Introduction
Double-strand break repair is mediated by two major
repair pathways, homologous recombination (HR) or non-homologous
end joining (NHEJ; reviewed in ref. 1). In mammalian cells more than 90% of
double-strand breaks are repaired by NHEJ. Impairment of either
pathway is associated with cell cycle arrest, cell death, genomic
instability and cancer (2). Human
diseases such as Nijmegen breakage syndrome (NBS) due to mutations
in the NBS1 gene result in defects in resection of double-strand
breaks (3). NBS1 functions as part
of the Mre11/Rad50/NBS1 (MRN) complex whose functions are not
restricted to HR but are also involved NHEJ (4).
NBS is a rare human autosomal recessive disorder
caused by hypomorphic mutations. This disorder is characterized by
growth retardation, immunodeficiency, microcephaly and cancer
predisposition. At the cellular level, NBS is characterized by
radiosensitivity, chromosomal breakage and defective cell cycle
checkpoints. NBS1 null mutations result in early embryonic
lethality (5), but NBS1
hypomorphic mutants are viable (6). Cells from these mice are defective in
S phase and G2/M checkpoints. Heterozygous mice with an NBS1 null
mutation in addition to homozygous animals with hypomorphic
mutations are predisposed to cancer. Conditional NBS1 mutant mice
have been characterized (7). For
example, neuronal inactivation of NBS1 results in chromosomal
breaks, microcephaly, growth retardation, cerebellar defects and
ataxia. The MRN complex is essential for maintaining genomic
integrity, cell viability and checkpoint activation.
MRN polymorphisms have been associated with
increased risk of breast cancer (8–10).
MRN expression was reduced in the majority of breast tumors
(11). Low expression of MRN
correlated with increased histologic grade and estrogen receptor
negativity. Response to radiotherapy correlated with high
expression of the MRN complex. Patients with high numbers of
ionizing radiation induced NBS1 foci had aggressive breast cancer
phenotypes (12,13).
Estradiol has been shown to markedly enhance
proliferation of mammary gland epithelium and estrogen receptor
(ER) α positive (+) breast cancer cells (14). ER is a member of a large family of
ligand dependent transcription factors that include steroid,
retinoid, thyroid and vitamin D receptors. ER have functional
domains for DNA binding, ligand binding, dimerization, and
transcriptional activation. Nuclear receptors such as ER require
coactivator proteins such as CREB binding protein (CBP) and steroid
receptor coactivator 1 (SRC1) to activate target gene transcription
(15). We previously demonstrated
that estradiol protected ER+ breast cancer cell lines
against double-strand breaks and cell death (16). Ectopic ER expression was sufficient
to produce these effects and this protection involved the
coactivator CBP. We now demonstrate that this protection from
double-strand break damage is mediated via regulation by c-myc, p53
and coactivators in intron 1 of the NBS1 gene.
Materials and methods
Cell culture and stable transfection
The human mammary epithelial and breast cancer cell
lines used in this study were purchased from the American Type
Culture Collection and cultured in Dulbecco’s modified Eagle’s
medium without phenol red, 10% charcoal-resin treated fetal bovine
serum, and 40 μg/ml gentamicin in a humidified atmosphere of
5% CO2 at 37°C. Cultures were treated with 100 nM E2 for
4 h, 3 Gy ionizing radiation, combined E2 and radiation or vehicle.
For some experiments, cells were transfected with 2 μg
c-myc, CBP, SRC1 or neomycin resistance plasmid using Lipofectamine
according to manufacturer’s recommendations (Invitrogen, Carlsbad,
CA). Cells were selected in 400 μg/ml G418 for 14 days.
Resistant clones were picked for expansion and characterization.
For inhibition of gene expression experiments, cells were
transfected with siRNA to ERα, Mre11, Rad50, NBS1 or control siRNA
according to manufacturer’s protocol (Dharmacon, Lafayette,
CO).
