Contents
Introduction
Prodrug strategies in cancer treatment
Cathepsin B (Cat B) as a prodrug-activating
enzyme
Cat B-cleavable DOX prodrugs
Conclusions
Introduction
Chemotherapy is a major therapeutic approach for the
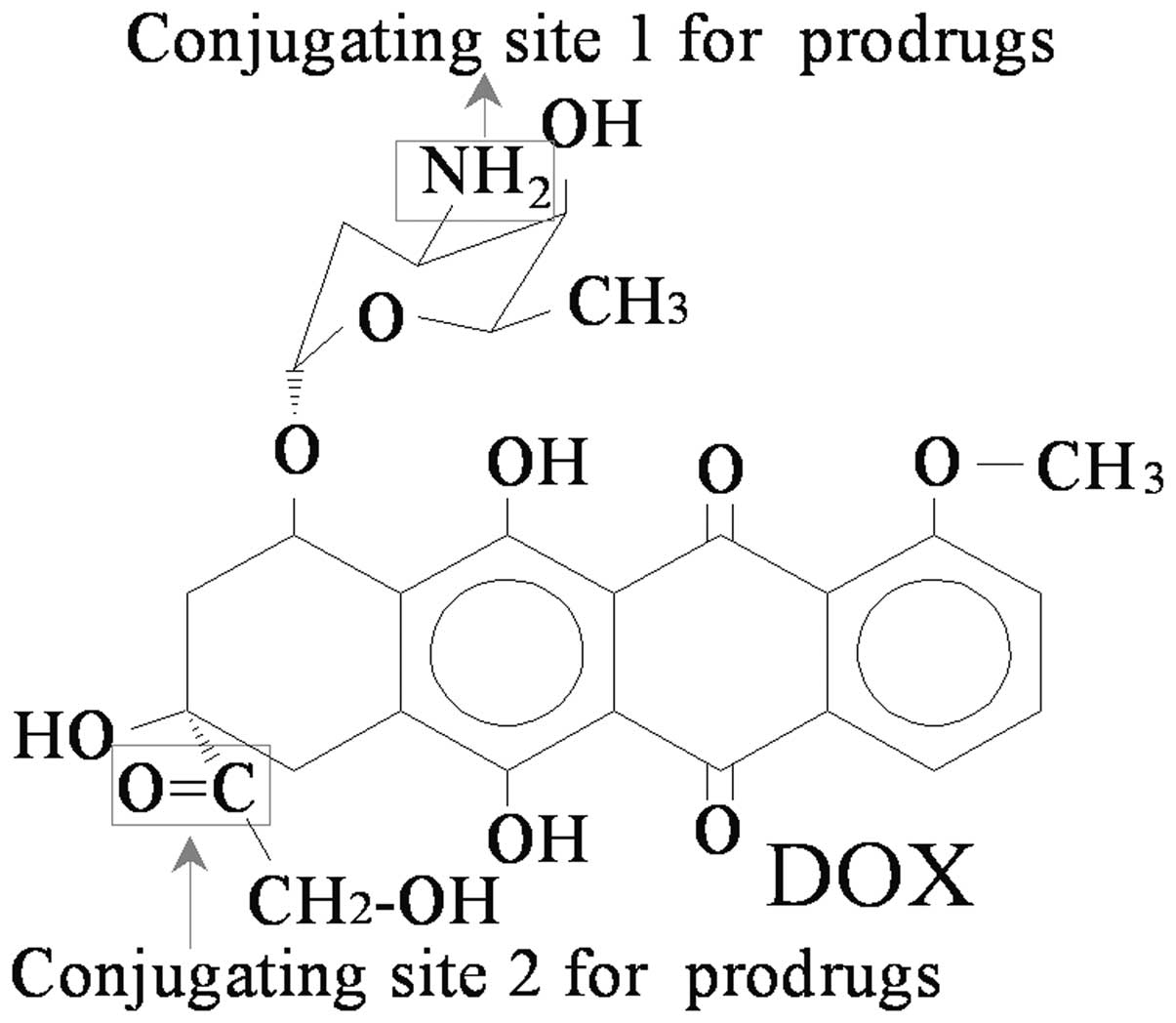
treatment of cancer. Doxorubicin (DOX; Fig. 1), an anthracycline isolated from
Streptomyces strains, is one of the most effective
anticancer drugs used for the treatment of hematological
malignancies and a broad range of solid tumors, including lymphoma,
Kaposi’s sarcoma, bone tumors, as well as stomach, breast and
ovarian cancers (1,2). DOX in its salt form is readily
distributed into almost all tissues and intracellular compartments
via passive diffusion or active transport following intravenous
administration, resulting in indiscriminative toxic effects on all
cells exposed to it. Therefore, the clinical application of DOX is
limited by its dose-dependent side-effects, such as bone marrow
toxicity, cardiotoxicity, nephrotoxicity and hepatotoxicity.
To reduce the side-effects of this drug, significant
efforts have been made to develop DOX derivatives and analogs with
less toxic effects and improved pharmacological properties. Several
strategies have been investigated in clinical and preclinical
trials, including various methods of administration, combinations
with other chemotherapeutic drugs [e.g., adriamycin, bleomycin,
vinblastine and dacarbazine (ABVD), cyclophosphamide,
hydroxydaunomycin, oncovin and prednisone (CHOP)] (3), the addition of antioxidant nutrients
(4) and cardioprotectors (5–7), the
development of liposomes (8) and
nanoparticles (9), the effects of
acute exercise (10) and the
development of prodrugs (11–13).
In this review, we focused on the DOX prodrug strategies.
Prodrug strategies in cancer treatment
Prodrugs are derivatives of drugs which remain
inactive in their prototype form but are metabolized in the body to
generate the active drugs at the site of action. They are
particularly useful in the development of novel antitumor
chemotherapeutic drugs, leading to reduced toxicity, improved
specificity and the avoidance of multidrug resistance (14,15).
The use of prodrugs for targeted therapy is usually based on
tumor-associated cell surface markers, such as antigens or
receptors, whose expression differs between normal and cancer cells
(16,17). Several prodrug strategies have been
pursued, including active and passive targeting approaches with
antibodies, serum proteins, liposomes and synthetic polymers
(18–22). There have been some classic and
clinically successful prodrugs, such as capecitabine, an
enzyme-activated prodrug, which is converted into 5-fluoro uridine
or 5-fluoro-2-deoxyuridine in tumor cells to achieve targeted
cytotoxicity (23).
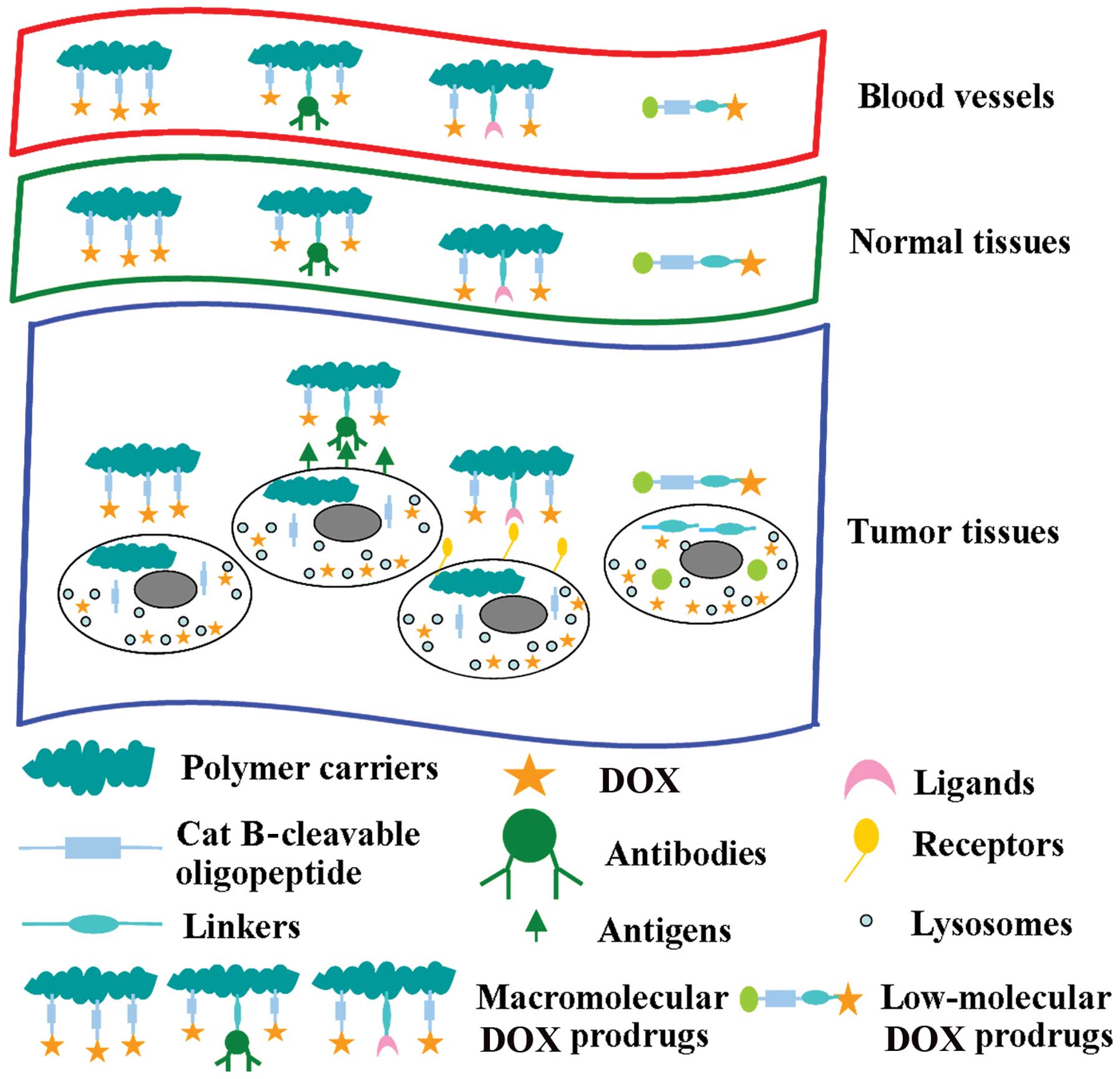
Prodrugs can be divided into high- and low-molecular
weight drugs in terms of molecular weight (Mw). The former are
internalized by passive or active endocytosis and ultimately become
localized in the lysosomal components of cells, while the latter
usually enter cells mainly by diffusion (24). The Mw and biodistribution of drugs
have important impacts on antitumor efficacy. Macromolecular drugs
accumulate in tumor tissues due to the enhanced permeability and
retention effect (25–27). A Mw below the renal threshold
(∼50,000 g/mol) is rapidly lost from the circulation; therefore,
macro-molecular weight drugs may have increased intravascular
half-lives, resulting in an increased therapeutic efficacy
(27). N-(2-hydroxypropyl)
methacrylamide (HPMA), known as one of the most widely used
prototypic polymeric drug carriers, was first used to synthesize
polymeric drugs in the 1970s, due to its non-immunogenic and
non-toxic properties and long circulating half-life (28,29).
