrated one of the mechanisms underlying the synergistic
effects of ZOL and IR. Due to inhibition of small GTPase
prenylation, ZOL inhibited phosphorylation of AKT and ERK1/2, along
with the transcriptional activity of NF-κB, and synergistic effects
were obtained. Furthermore, SE I was postulated to be the most
effective treatment because adaptive resistance to IR occurred
within a few hours. The increased ROS generation was also one of
the mechanisms underlying the combined effects. Although further
studies of the in vivo effects are required, these results
raise the possibility that the combination of ZOL and radiation may
represent a promising type of therapy for fibrosarcoma.
Abbreviations:
|
ZOL
|
zoledronic acid;
|
|
IR
|
ionizing radiation;
|
|
ROS
|
reactive oxygen species;
|
|
BPs
|
bisphosphonates;
|
|
DCFH-DA
|
2,7-dichlorodihydrofluorescein
diacetate;
|
|
DMSO
|
dimethylsulfoxide
|
Acknowledgements
This study was supported by JSPS
KAKENHI (Grant-in-Aid for Scientific Research C: 23592196 to
HM).
References
|
1.
|
Pritchard DJ, Sim FH, Ivins JC, Soule EH
and Dahlin DC: Fibrosarcoma of bone and soft tissues of the trunk
and extremities. Orthop Clin North Am. 8:869–881. 1977.PubMed/NCBI
|
|
2.
|
Pritchard DJ, Soule EH, Taylor WF and
Ivins JC: Fibrosarcoma - a clinicopathologic and statistical study
of 199 tumors of the soft tissues of the extremities and trunk.
Cancer. 33:888–897. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Scott SM, Reiman HM, Pritchard DJ and
Ilstrup DM: Soft tissue fibrosarcoma. A clinicopathologic study of
132 cases. Cancer. 64:925–931. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Russell RG and Rogers MJ: Bisphosphonates:
from the laboratory to the clinic and back again. Bone. 25:97–106.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Green JR: Antitumor effects of
bisphosphonates. Cancer. 97:840–847. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Lee MV, Fong EM, Singer FR and Guenette
RS: Bisphosphonate treatment inhibits the growth of prostate cancer
cells. Cancer Res. 61:2602–2608. 2001.PubMed/NCBI
|
|
7.
|
Kubo T, Shimose S, Matsuo T, et al:
Inhibitory effects of a new bisphosphonate, minodronate, on
proliferation and invasion of a variety of malignant bone tumor
cells. J Orthop Res. 24:1138–1144. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Koto K, Murata H, Kimura S, et al:
Zoledronic acid inhibits proliferation of human fibrosarcoma cells
with induction of apoptosis, and shows combined effects with other
anticancer agents. Oncol Rep. 24:233–239. 2010.
|
|
9.
|
Horie N, Murata H, Nishigaki Y, et al: The
third-generation bisphosphonates inhibit proliferation of murine
osteosarcoma cells with induction of apoptosis. Cancer Lett.
238:111–118. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Horie N, Murata H, Kimura S, et al:
Combined effects of a third-generation bisphosphonate, zoledronic
acid with other anticancer agents against murine osteosarcoma. Br J
Cancer. 96:255–261. 2007. View Article : Google Scholar
|
|
11.
|
Koto K, Horie N, Kimura S, et al:
Clinically relevant dose of zoledronic acid inhibits spontaneous
lung metastasis in a murine osteosarcoma model. Cancer Lett.
274:271–278. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Jagdev SP, Coleman RE, Shipman CM, Rostami
HA and Croucher PI: The bisphosphonate, zoledronic acid, induces
apoptosis of breast cancer cells: evidence for synergy with
paclitaxel. Br J Cancer. 84:1126–1134. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Kubo T, Shimose S, Matsuo T, Sakai A and
Ochi M: Efficacy of a nitrogen-containing bisphosphonate,
minodronate, in conjunction with a p38 mitogen activated protein
kinase inhibitor or doxorubicin against malignant bone tumor cells.