DNA damage and apoptosis analysis
DNA damage was quantitated by single cell gel
electrophoresis. Treated cells were mixed with 0.5% low melting
point agarose and added to microscope slides coated with 1.5%
agarose. Cells were alkali denatured (pH 13.0), subjected to
electrophoresis at 0.86 V/cm for 25 min and stained with ethidium
bromide. The tail moment (DNA migration × tail intensity) of 50
randomly selected cells was analyzed from each slide using imaging
software. For apoptosis assays, human mammary epithelial and breast
cancer cell cultures were fixed with 70% ethanol at −20°C for 30
min and washed with PBS. Cultures of mouse mammary epithelial cells
were incubated with terminal deoxynucleotidyl transferase and
fluorescein conjugated dUTP at 37°C for 30 min followed by washing
in PBS. The percentage of apoptotic cells was determined by flow
cytometry.
Western blot analysis
Protein was extracted in 1X Laemmli buffer from
treated human mammary epithelial and breast cancer cell lines.
Total cellular protein (75 μg) was separated by SDS-PAGE on
10% resolving gels under denaturing and reducing conditions.
Separated proteins were electroblotted to PVDF membranes according
to manufacturer’s recommendations (Roche Applied Science,
Indianapolis, IN). Blots were incubated with antibodies to ERα,
Mre11, Rad50, NBS1, c-myc, CBP, SRC1 or β-actin for 16 h at 4°C.
After washing in Tris buffered saline containing 0.1% Tween-20
(TBST, pH 7.4), blots were incubated for 30 min at room temperature
with anti-IgG secondary antibody conjugated to horseradish
peroxidase. Following extensive washing in TBST, bands were
visualized by the enhanced chemiluminescence method (Roche Applied
Science). Bands were quantitated by laser densitometry.
Chromatin immunoprecipitation
Treated human mammary epithelial or breast cancer
cells were fixed in 1% formaldehyde for 10 min at room temperature.
Cells were washed in PBS and lysed in immunoprecipitation buffer
containing protease inhibitors for 30 min at 4°C, sheared and
centrifuged at 10,000 × g for 10 min. Supernatants were cleared
with 2 μg sheared salmon sperm DNA, 20 μl preimmune
serum, and 20 μl protein A/G sepharose beads for 2 h at 4°C.
Aliquots of the supernatant were used as input DNA for
normalization. Immunoprecipitation using anti-myc, -p53, -CBP,
-SRC1 or -acetylated histone H3 antibodies (Santa Cruz
Biotechnology, Santa Cruz, CA) was performed overnight at 4°C.
Preimmune IgG was used as the negative control antibody.
Immunoprecipitates were washed extensively in immunoprecipitation
buffer, resuspended in 10 mM Tris-HCl, 1 mM EDTA (TE, pH 8.0) and
incubated at 65°C for 6 h to reverse crosslinks. The supernatants
were extracted with phenol/chloroform and ethanol precipitated.
Following washing in 70% ethanol, pellets were dried and suspended
in 50 μl TE. For real-time PCR, 1 μl of template was
amplified in buffer containing 10 mM Tris-HCl (pH 8.3), 50 mM KCl,
2.5 mM MgCl2, 200 nM each dNTP and 100 ng each primer
(5′-GATAACCCTTTCCCACTGATTG-3′ and 5′-GAGAACTGCTTGAACCCAG-3′)
flanking the myc and p53 binding sites in the NBS1 first intron
(accession AY566246; 3024–3029 bp and 3124–3134 bp, respectively).
The optimized cycle parameters were one cycle at 94°C for 3 min
followed by 25 cycles of 94°C for 25 sec, 58°C for 60 sec, 72°C for
60 sec and one final cycle at 72°C for 10 min (iCycler,
Bio-Rad).
Nucleosomal mapping
Nuclei were isolated from parental human breast
cancer cell lines treated with E2, IR, E2+IR, or vehicle. Chromatin
was digested to mononucleosomal form with micrococcal nuclease
(Roche Applied Science). The digestion was stopped by addition of
50 mM EDTA. Nuclei were lysed in 1% SDS and treated with 0.1 mg
proteinase K overnight at 37°C. DNA was purified by
phenol/chloroform extraction and ethanol precipitation. DNA was
suspended in TE buffer and analyzed by agarose gel electrophoresis
to ensure digestion to mononucleosomal fragments. These fragments
were eluted from the gel and used as PCR templates to determine
nucleosomal occupancy of NBS1 intron 1. Undigested genomic DNA was
used as the positive control and template free samples were used as
the negative control.