It has been demonstrated that an HPMA-copolymer Mw of 200,000 to
600,000 g/mol is desirable for the efficient passive targeting of
solid tumors (30). Prodrugs
bearing HPMA have been developed in preclinical studies and include
caplostatin (31,32), P-GDM (33,34)
and P-HYD-IgG (35), as well as in
phase I/II clinical studies and included HPMA
copolymer-Gly-Phe-Leu-Gly-doxorubicin (PK1) (36–39),
galactosamine-targeted poly(HPMA)-doxorubicin (PK2) (40–42),
PK3 (36), PNU166945 (43), AP5346 (44–48)
and AP5280 (49–51).
Cathepsin B (Cat B) as a prodrug-activating
enzyme
Some tumor-associated enzymes, such as proteases,
glucuronidases or carboxylesterases, expressed intra- or
extracellularly in cancer cells, can release or activate prodrugs.
Cat B, a lysosomal cysteine protease in normal cells and tissues,
is considered to be one of the best examples of intracellular
proteases. It is highly upregulated in malignant tumors and
premalignant lesions at the mRNA and protein levels (52). Cat B is localized in perinuclear
vesicles, presumably lysosomes in normal cells. However, in tumor
cells and oncogene-transformed cells, Cat B is localized in
perinuclear vesicles and vesicles throughout the cytoplasm and at
the cell periphery (53).
Pericellular Cat B participates in degrading processes associated
with tumor proliferation, invasion and metastasis. Moreover,
exposure to DOX can induce a time- and dose-dependent upregulation
of Cat B expression at the mRNA and protein levels (5).
Cat B cleaves Leu, Arg-Arg, Ala-Leu, Phe-Arg,
Phe-Lys, Ala-Phe-Lys, Gly-Leu-Phe-Gly, Gly-Phe-Leu-Gly and
Ala-Leu-Ala-Leu (18,54–58).
There are several low- and high-Mw DOX prodrugs that can be
activated by Cat B. Furthermore, DOX immunoconjugates, in which DOX
is linked to a carcinoma-specific antibody through Cat B-cleavable
oligopeptides, have also been designed (59). All of these conjugates have shown
rapid and almost quantitative DOX release in the presence of Cat B.
The rate of DOX release depends on the length and structure of the
spacer. The tetrapeptide, Gly-Phe-Leu-Gly, has been found to be one
of the most suitable spacers. In this regard, the steric
interaction between the peptide substrate and Cat B has a
significant impact on the release of DOX from prodrugs (60). Therefore, to decrease the steric
interaction, it is necessary to integrate a self-immolative spacer,
such as para-aminobenzyloxycarbonyl (PABC) between the drug and the
oligopeptide substrate.
Cat B-cleavable DOX prodrugs
Examples of Cat B-cleavable DOX prodrugs are
illustrated in Fig. 2 and
summarized in Table I.
 | Table I.List of Cat B-cleavable DOX
prodrugs. |
Table I.
List of Cat B-cleavable DOX
prodrugs.
| Name | Biodegradable
spacer | Mw (g/mol) | DOX proportion | Current status | MTD | Refs. |
|---|
| DOX | None | 543.5 | 100% | Clinical
therapy | 60–80
mg/m2 | (69) |
| PK1 |
Gly-Phe-Leu-Gly | 30,000 | 8 (wt%) | Phase II | 320
mg/m2 | (36,38,39,
61–71) |
| PK2 |
Gly-Phe-Leu-Gly | 27,000 | 8 (wt%) | Phase I/II | 160
mg/m2 | (10,11,40,
41,72–76) |
| P-DOX |
Gly-Phe-Leu-Gly |
22,000–1,230,000 | NA | Preclinical | ND | 26,77,78 |
|
P-(GFLG)-DOX-Ab |
Gly-Phe-Leu-Gly | 270,000 | 3.3 (wt%) | Preclinical | ND | (59,79–81) |
|
P-(GFLG-DOX)-GalN |
Gly-Phe-Leu-Gly | 25,000/46,000 | 5.6/1.5 (wt%) | Preclinical | ND | (59,82,90) |
|
P-(GFLG-DOX)-Lac |
Gly-Phe-Leu-Gly | 20,000–32,000 | 1.4 mol% | Preclinical | ND | (90) |
|
P-(GFLG-DOX)-TriGal |
Gly-Phe-Leu-Gly | 20,000–32,000 | 2.1 mol% | Preclinical | ND | (90) |
| Ma-GFLG-DOX |
Gly-Phe-Leu-Gly | NA | NA | Preclinical | ND | (91,92) |
|
D2-GFLG-P(DOXH) |
Gly-Phe-Leu-Gly | 215,000 | 9.2 (wt%) | Preclinical | ND | (91,93) |
| HMW1D |
Gly-Phe-Leu-Gly | 115,000 | 7.4 (wt%) | Preclinical | ND | (93) |
| TET1D |
Gly-Phe-Leu-Gly | 19,600 | 10.5 (wt%) | Preclinical | ND | (93) |
|
EMC-Arg-Arg-Ala-Leu-Ala-Leu-DOX |
Ala-Leu-Ala-Leu | NA | NA | Preclinical | ND | (94–96) |
|
Ac-Phe-Lys-PABC-DOX | Phe-Lys | 1045.5 | 52.0 (wt%) | Preclinical | ND | (12) |
|
EMC-Phe-Lys-PABC-DOX | Phe-Lys | 1133 | 50.0 (wt%) | Preclinical | ND | (2,18,104) |
| PG-Phe-Lys-DOX | Phe-Lys | 1207.8 | 45.0 (wt%) | Preclinical | ND | (18,41,105) |
|
Z-Phe-Lys-PABC-DOX | Phe-Lys | 1074.0 | 50.6 (wt%) | Preclinical | ND | (104) |
|
BR96-SC-Phe-Lys-PABC-DOX | Phe-Lys | NA | NA | Preclinical | ND | (104) |
DOX prodrugs containing the tetrapeptide
Gly-Phe-Leu-Gly
The tetrapeptide, Gly-Phe-Leu-Gly, has been proven
to be the most effective with respect to both plasma stability and
rapid hydrolysis in the presence of Cat B. Therefore, many DOX
prodrugs are based on this tetrapeptide.
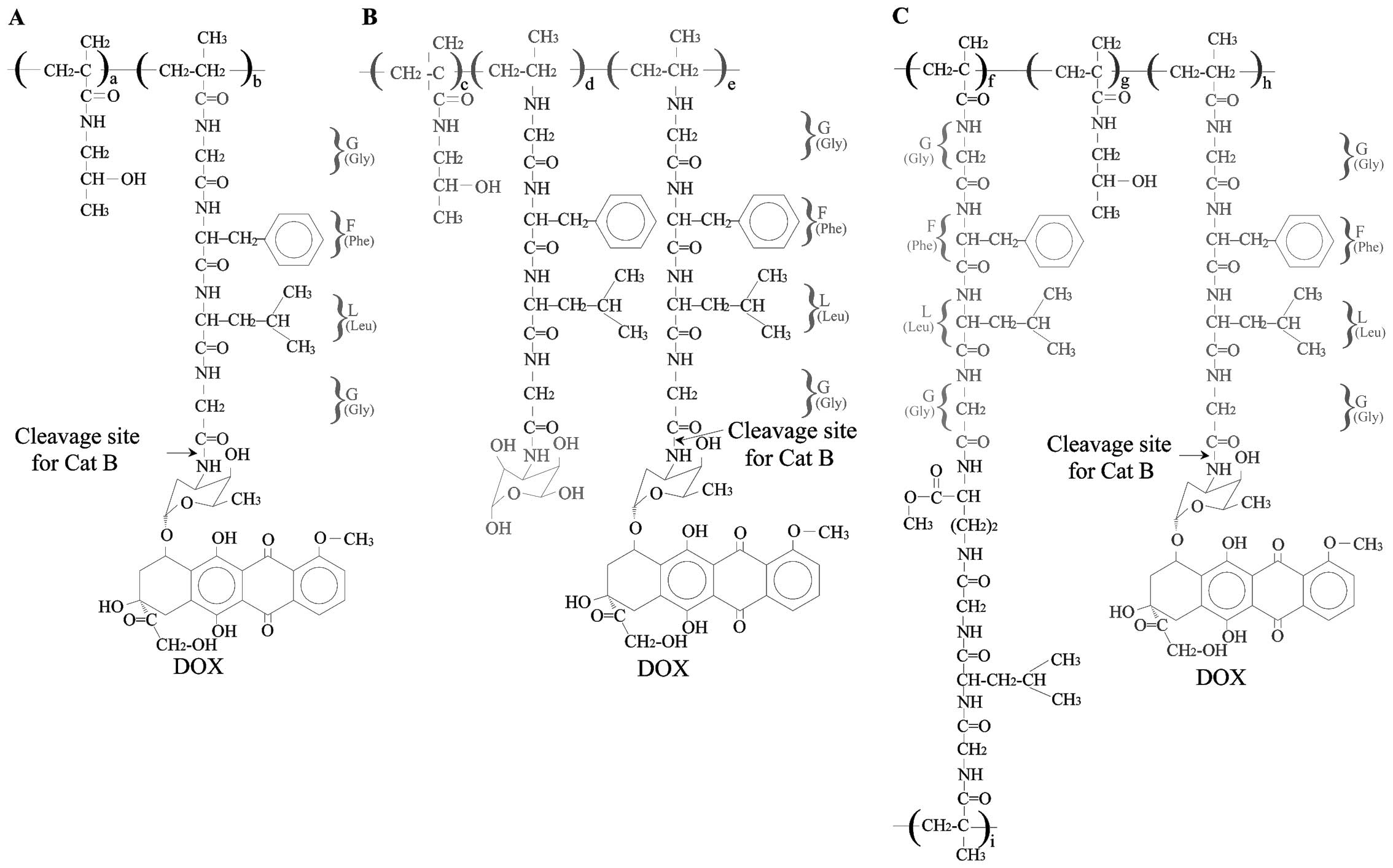
PK1
PK1 [FCE28068; P(GFLG)-ADR; DOX-HPMA;
doxorubicin-HPMA copolymer conjugate; HPMA-doxorubicin, 8 wt% DOX;
Fig. 3A], a polymeric prodrug of
Mw ∼30,000 g/mol, was the first macromolecular prodrug to enter
clinical trials, and has reached phase II clinical trials.