Cancer Chemother Pharmacol. 62:111–116. 2008. View Article : Google Scholar
|
|
14.
|
Ottewell PD, Monkkonen H, Jones M, Lefley
DV, Coleman RE and Holen I: Antitumor effects of doxorubicin
followed by zoledronic acid in a mouse model of breast cancer. J
Natl Cancer Inst. 100:1167–1178. 2008. View Article : Google Scholar
|
|
15.
|
Ural AU, Avcu F, Candir M, Guden M and
Ozcan MA: In vitro synergistic cytoreductive effects of zoledronic
acid and radiation on breast cancer cells. Breast Cancer Res.
8:R522006. View
Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Algur E, Macklis RM and Hafeli UO:
Synergistic cytotoxic effects of zoledronic acid and radiation in
human prostate cancer and myeloma cell lines. Int J Radiat Oncol
Biol Phys. 61:535–542. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Milas L, Hunter NR, Mason KA, Kurdoglu B
and Peters LJ: Enhancement of tumor radioresponse of a murine
mammary carcinoma by paclitaxel. Cancer Res. 54:3506–3510.
1994.PubMed/NCBI
|
|
18.
|
Tishler RB, Geard CR, Hall EJ and Schiff
PB: Taxol sensitizes human astrocytoma cells to radiation. Cancer
Res. 52:3495–3497. 1992.PubMed/NCBI
|
|
19.
|
Acharya A, Das I, Chandhok D and Saha T:
Redox regulation in cancer: a double-edged sword with therapeutic
potential. Oxid Med Cell Longev. 3:23–34. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Sedelnikova OA, Redon CE, Dickey JS,
Nakamura AJ, Georgakilas AG and Bonner WM: Role of oxidatively
induced DNA lesions in human pathogenesis. Mutat Res. 704:152–159.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Ahmed KM and Li JJ: NF-kappa B-mediated
adaptive resistance to ionizing radiation. Free Radic Biol Med.
44:1–13. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Hansen MB, Nielsen SE and Berg K:
Re-examination and further development of a precise and rapid dye
method for measuring cell growth/cell kill. J Immunol Methods.
119:203–210. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ram R, Uziel O, Eldan O, et al: Ionizing
radiation up-regulates telomerase activity in cancer cell lines by
post-translational mechanism via ras/phosphatidylinositol
3-kinase/Akt pathway. Clin Cancer Res. 15:914–923. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Zingg D, Riesterer O, Fabbro D, Glanzmann
C, Bodis S and Pruschy M: Differential activation of the
phosphatidylinositol 3’-kinase/Akt survival pathway by ionizing
radiation in tumor and primary endothelial cells. Cancer Res.
64:5398–5406. 2004.
|
|
25.
|
Wang CY, Mayo MW and Baldwin AS Jr: TNF-
and cancer therapy-induced apoptosis: potentiation by inhibition of
NF-kappaB. Science. 274:784–787. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Ryu K, Murata H, Koto K, et al: Combined
effects of bisphosphonate and radiation on osteosarcoma cells.
Anticancer Res. 30:2713–2720. 2010.PubMed/NCBI
|
|
27.
|
Morgan WF and Murnane JP: A role for
genomic instability in cellular radioresistance? Cancer Metastasis
Rev. 14:49–58. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Morgan WF: Is there a common mechanism
underlying genomic instability, bystander effects and other
nontargeted effects of exposure to ionizing radiation? Oncogene.
22:7094–7099. 2003. View Article : Google Scholar
|
|
29.
|
Ch’ang HJ, Maj JG, Paris F, et al: ATM
regulates target switching to escalating doses of radiation in the
intestines. Nat Med. 11:484–490. 2005.PubMed/NCBI
|
|
30.
|
Stecca C and Gerber GB: Adaptive response
to DNA-damaging agents: a review of potential mechanisms. Biochem
Pharmacol. 55:941–951. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Spitz DR, Azzam EI, Li JJ and Gius D:
Metabolic oxidation/reduction reactions and cellular responses to
ionizing radiation: a unifying concept in stress response biology.
Cancer Metastasis Rev. 23:311–322. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Schieven GL, Kirihara JM, Myers DE,
Ledbetter JA and Uckun FM: Reactive oxygen intermediates activate
NF-kappa B in a tyrosine kinase-dependent mechanism and in
combination with vanadate activate the p561ck and p59fyn tyrosine
kinases in human lymphocytes. Blood. 82:1212–1220. 1993.
|
|
33.
|
Guo G, Wang T, Gao Q, et al: Expression of
ErbB2 enhances radiation-induced NF-kappaB activation. Oncogene.