Transient transfection, double-strand
break repair and NBS1 intron 1 analysis
pACT-luc luciferase vector was digested with
XbaI and BstEII or EcoRV and XhoI
restriction enzymes and purified by gel electrophoresis followed by
end-filling with Klenow fragment of DNA polymerase (17). A total of 1 μg of each
linear plasmid was transiently transfected into triplicate cultures
of 50% confluent human breast cancer cell lines using Lipofectamine
according to the manufacturer’s recommendations (Invitrogen). The
728 bp homology between the two plasmids can reconstitute
luciferase activity which correlates with DNA double-strand break
repair activity. Undigested pACT-luc vector was used as the
positive control and 1 μg β-galactosidase expression plasmid
was used to normalize for transfection efficiency. In separate
experiments, triplicate cultures of 50% confluent cells were
transiently transfected with 2 μg of pGL3 luciferase
reporter vector containing 5′ flanking constructs of the NBS1
promoter, exon 1 and intron 1 (−360/+1076), lacking the promoter
(−17/+1076) or lacking intron 1 (−360/+88). Intron 1 was cloned
into pGL3 vector and transiently transfected with 1 μg
c-myc, p53, CBP, SRC1 or blank expression plasmids. Separate
cultures were transfected with pGL3 vector containing point
mutations in the intron 1 myc (CACcaGC) or p53 (GGGgccGCTCC)
binding sites. Cultures were treated with E2, ionizing radiation or
vehicle for 24 h. Cells were harvested and reporter gene activity
determined using a commercially available kit and luminometer
(Applied Biosystems, Carlsbad, CA). Luciferase activity was
normalized to β-galactosidase levels for each sample. Statistical
significance was determined by ANOVA.
Results
We previously determined that estradiol (E2)
treatment decreased DNA damage and increased survival of
ER+ human breast cancer cell lines exposed to ionizing
radiation (IR) (16). To determine
the mechanism of this protection, we treated ER+ and
ER− human breast cancer cell lines with E2, IR or E2
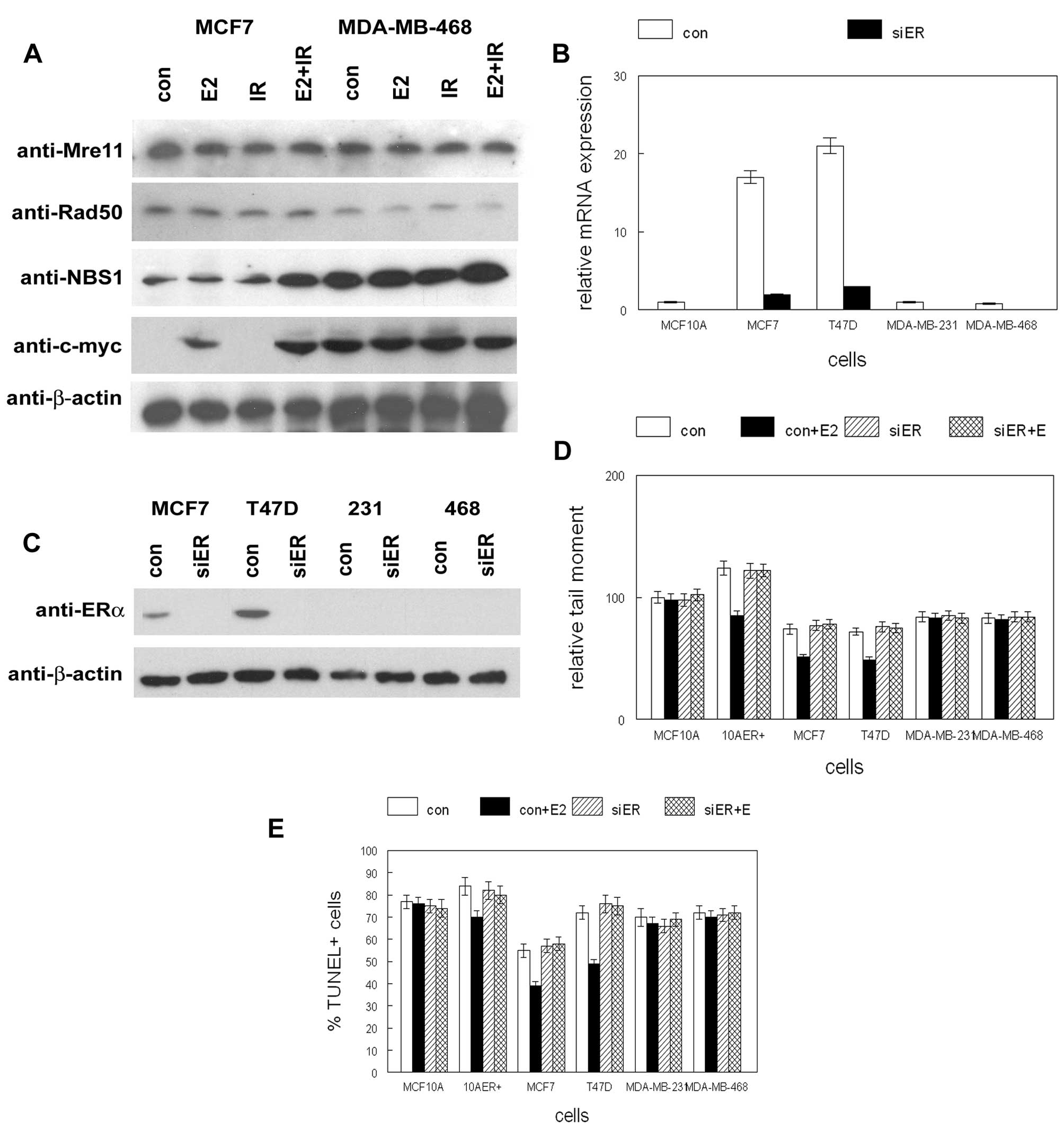
followed by IR. As shown in Fig.
1A, NBS1 protein expression was induced by 5-fold in
ER+ MCF7 cells when treated with E2 followed by IR. No
NBS1 expression changes were observed in ER− MDA-MB-468
cells. No changes in expression of other MRN gene products (Mre11,