Preclinical studies using tumor cells, including
L1210 leukemia (61–64), A2780 and DOX-resistant A2780/AD
ovarian carcinoma cells, have shown that PK1 can partially avoid
the ATP-driven P-glycoprotein (Pgp) efflux pump compared with free
DOX (65–67). The IC50 doses of free
DOX and PK1 account for the differences in the mechanisms of
cellular uptake (65). In
preclinical studies using animal models, including B16F10 melanoma,
L1210 leukemia, M5076, LS174T human colorectal xenografts (64) and sensitive and resistant human
ovarian carcinoma models (68),
PK1 has shown enhanced efficacy. The release of DOX from PK1 in
vitro and in vivo using HPLC analysis has shown only a
single peak, representing DOX (64). PK1 does not release DOX in the
plasma and the covalently-bound drug is biologically inactive
following intravenous administration.
Phase I clinical studies on patients with solid
tumors, including colorectal, breast, biliary tract, pancreatic,
urinary tract, head/neck, non-small cell lung (NSCL), mesothelioma
and stomach cancers, have shown that the maximum tolerated dose
(MTD) for PK1 is 320 mg/m2, which is 4- to 5-fold higher
than the usual clinical dose of free DOX (60–80 mg/m2)
(69). PK1 decreases non-specific
organ toxicities by several folds and allows the active drug to be
delivered intracellularly, while maintaining antitumor activity
(36,39). Phase II studies using PK1 have
shown decreased toxicity with evident activity in breast, NSCL and
colorectal cancers. Furthermore, SPECT and γ-camera imaging with
123I-labelled drugs have shown obvious tumor
accumulation in two metastatic breast cancers (38). PK1 and free DOX greatly differ in
their antiproliferative effects and cell death signals in EL-4
cancer cells; treatment with free DOX greatly increases p38
phosphorylation, while PK1 increases it only slightly; PK1 also
significantly increases ERK phosphorylation, while free DOX
slightly decreases it (70).
In addition, polymer-directed enzyme prodrug therapy
(PDEPT) combining HPMA copolymer-Cat B and PK1 has shown activity
against a COR-L23 xenograft, whereas PK1 alone has not and in
B16F10 melanoma tumors PDEPT has been shown to more effective than
either PK1 or free DOX alone (71).
PK2
PK2 (FCE28069, 27,000 g/mol, 8 wt% DOX; Fig. 3B), the only targeted polymer
conjugate containing galactosamine to enter clinical trials, is
designed to target the asialoglycoprotein receptor (ASGPR) which is
selectively expressed in hepatocytes and hepatoma cell lines
(40,72). Pharmacokinetic studies using PK2 in
rats and mice have shown effective liver targeting with >70% of
the released DOX being selectively targeted to the liver following
intravenous administration (73,74).
Preclinical studies using rats have demonstrated that PK2 displays
a ∼5-fold reduction in cardiotoxicity as opposed to free DOX
following intravenous or intraperitoneal administration at various
doses (11,64). Furthermore, antitumor activity has
been shown to be improved in rodent tumor models (69).
In a pivotal study on a patient with multifocal
hepato-cellular carcinoma, HPLC and 123I-based imaging
showed the biphasic clearance of PK2 from the plasma (half-life,
78±1 and 990±15 min) and ∼30% of the delivered drug accumulated in
the liver at 24 h. Moreover, SPECT analysis showed that the
radioactivity concentration was 3- to 4-fold higher in peritu-moral
liver tissue than in the tumor tissue itself (40).
Phase I/II trials have shown that the MTD of PK2 is
160 mg/m2 (DOX equivalent) and several hepatocellular
carcinoma patients have displayed partial responses and/or stable
disease (41). γ-camera imaging
and CT scanning have revealed that 15–20% of total PK2 is retained
in the liver and is mostly concentrated in normal liver tissue
(normal versus tumor tissue, 5:1), suggesting that the
galactosamine-targeted polymer is mainly delivered to normal
regions of the liver due to the increased ASGPR expression in the
normal liver (75) and the
phagocytosis by Kupffer cells with ‘galactose particle’ receptor
expression (76). Despite this
disparity in PK2 distribution, the drug concentration in tumor
tissue was still 12- to 50-fold higher than it would have been with
the administration of free DOX alone.
HPMA copolymer-doxorubicin conjugates
(P-DOX)
P-DOX conjugates (Fig.
3C) (77,78) contain the oligopeptide
Gly-Phe-Leu-Gly and the
N2,N5-bis(N-methacryloyl-glycylphenylalanyl-leucyl-glycyl)
ornithine cross-linker, which permits the synthesis of P-DOX
conjugates with various Mws, from 22 to 1230 kDa. The clearance
rate of P-DOX from the blood is Mw-dependent and is much slower
than that of free DOX (26,77).
The therapeutic efficacy has been shown to increase as the Mw of
P-DOX increases in nude mice bearing subcutaneous OVCAR-3
xenografts. The low residual concentration of P-DOX in tissues
(apart from tumors) helps to avoid potential long-term side-effects
(77). The toxicity against
hematopoietic precursors and normal lymphocytes of inbred mice is
considerably decreased (78).
HPMA copolymer-DOX-OV-TL16
[P-(GFLG)-DOX-Ab]
P-(GFLG)-DOX-Ab (270,000 g/mol, 3.3 wt% DOX;
Fig. 4A) is recognized by the OA3
antigen, which plays a role in membrane transport and/or signal
transduction for its multimembrane-spanning domain structure
(59,79–81).
The P-(GFLG)-DOX-Ab is rapidly absorbed by OVCAR-3 cells and
transported into their lysosomal compartment. DOX is subsequently
released from the conjugate at the site with a degradable GFLG
spacer, diffused via the lysosomal membrane and accumulates in the
cell nuclei (80). Preliminary
data on the relative retention of DOX in MDR (A2780/AD) cells have
indicated a higher intracellular DOX concentration after incubation
with HPMA copolymer-DOX conjugate compared with free DOX (59).
HPMA
copolymer-Gly-Phe-Lys-Gly-DOX-N-acylated galactosamine
[P-(GFLG)-DOX-GalN]
P-(GFLG)-DOX-GalN (25,000 g/mol; Fig. 4B), contains N-acylated
galactosamine (GalN), which was designed to be recognized by ASGPR
in HepG2 human hepatocellular carcinoma cells (59,82)
and individual members of the galectin family (e.g., galectin-3) in
human colon adenocarcinoma (83,84).
Galectin-3 is expressed in normal tissues and highly expressed in
neoplastic tissues (85–87); although the exact opposite has been
shown to occur (88,89). In SW-480 and SW-620 cells, the
presence of galectin-3 on the cell surface has been demonstrated by
flow cytometry; however, it has not been detected on the surface of
Colo-205 cells. The cellular cytotoxicity of P-(GFLG)-DOXGalN
determined by MTT assay has been shown to be ∼10-fold higher than
P-GFLG-DOX and 10-fold higher in Colo-205 cells than in SW-480 and
SW-620 cells (90). This suggests
the participation of other galectins, such as galectin-1, -4, -7 or
-8, in P-(GFLG)-DOX-GalN targeting.
Lactose-containing HPMA
copolymer-doxorubicin conjugate [P-(GFLG-DOX)-lac]
P-(GFLG-DOX)-lac (Fig.
4C) (90), can also be
biorecognized by galectin-3 on the surface of colon cancer cells.
The in vitro cytoxicity determined by MTT assay is higher
than that of the non-glycosylated P-(GFLG)-DOX product and almost
1,000-fold lower than that of free DOX in HepG2 human
hepatocellular carcinoma cells and Colo-205, SW-480 and SW-620
colon adenocarcinoma cells.
Trivalent galactose-containing HPMA
copolymerdoxorubicin conjugate [P-(GFLG-DOX)-TriGal]
P-(GFLG-DOX)-TriGal (Fig. 4D) contains trivalent galactose,
which can also be biorecognized by galectin-3 on the surface of
colon adenocarcinoma cells. The cytotoxicity of the
P-(GFLGDOX)-TriGal has been shown to be at least 10-fold higher
than that of the non-glycosylated P-(GFLG)-DOX product in Colo-205,
SW-480 and SW-620 colon adenocarcinoma cells (90).