23:535–545. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Blonska M, You Y, Geleziunas R and Lin X:
Restoration of NF-kappaB activation by tumor necrosis factor alpha
receptor complex-targeted MEKK3 in receptor-interacting
protein-deficient cells. Mol Cell Biol. 24:10757–10765. 2004.
View Article : Google Scholar
|
|
35.
|
Chen G and Goeddel DV: TNF-R1 signaling: a
beautiful pathway. Science. 296:1634–1635. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Dent P, Yacoub A, Fisher PB, Hagan MP and
Grant S: MAPK pathways in radiation responses. Oncogene.
22:5885–5896. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Tang G, Minemoto Y, Dibling B, et al:
Inhibition of JNK activation through NF-kappaB target genes.
Nature. 414:313–317. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Chen X, Shen B, Xia L, et al: Activation
of nuclear factor kappaB in radioresistance of TP53-inactive human
keratinocytes. Cancer Res. 62:1213–1221. 2002.PubMed/NCBI
|
|
39.
|
Kim IA, Bae SS, Fernandes A, et al:
Selective inhibition of Ras, phosphoinositide 3 kinase, and Akt
isoforms increases the radiosensitivity of human carcinoma cell
lines. Cancer Res. 65:7902–7910. 2005.PubMed/NCBI
|
|
40.
|
Toulany M, Kasten-Pisula U, Brammer I, et
al: Blockage of epidermal growth factor
receptor-phosphatidylinositol 3-kinase-AKT signaling increases
radiosensitivity of K-RAS mutated human tumor cells in vitro by
affecting DNA repair. Clin Cancer Res. 12:4119–4126. 2006.
View Article : Google Scholar
|
|
41.
|
Toulany M, Kehlbach R, Florczak U, et al:
Targeting of AKT1 enhances radiation toxicity of human tumor cells
by inhibiting DNA-PKcs-dependent DNA double-strand break repair.
Mol Cancer Ther. 7:1772–1781. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Mandic A, Viktorsson K, Heiden T, Hansson
J and Shoshan MC: The MEK1 inhibitor PD98059 sensitizes C8161
melanoma cells to cisplatin-induced apoptosis. Melanoma Res.
11:11–19. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Smalley KS and Eisen TG: Farnesyl
thiosalicylic acid inhibits the growth of melanoma cells through a
combination of cytostatic and pro-apoptotic effects. Int J Cancer.
98:514–522. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Spry M, Scott T, Pierce H and D’Orazio JA:
DNA repair pathways and hereditary cancer susceptibility syndromes.
Front Biosci. 12:4191–4207. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Cooke MS, Evans MD, Dizdaroglu M and Lunec
J: Oxidative DNA damage: mechanisms, mutation, and disease. FASEB
J. 17:1195–1214. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Dhar A, Young MR and Colburn NH: The role
of AP-1, NF-kappaB and ROS/NOS in skin carcinogenesis: the JB6
model is predictive. Mol Cell Biochem. 234–235:185–193.
2002.PubMed/NCBI
|
|
47.
|
Hunt CR, Sim JE, Sullivan SJ, et al:
Genomic instability and catalase gene amplification induced by
chronic exposure to oxidative stress. Cancer Res. 58:3986–3992.
1998.PubMed/NCBI
|
|
48.
|
Wong GH: Protective roles of cytokines
against radiation: induction of mitochondrial MnSOD. Biochim
Biophys Acta. 1271:205–209. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Pommier Y, Barcelo JM, Rao VA, et al:
Repair of topoisomerase I-mediated DNA damage. Prog Nucleic Acid
Res Mol Biol. 81:179–229. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Sordet O, Redon CE, Guirouilh-Barbat J, et
al: Ataxia telangiectasia mutated activation by transcription- and
topoisomerase I-induced DNA double-strand breaks. EMBO Rep.
10:887–893. 2009. View Article : Google Scholar : PubMed/NCBI
|