Rad50) in response to E2 or IR were observed. Similar results were
observed in ER+ T47D and ER− MDA-MB-231 cells
(data not shown). These results indicate that both E2 and IR were
required to induce NBS1 expression in ER+ breast cancer
cell lines.
To determine if E2 mediated protection from
double-strand break damage was dependent on ER, we transfected
ER+ and ER− human mammary epithelial and
breast cancer cell lines with siRNA to ER. Expression of ER mRNA
and protein following siRNA transfection is shown in Fig. 1B and C. ER expression was reduced
in ER+ MCF7 and T47D cells by >90% following siRNA
transfection. Treatment of ER+ cells with E2 reduced
double-strand break damage by 25–30% following IR as determined by
tail moment (p<0.04; Fig. 1D).
This protective effect was abolished by ER siRNA transfection and
was not observed in ER− cells. Treatment of
ER+ cells with E2 reduced apoptosis by similar magnitude
as determined by TUNEL assay (p<0.03; Fig. 1E). This effect was completely
inhibited by ER siRNA transfection and was not observed in
ER− cells. These results indicate that the protective
effect of E2 against double-strand break damage was mediated by
ER.
Pretreatment with E2 prior to ionizing radiation
induced NBS1 expression in ER+ human mammary epithelial
and breast cancer cells (Fig. 1).
To determine the role of the MRN complex in mediating this
response, we transfected these cells with siRNAs to Mre11, Rad50 or
NBS1. Expression of these gene products following siRNA
transfection is shown in Fig. 2A and
B. Expression of Mre11, Rad50 or NBS1 was reduced by 90% in
siRNA transfected cultures. Transfection of siRNAs to Mre11, Rad50,
or NBS1 inhibited recombination of luciferase plasmids by 70–80%
(p<0.002; Fig. 2C), providing
functional confirmation of reduced expression of these gene
products. To determine the effects of MRN gene product inhibition,
we exposed siRNA transfected cells to E2 or vehicle followed by IR.
As shown in Fig. 2D, NBS1 siRNA
blocked the protective effects of E2 against double-strand break
damage. These effects were observed only in ER+ cells.
Similar effects of NBS1 siRNA were observed on E2 mediated
protection against ionizing radiation induced apoptosis (Fig. 2E). These results indicate that NBS1
mediates the E2 protective effects against ionizing radiation
induced double-strand break damage and apoptosis.