N-Methacryloyl-glycyl)-dl-phenylalanyl-leucyl-glycyl-DOX
(Ma-GFLG-DOX)
Ma-GFLG-DOX contains the tetrapeptide,
Gly-Phe-Leu-Gly (91,92). It remains quite stable in buffer at
pH 7.4 (model of the bloodstream), but releases DOX either under
mild acidic conditions or in the presence of Cat B (rich in the
tumor microenvironment).
PAMAM dendrimers
(D-NH2)-Gly-Phe-Leu-Gly-HPMA-doxorubicin (D2-GFLG-P-DOX)
D2-GFLG-P-DOX (215,000 g/mol, 9.2 wt% DOX), which is
attached to DOX via a pH-sensitive hydrazone bond (91,93),
was prepared by grafting the semitelechelic HPMA copolymers, which
have Mws below the renal threshold, onto a PAMAM dendrimer core via
a biodegradable linkage GFLG oligopeptide. An in vitro study
using phosphate buffers at pH 5.0 or 7.4 at 37°C (hydrazone
conjugates) and in a Cat B-containing (5×10−7 M)
phosphate buffer at 37°C (amide conjugates) showed that the
presence of Cat B increased the rate of DOX release (91).
HMW1D
HMW1D (115,000 g/mol, 7.4 wt% DOX), a branched
polymer prodrug, contains water-soluble polymer drug carriers, HPMA
copolymers, and a biodegradable oligopeptide sequence, GFLG,
linking shorter polymer chains (Mw, 20,000 g/mol) into a high-Mw
structure (Mw, 110,000 g/mol) to enhance the passive accumulation
of the drug by increasing its Mw. An in vitro study showed
that this pH-sensitive prodrug (HMW1D) can be degraded by Cat B
(5×10−7 M), 37°C, pH 6.0 (93).
TET1D
TET1D (19,600 g/mol, 10.5 wt% DOX; Fig. 5) (93) a non-targeted polymer-bound
doxorubicin conjugate, contains a hydrazone bond, which
significantly improves the rate of DOX release, compared with that
of classical HPMA polymer prodrugs bearing DOX attached via amide
bonds limited to maximum 8–9 wt%. An in vitro study using
T-splenocytes and mouse EL-4 T cell lymphoma cells showed that the
toxicity of TET1D is much higher compared with that of similar
classic conjugates and an in vivo study using EL4 T cell
lymphoma mice C57BL/10 showed that the antitumor activity was also
significantly increased. An in vitro study showed that TET1D
can be cleaved by Cat B; however, Cat B is not essential in the
release of DOX, for it also contains a pH-sensitive spacer which is
stable under physiological conditions (pH 7.4, e.g., blood) and
hydrolytically degradable in a mild acidic environment (pH 5.0,
e.g., endosome) (93).
DOX prodrugs containing the tetrapeptide,
Ala-Leu-Ala-Leu 6-Maleimidocaproic acid-Arg-Arg-Ala-Leu-Ala-Leu-DOX
(EMC-Arg-Arg-Ala-Leu-Ala-Leu-DOX)
EMC-Arg-Arg-Ala-Leu-Ala-Leu-DOX bears maleimide
(94), which can rapidly and
selectively react in situ with the cysteine-34 position of
circulating albumin after intravenous administration and release
the drug at the tumor site (95,96).
Albumin is a promising drug carrier due to its passive accumulation
in solid tumors, which have a high metabolic turnover,
angiogenesis, hypervasculature, defective vascular architecture and
impaired lymphatic drainage (97).
Albumin has non-toxic, non-immunogenic, biocompatible and
biodegradable properties (98) and
has demonstrated preferential tumor uptake in various tumor
xenograft animal models (99). The
antitumor efficacy of EMC-Arg-Arg-Ala-Leu-Ala-Leu-DOX has been
shown to be comparable to that of free DOX in a M-3366 breast
cancer xenograft model at equivalent doses (94). Moreover, the albumin-binding DOX
prodrug, DOX-EMCH (INNO-206), has been examined in clinical trials
(100,101).
DOX prodrugs containing the dipeptide,
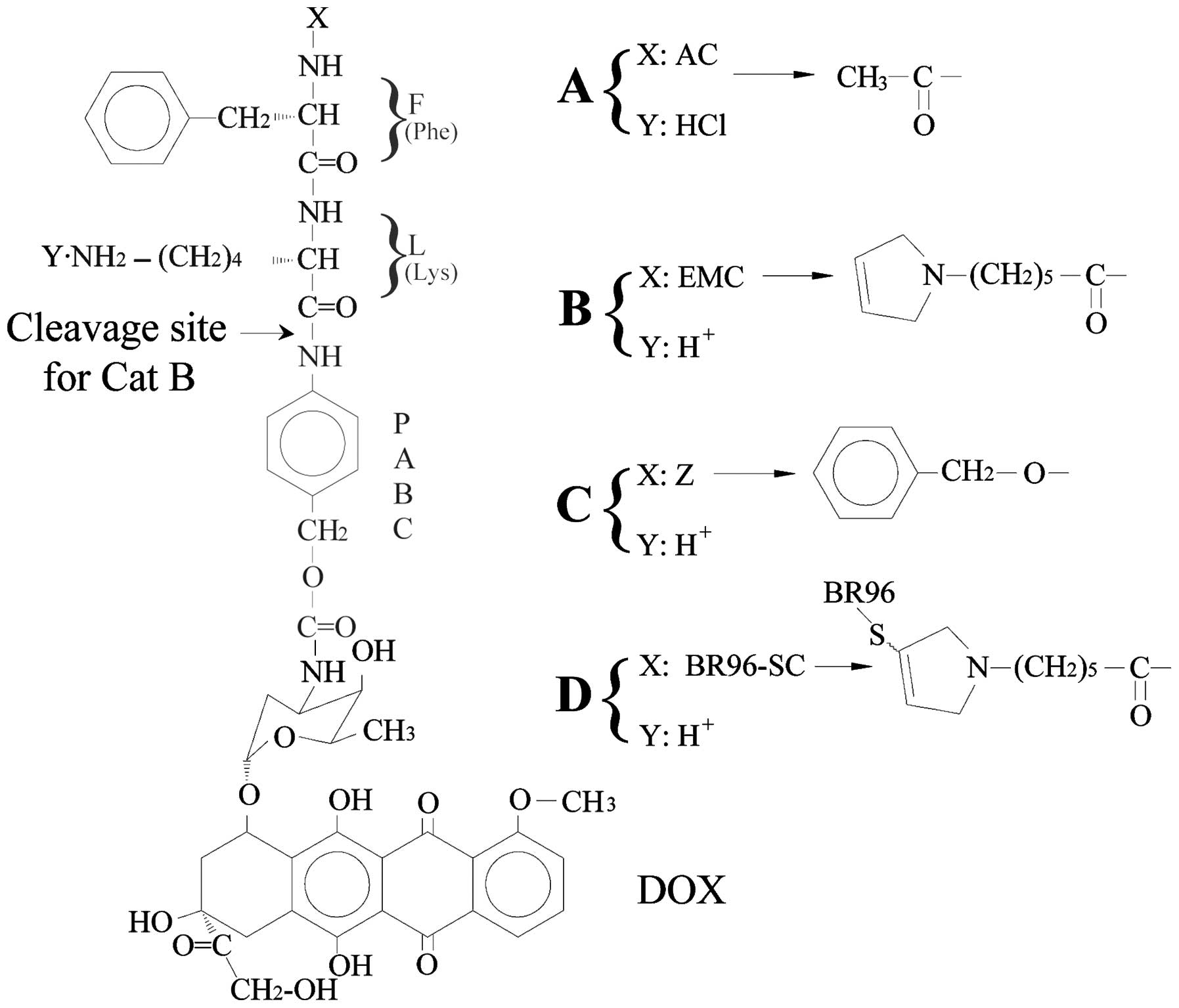
Phe-Lys Ac-Phe-Lys-PABC-DOX
Ac-Phe-Lys-PABC-DOX (PDOX, 1045.5 g/mol, 52.0% DOX,
Fig. 6A) contains the dipeptide,
Phe-Lys, which is specific for Cat B and the self-immolative
spacer, PABC (12,102–104). An in vivo study using a
nude mice model of gastric cancer with peritoneal carcinomatosis
showed that, compared with free DOX, PDOX (16 mg/kg, twice that of
DOX in terms of equal molecular content) produced better antitumor
effects in terms of experimental peritoneal carcinomatosis index
(ePCI) (Fig. 7A) and body weight
(Fig. 7D), and reduced liver
(Fig. 7B and C), kidney (Fig. 7E and F) and heart (Fig. 7G–I) toxicities (12).