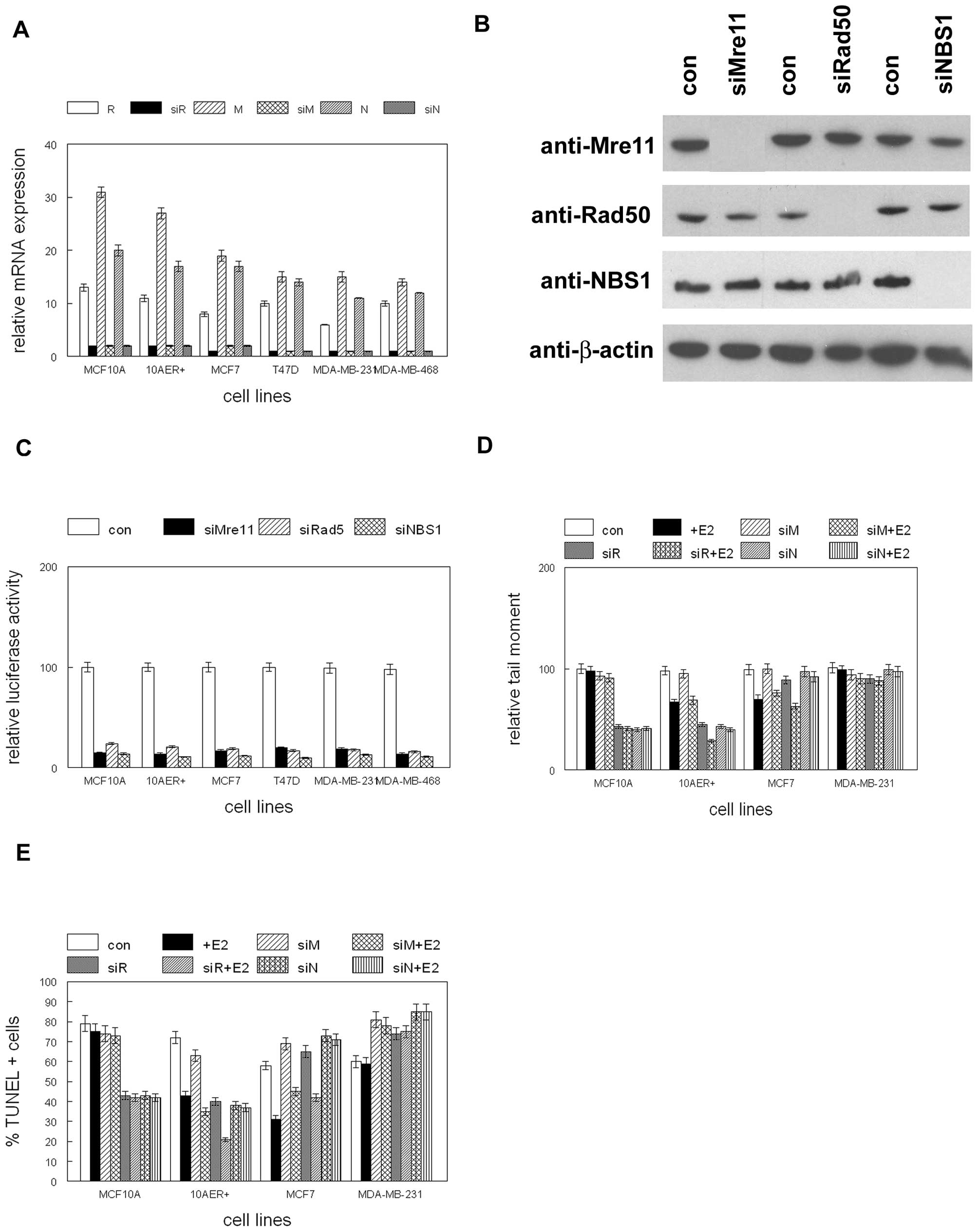 | Figure 2siRNA inhibits expression of MRN gene
products. (A) MCF10A, MCF10A stably expressing ER
(10AER+), and human breast cancer cell lines were
transfected with siRNA to Mre11 (siM), Rad50 (siR), NBS1 (siN) or
control siRNAs (M, R, N) followed by qRT-PCR. (B) MCF10A, MCF10A
stably expressing ER (10AER+), and human breast cancer
cell lines were transfected with siRNA to Mre11 (siMre11), Rad50
(siRad50), NBS1 (siNBS1) or control siRNA (con) followed by western
blot analysis with antibodies indicated at left. (C) siRNAs to MRN
gene products inhibits double-strand break repair. MCF10A, MCF10A
stably expressing ER (10AER+), and human breast cancer
cell lines were transfected with siRNA to Mre11 (siMre11), Rad50
(siRad50), NBS1 (siNBS1) or control siRNA (con) and digested
luciferase reporter vector. (D) NBS1 mediates E2 protection from IR
mediated DNA damage. MCF10A, MCF10A stably expressing ER
(10AER+), and human breast cancer cell lines were
transfected with siRNA to Mre11 (siM), Rad50 (siR), NBS1 (siN) or
control siRNA (con), treated with E2 (+E2) or vehicle, and exposed
to ionizing radiation. Relative tail moment is shown. (E) NBS1
mediates E2 protection from IR mediated apoptotic cell death.
MCF10A, MCF10A stably expressing ER (10AER+), and human
breast cancer cell lines were transfected with siRNA to Mre11
(siM), Rad50 (siR), NBS1 (siN) or control siRNA (con), treated with
E2 (+E2) or vehicle, and exposed to ionizing radiation. Percent of
apoptotic cells is shown. Error bars indicate SEM of three
independent experiments. |
To determine if NBS1 induction was mediated by
transcription, we transiently transfected a reporter construct
containing the 5′ flanking, exon 1 and intron 1 regions of the gene
into human breast cancer cell lines prior to E2 and IR treatment.
As shown in Fig. 3A, the
combination of E2 and IR induced reporter activity by 5-fold in
ER+ MCF7 cells (p<0.01). Deletion of the proximal
promoter region had no effect on reporter activity, but removal of
intron 1 completely abolished luciferase expression. These effects
were not observed in ER− MDA-MB-231 cells. In
silico analysis of potential transcription factor binding sites
in NBS1 intron 1 mediating E2 and DNA damage responses revealed a
myc and p53 site in close proximity. To determine relative
occupancy of the myc and p53 binding sites in the NBS1 intron 1 in
response to E2 and IR we performed chromatin immunoprecipitation.
As shown in Fig. 3B, the
combination of E2 and IR enhanced binding of c-myc to this region
of NBS1 intron 1 by 4-fold in ER+ human breast
epithelial cell lines (p<0.02). E2 or IR alone had no effect on
c-myc binding to the NBS1 intron 1. ER− cells showed no
binding of c-myc to the NBS1 intron 1. Similarly the combination of
E2 and IR induced p53 binding to this region by 4–7-fold in
ER+ cells (p<0.01; Fig.
3C). E2 or IR alone had no effect on p53 binding to NBS1 intron
1. ER− cells showed no binding of p53 to the NBS1 intron
1. NBS1 intron 1 activity was strongly induced by the combination
of E2 and IR (2–6-fold in ER+ cells; p<0.05; Fig. 3D). E2 or IR alone had no effect on
NBS1 intron 1 activity and no induction was observed in
ER− cells. Mutation of the myc or p53 binding sites
abolished the inductive effects of E2 and IR on NBS1 intron 1
activity (Fig. 3E and F).
Transient overexpression of c-myc or p53 alone failed to activate
NBS1 intron 1 activity (Fig. 3G).
These results indicate that E2 and IR were required to activate the
NBS1 intron 1 via cooperative c-myc and p53 binding to their
cognate binding sites.