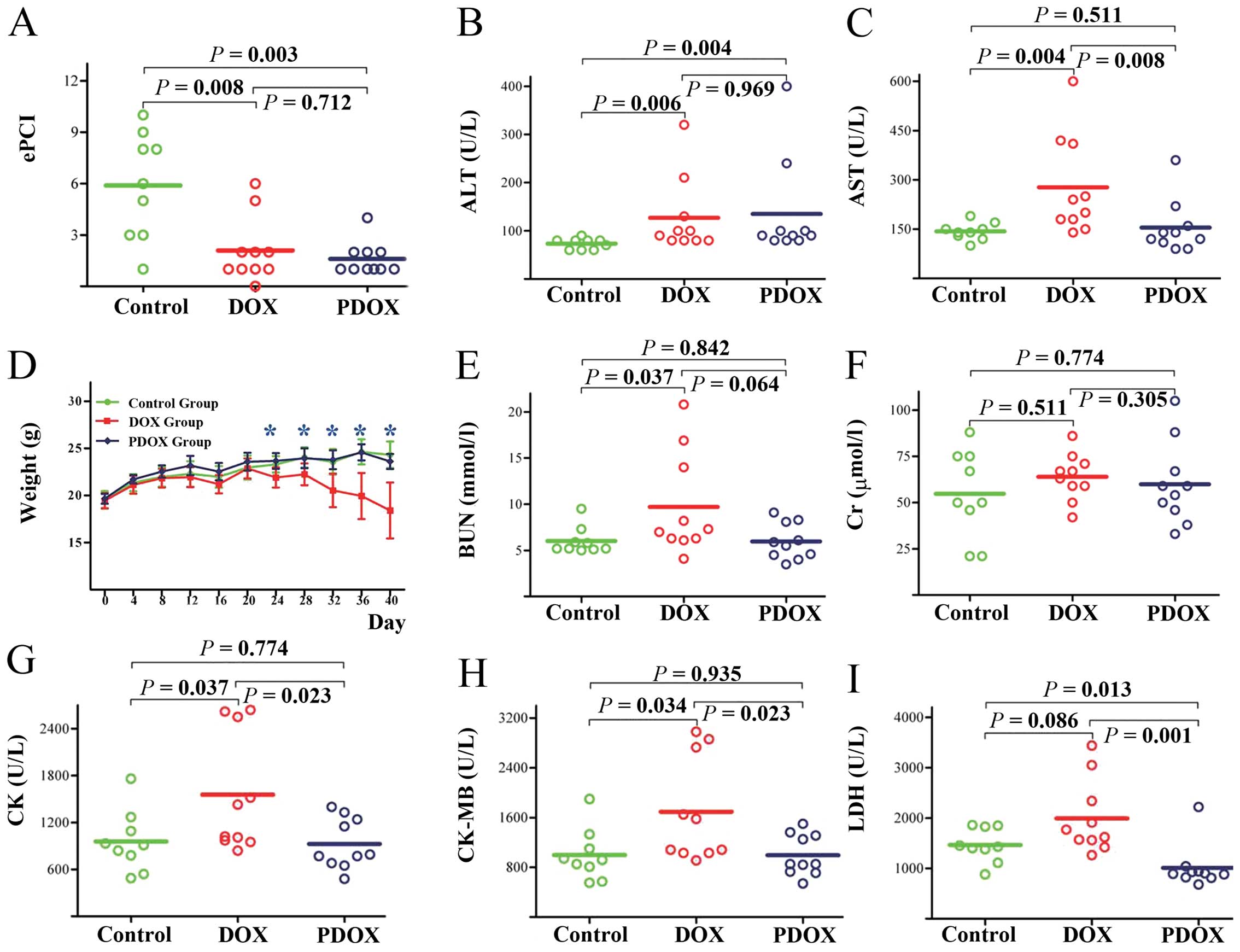 | Figure 7.Cat B-cleavable prodrug
Ac-Phe-Lys-PABC-DOX (PDOX) enhances treatment efficacy and reduces
toxicity in treating gastric cancer with peritoneal carcinomatosis
[modified from a previous study (12)]. (A) Effects of DOX and PDOX on a
peritoneal carcinomatosis model are shown with the detailed
experimental peritoneal carcinomatosis index (ePCI) score; both DOX
and PDOX significantly reduced the ePCI. PDOX reduced general
toxicity and toxicity to the liver, kidney and the heart in
particular. (D) Nude mice in the PDOX group had similar body
weights to those in the control group throughout the study period,
while nude mice in the DOX group showed a progressive decrease in
body weight after 4 doses of intraperitoneal injection. Effects of
PDOX and DOX on major liver and renal function parameters are shown
in (B) ALT, (C) AST, (E) BUN and (F) Cr. PDOX significantly
decreased hepatotoxicity compared with DOX in terms of AST. PDOX
significantly decreased myocardial toxicity compared with DOX by
reducing (G) CK, (H) CK-MB and (I) LDH. ALT, alanine
aminotransferase; AST, aspartate aminotransferase; BUN, blood urea
nitrogen; Cr, creatinine; CK, creatine kinase; CK-MB, creatine
kinase-MB isoenzyme; LDH, lactate dehydrogenase. |
ε-maleimidocaproic acid-Phe-Lys-PABC-DOX
(EMC-Phe-Lys-PABC-DOX)
EMC-Phe-Lys-PABC-DOX (Fig. 6B) (2,18,104) has exhibited dramatic differences
in antitumor activity between in vitro and in vivo
studies. An in vitro cytotoxicity study using the pancreatic
tumor cell line, AsPC1 LN, and the melanoma cancer cell line,
MDA-MB-231 LN, showed that DOX was ∼6-fold more active than the
prodrug. However, an in vivo study using a breast cancer
xeno-graft nude mice model of MDA-MB-435 cells showed that the
prodrug exhibited superior antitumor activity (tumor size, 15% of
that in nude mice treated with the vehicle) compared to DOX (tumor
size, 49% of that in nude mice treated with the vehicle) in an
equitoxic comparison (2).
PG-Phe-Lys-DOX
Hyperbranched polyglycerol-Phe-Lys-DOX
(PG-Phe-Lys-DOX, 45% DOX) (18,41,105), contains the dipeptide, Phe-Lys,
and hyperbranched polyglycerol. The drug release of the conjugates
suggested an effective cleavage of PG-Phe-Lys-DOX and release of
DOX in the presence of Cat B. The IC50 of PG-Phe-Lys-DOX
in the breast cancer cell line, MDA-MB-231, and the pancreatic
carcinoma cell line, AsPC1, was 1.10±0.4 and 2.4±0.6 μM,
respectively, both of which were lower than that of free DOX
(105).
Z-Phe-Lys-PABC-DOX
Benzyloxycarbonyl-Phe-Lys-PABC-DOX
(Z-Phe-Lys-PABC-DOX; Fig. 6C), is
stable in human plasma and rapidly releases DOX in the presence of
Cat B at 37°C, pH 5.0 (half-life, 8 min), which is 30-fold faster
than that of the Val-Cit conjugate. On the other hand, the release
rate is significantly faster than Z-Phe-Lys-DOX, suggesting that a
self immolative spacer, such as PABC, is helpful for DOX release
from conjugates (104).
BR96-SC-Phe-Lys-PABC-DOX
BR96-SC-Phe-Lys-PABC-DOX (Fig. 6D) contains the chimeric monoclonal
antibody, BR96, that binds specifically to a
Lewisy-related, tumor-associated antigen expressed on
the surface of many human carcinoma cells. An in vitro study
using human carcinomal cell lines expressing varying levels of the
BR96 antigen showed that the cytotoxicity of BR96-Phe-Lys-PABC-DOX
was directly related to the level of antigen expression on the cell
membrane: the higher level of BR96 antigen, the higher the
sensitivity to BR96-Phe-Lys-PABC-DOX. The cytotoxicity of
BR96-Phe-Lys-PABC-DOX in high BR96 antigen-expressing cell lines is
higher than that of the non-binding IgG-SC-Phe-Lys-PABC-DOX
conjugate (>220-fold), confirming its BR96 antigen specificity
(104).
Other DOX prodrugs containing
dipeptides
Dubowchik et al(104) and de Groot et al(106) synthesized a series of other DOX
prodrugs containing the dipeptides, Phe-Lys, Ala-Lys or Phe-Arg,
including Z-Phe-Lys-PABC-DOX•HCl, MC-Phe-Lys(MMT)-PABC-DOX,
MC-Phe-Lys-PABC-DOX•Cl2CHCO2H,
Z-Phe-Lys(alloc)-DOX, Z-Phe-Lys-DOX•HCl, Z-Ala-Lys(alloc)-PABC-DOX,
Z-Ala-Lys-PABC-DOX•HCl, Z-Phe-Arg(NO2)-PABC-DOX,
Z-Phe-Arg(Ts)-PABC-DOX, Fmoc-Phe-Lys(Aloc)-PABC-DOX and
H-Phe-Lys(Aloc)-PABC-DOX. However, data regarding their antitumor
activity are lacking.
Conclusions
Over the past few decades, significant efforts have
been made to develop antitumor prodrugs with increased efficacy and
decreased toxicity. Numerous DOX prodrugs have been synthesized by
structure modification strategies. Cat B-cleavable DOX prodrugs
release the free drugs in the presence of Cat B and in a subacidic
environment. A number of in vitro cancer cell studies and
in vivo tumor xenograft studies have demonstrated Cat
B-cleavable DOX prodrugs to be less toxic in vitro and more
effective in vivo, demonstrating the role of Cat B.
However, there remain many challenges and questions.
The majority of the studies mentioned in this review are in a very
early preclinical stage with little information on physicochemical
properties, cytotoxicity and antitumor efficacy in tumor cells and
xenografts. The subcellular distribution of the prodrugs, the free
drugs released and the antitumor mechanisms remain unclear. Further
studies are warranted and should focus on preclinical and clinical
evaluation of existing prodrugs, rather than synthesizing novel
drug candidates in this field.
Abbreviations:
|
Cat B
|
cathepsin B
|
|
DOX
|
doxorubicin
|
|
HPMA
|
N-(2-hydroxypropyl)methacrylamide
|
|
PK1
|
HPMA
copolymer-Gly-Phe-Leu-Gly-doxorubicin
|
|
PK2
|
galactosamine-targeted
poly(HPMA)-doxorubicin
|
|
P-DOX
|
HPMA copolymer-doxorubicin
conjugates
|
|
P-(GFLG)-DOX-Ab
|
HPMA copolymer-DOX-OV-TL16
|
|
P-(GFLG)-DOX-GalN
|
HPMA
copolymer-Gly-Phe-Lys-Gly-DOX-N-acylated galactosamine
|
|
P-(GFLG-DOX)-lac
|
lactose-containing HPMA
copolymer-doxorubicin conjugate
|
|
P-(GFLG-DOX)-TriGal
|
trivalent galactose-containing HPMA
copolymer-doxorubicin conjugate
|
|
Ma-GFLG-DOX
|
(N-methacryloyl-glycyl)-dl-phenylalanyl-leucylglycyl-DOX
|
|
D2-GFLG-P-DOX
|
PAMAM dendrimers
(D-NH2)-Gly-Phe-Leu-Gly-HPMA-doxorubicin
|
|
EMC-Arg-Arg-Ala-Leu-Ala-Leu-DOX
|
6-maleimidocaproic
acid-Arg-Arg-Ala-Leu-Ala-Leu-DOX
|
|
EMC-Phe-Lys-PABC-DOX
|
ε-maleimidocaproic
acid-Phe-Lys-PABC-DOX
|
|
PG-Phe-Lys-DOX
|
hyperbranched
polyglycerol-Phe-Lys-DOX
|
|
Z-Phe-Lys-PABC-DOX
|
benzyloxycarbonyl-Phe-Lys-PABC-DOX
|
Acknowledgements
This study was supported by the State
Key Research Project on Infectious Diseases (2012ZX10002012-012)
and the National Natural Science Foundation of China (no. 81171396)
and National University Students Innovation Training Project of
China (101048639).
References
|
1.
|
Gianni L, Grasselli G, Cresta S, Locatelli
A, Vigano L and Minotti G: Anthracyclines. Cancer Chemother Biol
Response Modif. 21:29–40. 2003. View Article : Google Scholar
|
|
2.
|
Abu Ajaj K, Graeser R, Fichtner I and
Kratz F: In vitro and in vivo study of an albumin-binding prodrug
of doxorubicin that is cleaved by cathepsin B. Cancer Chemother
Pharmacol. 64:413–418. 2009.PubMed/NCBI
|
|
3.
|
Ogura M: Adriamycin (doxorubicin). Gan To
Kagaku Ryoho. 28:1331–1338. 2001.(In Japanese).
|
|
4.
|
Granados-Principal S, Quiles JL,
Ramirez-Tortosa CL, Sanchez-Rovira P and Ramirez-Tortosa MC: New
advances in molecular mechanisms and the prevention of adriamycin
toxicity by antioxidant nutrients. Food Chem Toxicol. 48:1425–1438.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Herman EH, Ferrans VJ, Jordan W and
Ardalan B: Reduction of chronic daunorubicin cardiotoxicity by
ICRF-187 in rabbits. Res Commun Chem Pathol Pharmacol. 31:85–97.
1981.PubMed/NCBI
|
|
6.
|
Wexler LH, Andrich MP, Venzon D, et al:
Randomized trial of the cardioprotective agent ICRF-187 in
pediatric sarcoma patients treated with doxorubicin. J Clin Oncol.
14:362–372. 1996.PubMed/NCBI
|
|
7.
|
Lipshultz SE: Dexrazoxane for protection
against cardiotoxic effects of anthracyclines in children. J Clin
Oncol. 14:328–331. 1996.PubMed/NCBI
|
|
8.
|
Cattel L, Ceruti M and Dosio F: From
conventional to stealth liposomes: a new frontier in cancer
chemotherapy. Tumori. 89:237–249. 2003.PubMed/NCBI
|
|
9.
|
Li J, Wu C, Dai Y, Zhang R, Wang X, Fu D
and Chen B: Doxorubicin-CdS nanoparticles: a potential anticancer
agent for enhancing the drug uptake of cancer cells. J Nanosci
Nanotechnol. 7:435–439. 2007.PubMed/NCBI
|
|
10.
|
Ascensao A, Lumini-Oliveira J, Machado NG,
et al: Acute exercise protects against calcium-induced cardiac
mitochondrial permeability transition pore opening in
doxorubicin-treated rats. Clin Sci. 120:37–49. 2011. View Article : Google Scholar
|
|
11.
|
Yeung TK, Hopewell JW, Simmonds RH, et al:
Reduced cardiotoxicity of doxorubicin given in the form of
N-(2-hydroxypropyl) methacrylamide conjugates: and experimental
study in the rat. Cancer Chemother Pharmacol. 29:105–111. 1991.
View Article : Google Scholar
|
|
12.
|
Shao LH, Liu SP, Hou JX, et al: Cathepsin
B cleavable novel prodrug Ac-Phe-Lys-PABC-ADM enhances efficacy at
reduced toxicity in treating gastric cancer peritoneal
carcinomatosis: an experimental study. Cancer. 118:2986–2996. 2011.
View Article : Google Scholar
|
|
13.
|
Kratz F, Warnecke A, Schmid B, Chung DE
and Gitzel M: Prodrugs of anthracyclines in cancer chemotherapy.
Curr Med Chem. 13:477–523. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Muller MB, Keck ME, Binder EB, et al:
ABCB1 (MDR1)-type P-glycoproteins at the blood-brain barrier
modulate the activity of the hypothalamic-pituitary-adrenocortical
system: implications for affective disorder.
Neuropsychopharmacology. 28:1991–1999. 2003. View Article : Google Scholar
|
|
15.
|
Gottesman MM, Fojo T and Bates SE:
Multidrug resistance in cancer: role of ATP-dependent transporters.
Nat Rev Cancer. 2:48–58. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Lu Y, Yang J and Sega E: Issues related to
targeted delivery of proteins and peptides. AAPS J. 8:E466–E478.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Juillerat-Jeanneret L and Schmitt F:
Chemical modification of therapeutic drugs or drug vector systems
to achieve targeted therapy: looking for the grail. Med Res Rev.
27:574–590. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Calderon M, Graeser R, Kratz F and Haag R:
Development of enzymatically cleavable prodrugs derived from
dendritic polyglycerol. Bioorg Med Chem Lett. 19:3725–3728. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Haag R and Kratz F: Polymer therapeutics:
concepts and applications. Angew Chem Int Ed Engl. 45:1198–1215.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Duncan R: Polymer conjugates as anticancer
nanomedicines. Nat Rev Cancer. 6:688–701. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Vicent MJ, Dieudonne L, Carbajo RJ and
Pineda-Lucena A: Polymer conjugates as therapeutics: future trends,
challenges and opportunities. Expert Opin Drug Deliv. 5:593–614.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Kiick KL: Materials science. Polymer
therapeutics. Science. 317:1182–1183. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Schilsky RL: Pharmacology and clinical
status of capecitabine. Oncology. 14:1297–1306; discussion
1309–1311, 2000.
|
|
24.
|
Basu SK: Receptor-mediated endocytosis of
macromolecular conjugates in selective drug delivery. Biochem
Pharmacol. 40:1941–1946. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Noguchi Y, Wu J, Duncan R, Strohalm J,
Ulbrich K, Akaike T and Maeda H: Early phase tumor accumulation of
macromolecules: a great difference in clearance rate between tumor
and normal tissues. Jpn J Cancer Res. 89:307–314. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Shiah JJ, Sun Y, Peterson CM and Kopecek
J: Biodistribution of free and N-(2-hydroxypropyl)methacrylamide
copolymer-bound mesochlorin e(6) and adriamycin in nude mice
bearing human ovarian carcinoma OVCAR-3 xenografts. J Control
Release. 61:145–157. 1999. View Article : Google Scholar
|
|
27.
|
Bogdanov A Jr, Wright SC, Marecos EM,
Bogdanova A, Martin C, Petherick P and Weissleder R: A
long-circulating co-polymer in ‘passive targeting’ to solid tumors.
J Drug Target. 4:321–330. 1997.PubMed/NCBI
|
|
28.
|
Kopecek J, Sprincl L and Lim D: New types
of synthetic infusion solutions. I. Investigation of the effect of
solutions of some hydrophilic polymers on blood. J Biomed Mater
Res. 7:179–191. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Sprincl L, Exner J, Sterba O and Kopecek
J: New types of synthetic infusion solutions. III. Elimination and
retention of poly-[N-(2-hydroxypropyl)methacrylamide] in a test
organism. J Biomed Mater Res. 10:953–963. 1976.PubMed/NCBI
|
|
30.
|
Etrych T, Kovar L, Strohalm J, Chytil P,
Rihova B and Ulbrich K: Biodegradable star HPMA polymer-drug
conjugates: biodegradability, distribution and anti-tumor efficacy.
J Control Release. 154:241–248. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Satchi-Fainaro R, Puder M, Davies JW, et
al: Targeting angio-genesis with a conjugate of HPMA copolymer and
TNP-470. Nat Med. 10:255–261. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Satchi-Fainaro R, Mamluk R, Wang L, et al:
Inhibition of vessel permeability by TNP-470 and its polymer
conjugate, caplostatin. Cancer Cell. 7:251–261. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Kasuya Y, Lu ZR, Kopeckova P, Minko T,
Tabibi SE and Kopecek J: Synthesis and characterization of HPMA
copolymeraminopropylgeldanamycin conjugates. J Control Release.
74:203–211. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Nishiyama N, Nori A, Malugin A, Kasuya Y,
Kopeckova P and Kopecek J: Free and
N-(2-hydroxypropyl)methacrylamide copolymer-bound geldanamycin
derivative induce different stress responses in A2780 human ovarian
carcinoma cells. Cancer Res. 63:7876–7882. 2003.
|
|
35.
|
Etrych T, Mrkvan T, Rihova B and Ulbrich
K: Star-shaped immunoglobulin-containing HPMA-based conjugates with
doxorubicin for cancer therapy. J Control Release. 122:31–38. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Vasey PA, Kaye SB, Morrison R, et al:
Phase I clinical and pharmacokinetic study of PK1
[N-(2-hydroxypropyl)methacrylamide copolymer doxorubicin]: first
member of a new class of chemo-therapeutic agents-drug-polymer
conjugates. Cancer Research Campaign Phase I/II Committee. Clin
Cancer Res. 5:83–94. 1999.
|
|
37.
|
Bilim V: Technology evaluation: PK1,
Pfizer/Cancer Research UK. Curr Opin Mol Ther. 5:326–330.
2003.PubMed/NCBI
|
|
38.
|
Seymour LW, Ferry DR, Kerr DJ, et al:
Phase II studies of polymer-doxorubicin (PK1, FCE28068) in the
treatment of breast, lung and colorectal cancer. Int J Oncol.
34:1629–1636. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Thomson AH, Vasey PA, Murray LS, Cassidy
J, Fraier D, Frigerio E and Twelves C: Population pharmacokinetics
in phase I drug development: a phase I study of PK1 in patients
with solid tumours. Br J Cancer. 81:99–107. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Julyan PJ, Seymour LW, Ferry DR, et al:
Preliminary clinical study of the distribution of HPMA copolymers
bearing doxorubicin and galactosamine. J Control Release.
57:281–290. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Seymour LW, Ferry DR, Anderson D, et al:
Hepatic drug targeting: phase I evaluation of polymer-bound
doxorubicin. J Clin Oncol. 20:1668–1676. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Seymour LW, Ulbrich K, Wedge SR, Hume IC,
Strohalm J and Duncan R: N-(2-hydroxypropyl)methacrylamide
copolymers targeted to the hepatocyte galactose-receptor:
pharmacokinetics in DBA2 mice. Br J Cancer. 63:859–866. 1991.
View Article : Google Scholar
|
|
43.
|
Meerum Terwogt JM, ten Bokkel Huinink WW,
Schellens JH, et al: Phase I clinical and pharmacokinetic study of
PNU166945, a novel water-soluble polymer-conjugated prodrug of
paclitaxel. Anticancer Drugs. 12:315–323. 2001.PubMed/NCBI
|
|
44.
|
Rice JR, Gerberich JL, Nowotnik DP and
Howell SB: Preclinical efficacy and pharmacokinetics of AP5346, a
novel diaminocyclohexane-platinum tumor-targeting drug delivery
system. Clin Cancer Res. 12:2248–2254. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Nowotnik DP and Cvitkovic E: ProLindac
(AP5346): a review of the development of an HPMA DACH platinum
polymer therapeutic. Adv Drug Deliv Rev. 61:1214–1219. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Campone M, Rademaker-Lakhai JM, Bennouna
J, Howell SB, Nowotnik DP, Beijnen JH and Schellens JH: Phase I and
pharmacokinetic trial of AP5346, a DACH-platinum-polymer conjugate,
administered weekly for three out of every 4 weeks to advanced
solid tumor patients. Cancer Chemother Pharmacol. 60:523–533. 2007.
View Article : Google Scholar
|
|
47.
|
Van der Schoot SC, Nuijen B, Sood P,
Thurmond KB II, Stewart DR, Rice JR and Beijnen JH: Pharmaceutical
development, quality control, stability and compatibility of a
parenteral lyophilized formulation of the investigational
polymer-conjugated platinum antineoplastic agent AP5346. Pharmazie.
61:835–844. 2006.
|
|
48.
|
Sood P, Thurmond KB II, Jacob JE, Waller
LK, Silva GO, Stewart DR and Nowotnik DP: Synthesis and
characterization of AP5346, a novel polymer-linked
diaminocyclohexyl platinum chemotherapeutic agent. Bioconjug Chem.
17:1270–1279. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Rademaker-Lakhai JM, Terret C, Howell SB,
et al: A Phase I and pharmacological study of the platinum polymer
AP5280 given as an intravenous infusion once every 3 weeks in
patients with solid tumors. Clin Cancer Res. 10:3386–3395. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Tibben MM, Rademaker-Lakhai JM, Rice JR,
Stewart DR, Schellens JH and Beijnen JH: Determination of total
platinum in plasma and plasma ultrafiltrate, from subjects dosed
with the platinum-containing N-(2-hydroxypropyl)methacrylamide
copolymer AP5280, by use of graphite-furnace Zeeman
atomic-absorption spectrometry. Anal Bioanal Chem. 373:233–236.
2002. View Article : Google Scholar
|
|
51.
|
Lin X, Zhang Q, Rice JR, Stewart DR,
Nowotnik DP and Howell SB: Improved targeting of platinum
chemotherapeutics. the antitumour activity of the HPMA copolymer
platinum agent AP5280 in murine tumour models. Eur J Cancer.
40:291–297. 2004.PubMed/NCBI
|
|
52.
|
Podgorski I and Sloane BF: Cathepsin B and
its role(s) in cancer progression. Biochem Soc Symp. 70:263–276.
2003.PubMed/NCBI
|
|
53.
|
Calkins CC, Sameni M, Koblinski J, Sloane
BF and Moin K: Differential localization of cysteine protease
inhibitors and a target cysteine protease, cathepsin B, by
immuno-confocal microscopy. J Histochem Cytochem. 46:745–751. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Kovar M, Strohalm J, Etrych T, Ulbrich K
and Rihova B: Star structure of antibody-targeted HPMA
copolymer-bound doxorubicin: a novel type of polymeric conjugate
for targeted drug delivery with potent antitumor effect. Bioconjug
Chem. 13:206–215. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Thanou M and Duncan R: Polymer-protein and
polymer-drug conjugates in cancer therapy. Curr Opin Investig
Drugs. 4:701–709. 2003.PubMed/NCBI
|
|
56.
|
Mai J, Waisman DM and Sloane BF: Cell
surface complex of cathepsin B/annexin II tetramer in malignant
progression. Biochim Biophys Acta. 1477:215–230. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Kratz F, Muller IA, Ryppa C and Warnecke
A: Prodrug strategies in anticancer chemotherapy. ChemMedChem.
3:20–53. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Trouet A, Masquelier M, Baurain R and
Deprez-De Campeneere D: A covalent linkage between daunorubicin and
proteins that is stable in serum and reversible by lysosomal
hydro-lases, as required for a lysosomotropic drug-carrier
conjugate: in vitro and in vivo studies. Proc Natl Acad Sci USA.
79:626–629. 1982. View Article : Google Scholar
|
|
59.
|
Omelyanenko V, Kopeckova P, Gentry C and
Kopecek J: Targetable HPMA copolymer-adriamycin conjugates.
Recognition, internalization and subcellular fate. J Control
Release. 53:25–37. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Carl PL, Chakravarty PK and
Katzenellenbogen JA: A novel connector linkage applicable in
prodrug design. J Med Chem. 24:479–480. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
61.
|
Seymour LW, Ulbrich K, Steyger PS,
Brereton M, Subr V, Strohalm J and Duncan R: Tumour tropism and
anti-cancer efficacy of polymer-based doxorubicin prodrugs in the
treatment of subcutaneous murine B16F10 melanoma. Br J Cancer.
70:636–641. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Duncan R, Kopeckova P, Strohalm J, Hume
IC, Lloyd JB and Kopecek J: Anticancer agents coupled to
N-(2-hydroxypropyl) methacrylamide copolymers. II. Evaluation of
daunomycin conjugates in vivo against L1210 leukaemia. Br J Cancer.
57:147–156. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
63.
|
Duncan R, Kopeckova-Rejmanova P, Strohalm
J, et al: Anticancer agents coupled to
N-(2-hydroxypropyl)methacrylamide copolymers. I. Evaluation of
daunomycin and puromycin conjugates in vitro. Br J Cancer.
55:165–174. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
64.
|
Hopewel JW, Duncan R, Wilding D and
Chakrabarti K: Preclinical evaluation of the cardiotoxicity of PK2:
a novel HPMA copolymer-doxorubicin-galactosamine conjugate
antitumour agent. Hum Exp Toxicol. 20:461–470. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Minko T, Kopeckova P, Pozharov V and
Kopecek J: HPMA copolymer bound adriamycin overcomes MDR1 gene
encoded resistance in a human ovarian carcinoma cell line. J
Control Release. 54:223–233. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
66.
|
Minko T, Kopeckova P and Kopecek J:
Chronic exposure to HPMA copolymer-bound adriamycin does not induce
multidrug resistance in a human ovarian carcinoma cell line. J
Control Release. 59:133–148. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
67.
|
Tijerina M, Fowers KD, Kopeckova P and
Kopecek J: Chronic exposure of human ovarian carcinoma cells to
free or HPMA copolymer-bound mesochlorin e6 does not induce
P-glycoprotein-mediated multidrug resistance. Biomaterials.
21:2203–2210. 2000. View Article : Google Scholar
|
|
68.
|
Minko T, Kopeckova P and Kopecek J:
Efficacy of the chemo-therapeutic action of HPMA copolymer-bound
doxorubicin in a solid tumor model of ovarian carcinoma. Int J
Cancer. 86:108–117. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
69.
|
Duncan R: Drug-polymer conjugates:
potential for improved chemotherapy. Anticancer Drugs. 3:175–210.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
70.
|
Kovar L, Strohalm J, Chytil P, et al: The
same drug but a different mechanism of action: comparison of free
doxorubicin with two different N-(2-hydroxypropyl)methacrylamide
copolymer-bound doxorubicin conjugates in EL-4 cancer cell line.
Bioconjug Chem. 18:894–902. 2007. View Article : Google Scholar
|
|
71.
|
Satchi R, Connors TA and Duncan R: PDEPT:
polymer-directed enzyme prodrug therapy. I. HPMA
copolymer-cathepsin B and PK1 as a model combination. Br J Cancer.
85:1070–1076. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
72.
|
Paul A, Vicent MJ and Duncan R: Using
small-angle neutron scattering to study the solution conformation
of N-(2-hydroxypropyl)methacrylamide copolymer-doxorubicin
conjugates. Biomacromolecules. 8:1573–1579. 2007. View Article : Google Scholar
|
|
73.
|
Pimm MV, Perkins AC, Strohalm J, Ulbrich K
and Duncan R: Gamma scintigraphy of a 123I-labelled
N-(2-hydroxypropyl) methacrylamide copolymer-doxorubicin conjugate
containing galactosamine following intravenous administration to
nude mice bearing hepatic human colon carcinoma. J Drug Target.
3:385–390. 1996.
|
|
74.
|
Duncan R, Seymour LC, Scarlett L, Lloyd
JB, Rejmanova P and Kopecek J: Fate of
N-(2-hydroxypropyl)methacrylamide copolymers with pendent
galactosamine residues after intravenous administration to rats.
Biochim Biophys Acta. 880:62–71. 1986. View Article : Google Scholar
|
|
75.
|
Virgolini I, Muller C, Klepetko W,
Angelberger P, Bergmann H, O’Grady J and Sinzinger H: Decreased
hepatic function in patients with hepatoma or liver metastasis
monitored by a hepatocyte specific galactosylated radioligand. Br J
Cancer. 61:937–941. 1990. View Article : Google Scholar
|
|
76.
|
Schlepper-Schafer J, Hulsmann D, Djovkar
A, Meyer HE, Herbertz L, Kolb H and Kolb-Bachofen V: Endocytosis
via galactose receptors in vivo. Ligand size directs uptake by
hepatocytes and/or liver macrophages. Exp Cell Res. 165:494–506.
1986.PubMed/NCBI
|
|
77.
|
Shiah JG, Dvorak M, Kopeckova P, Sun Y,
Peterson CM and Kopecek J: Biodistribution and antitumour efficacy
of long-circulating N-(2-hydroxypropyl)methacrylamide
copolymer-doxorubicin conjugates in nude mice. Eur J Cancer.
37:131–139. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
78.
|
Rihova B, Bilej M, Vetvicka V, Ulbrich K,
Strohalm J, Kopecek J and Duncan R: Biocompatibility of
N-(2-hydroxypropyl) methacrylamide copolymers containing
adriamycin. Immunogenicity and effect on haematopoietic stem cells
in bone marrow in vivo and mouse splenocytes and human peripheral
blood lymphocytes in vitro. Biomaterials. 10:335–342. 1989.
View Article : Google Scholar
|
|
79.
|
Omelyanenko V, Kopeckova P, Gentry C,
Shiah JG and Kopecek J: HPMA copolymer-anticancer drug-OV-TL16
antibody conjugates. 1. influence of the method of synthesis on the
binding affinity to OVCAR-3 ovarian carcinoma cells in vitro. J
Drug Target. 3:357–373. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
80.
|
Omelyanenko V, Gentry C, Kopeckova P and
Kopecek J: HPMA copolymer-anticancer drug-OV-TL16 antibody
conjugates. II. Processing in epithelial ovarian carcinoma cells in
vitro. Int J Cancer. 75:600–608. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
81.
|
Kunath K, Kopeckova P, Minko T and Kopecek
J: HPMA copolymer-anticancer drug-OV-TL16 antibody conjugates. 3.
The effect of free and polymer-bound adriamycin on the expression
of some genes in the OVCAR-3 human ovarian carcinoma cell line. Eur
J Pharm Biopharm. 49:11–15. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
82.
|
Jensen KD, Kopeckova P, Bridge JH and
Kopecek J: The cytoplasmic escape and nuclear accumulation of
endocytosed and microinjected HPMA copolymers and a basic kinetic
study in Hep G2 cells. AAPS PharmSci. 3:E322001. View Article : Google Scholar : PubMed/NCBI
|
|
83.
|
David A, Kopeckova P, Kopecek J and
Rubinstein A: The role of galactose, lactose and galactose valency
in the biorecognition of N-(2-hydroxypropyl)methacrylamide
copolymers by human colon adenocarcinoma cells. Pharm Res.
19:1114–1122. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
84.
|
David A, Kopeckova P, Rubinstein A and
Kopecek J: Enhanced biorecognition and internalization of HPMA
copolymers containing multiple or multivalent carbohydrate
side-chains by human hepatocarcinoma cells. Bioconjug Chem.
12:890–899. 2001. View Article : Google Scholar
|
|
85.
|
Irimura T, Matsushita Y, Sutton RC, et al:
Increased content of an endogenous lactose-binding lectin in human
colorectal carcinoma progressed to metastatic stages. Cancer Res.
51:387–393. 1991.
|
|
86.
|
Bresalier RS, Mazurek N, Sternberg LR,
Byrd JC, Yunker CK, Nangia-Makker P and Raz A: Metastasis of human
colon cancer is altered by modifying expression of the
beta-galactoside-binding protein galectin 3. Gastroenterology.
115:287–296. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
87.
|
Ohannesian DW, Lotan D, Thomas P, Jessup
JM, Fukuda M, Gabius HJ and Lotan R: Carcinoembryonic antigen and
other glycoconjugates act as ligands for galectin-3 in human colon
carcinoma cells. Cancer Res. 55:2191–2199. 1995.PubMed/NCBI
|
|
88.
|
Lotz MM, Andrews CW Jr, Korzelius CA, Lee
EC, Steele GD Jr, Clarke A and Mercurio AM: Decreased expression of
Mac-2 (carbohydrate binding protein 35) and loss of its nuclear
localization are associated with the neoplastic progression of
colon carcinoma. Proc Natl Acad Sci USA. 90:3466–3470. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
89.
|
Castronovo V, Campo E, van den Brule FA,
et al: Inverse modulation of steady-state messenger RNA levels of
two non-integrin laminin-binding proteins in human colon carcinoma.
J Natl Cancer Inst. 84:1161–1169. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
90.
|
David A, Kopeckova P, Minko T, Rubinstein
A and Kopecek J: Design of a multivalent galactoside ligand for
selective targeting of HPMA copolymer-doxorubicin conjugates to
human colon cancer cells. Eur J Cancer. 40:148–157. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
91.
|
Etrych T, Strohalm J, Chytil P, Cernoch P,
Starovoytova L, Pechar M and Ulbrich K: Biodegradable star HPMA
polymer conjugates of doxorubicin for passive tumor targeting. Eur
J Pharm Sci. 42:527–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
92.
|
Dvorak M, Kopeckova P and Kopecek J:
High-molecular weight HPMA copolymer-adriamycin conjugates. J
Control Release. 60:321–332. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
93.
|
Etrych T, Jelinkova M, Rihova B and
Ulbrich K: New HPMA copolymers containing doxorubicin bound via
pH-sensitive linkage: synthesis and preliminary in vitro and in
vivo biological properties. J Control Release. 73:89–102. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
94.
|
Schmid B, Chung DE, Warnecke A, Fichtner I
and Kratz F: Albumin-binding prodrugs of camptothecin and
doxorubicin with an Ala-Leu-Ala-Leu-linker that are cleaved by
cathepsin B: synthesis and antitumor efficacy. Bioconjug Chem.
18:702–716. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
95.
|
Kratz F, Warnecke A, Scheuermann K, et al:
Probing the cysteine-34 position of endogenous serum albumin with
thiol-binding doxorubicin derivatives. Improved efficacy of an
acid-sensitive doxorubicin derivative with specific albumin-binding
properties compared to that of the parent compound. J Med Chem.
45:5523–5533. 2002. View Article : Google Scholar
|
|
96.
|
Warnecke A and Kratz F:
Maleimide-oligo(ethylene glycol) derivatives of camptothecin as
albumin-binding prodrugs: synthesis and antitumor efficacy.
Bioconjug Chem. 14:377–387. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
97.
|
Kratz F and Beyer U: Serum proteins as
drug carriers of anti-cancer agents: a review. Drug Deliv.
5:281–299. 1998. View Article : Google Scholar
|
|
98.
|
Elzoghby AO, Samy WM and Elgindy NA:
Albumin-based nanoparticles as potential controlled release drug
delivery systems. J Control Release. 157:168–182. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
99.
|
Kratz F: Albumin as a drug carrier: design
of prodrugs, drug conjugates and nanoparticles. J Control Release.
132:171–183. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
100.
|
Lebrecht D, Geist A, Ketelsen UP,
Haberstroh J, Setzer B, Kratz F and Walker UA: The
6-maleimidocaproyl hydrazone derivative of doxorubicin (DOXO-EMCH)
is superior to free doxorubicin with respect to cardiotoxicity and
mitochondrial damage. Int J Cancer. 120:927–934. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
101.
|
Unger C, Haring B, Medinger M, Drevs J,
Steinbild S, Kratz F and Mross K: Phase I and pharmacokinetic study
of the (6-maleimidocaproyl)hydrazone derivative of doxorubicin.
Clin Cancer Res. 13:4858–4866. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
102.
|
Dubowchik GM and Firestone RA: Cathepsin
B-sensitive dipeptide prodrugs. 1. A model study of structural
requirements for efficient release of doxorubicin. Bioorg Med Chem
Lett. 8:3341–3346. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
103.
|
Dubowchik GM, Mosure K, Knipe JO and
Firestone RA: Cathepsin B-sensitive dipeptide prodrugs. 2. Models
of anti-cancer drugs paclitaxel (Taxol), mitomycin C and
doxorubicin. Bioorg Med Chem Lett. 8:3347–3352. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
104.
|
Dubowchik GM, Firestone RA, Padilla L, et
al: Cathepsin B-labile dipeptide linkers for lysosomal release of
doxorubicin from internalizing immunoconjugates: model studies of
enzymatic drug release and antigen-specific in vitro anticancer
activity. Bioconjug Chem. 13:855–869. 2002. View Article : Google Scholar
|
|
105.
|
Calderón M, Quadir MA, Strumia M and Haag
R: Functional dendritic polymer architectures as stimuli-responsive
nano-carriers. Biochimie. 92:1242–1251. 2010.PubMed/NCBI
|
|
106.
|
De Groot FM, Broxterman HJ, Adams HP, et
al: Design, synthesis and biological evaluation of a dual
tumor-specific motive containing integrin-targeted
plasmin-cleavable doxorubicin prodrug. Mol Cancer Ther. 1:901–911.
2002.
|