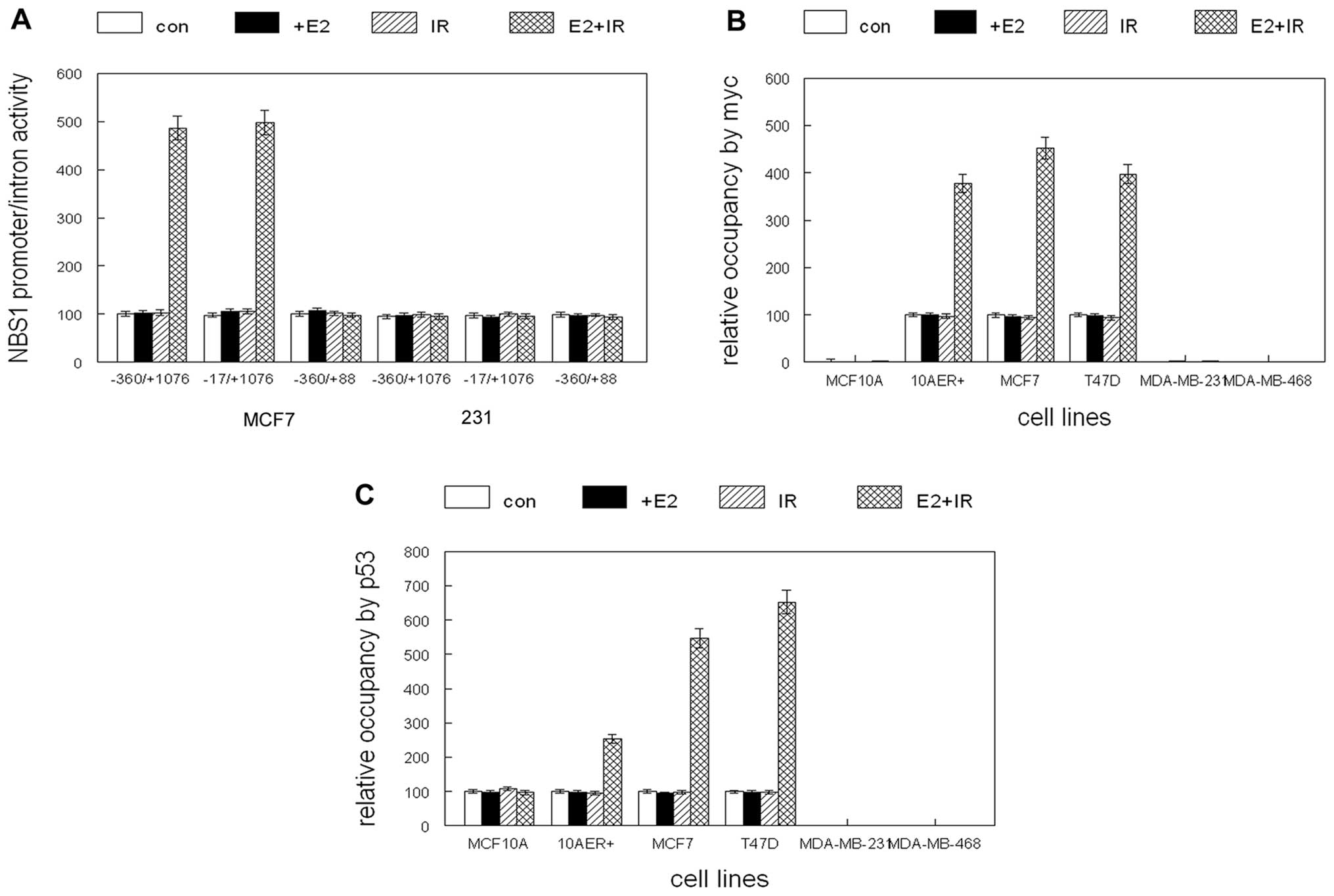 | Figure 3(A) Intron 1 mediates E2 and IR
induction of NBS1 gene expression. Constructs containing the NBS1
promoter, exon 1 and intron 1 (−360/+1076), lacking the promoter
(−17/+1076) or lacking intron 1 (−360/+88) were fused to the
luciferase reporter and transiently transfected into MCF7 or
MDA-MB-231 cells. (B) c-myc occupancy of NBS1 intron 1 is induced
by the combination of E2 and ionizing radiation in ER+
cells. MCF10A, MCF10A stably expressing ER (10AER+), and
human breast cancer cell lines were treated with E2 (+E2), ionizing
radiation (IR), combined E2 and IR (E2+IR), or vehicle (con) and
subjected to chromatin immunoprecipitation. Relative occupancy of
intron 1 by c-myc is shown. (C) p53 occupancy of NBS1 intron 1 is
induced by the combination of E2 and ionizing radiation in
ER+ cells. MCF10A, MCF10A stably expressing ER
(10AER+), and human breast cancer cell lines were
treated with E2 (+E2), ionizing radiation (IR), combined E2 and IR
(E2+IR), or vehicle (con) and subjected to chromatin
immunoprecipitation. Relative occupancy of intron 1 by p53 is
shown. (D) NBS1 intron activity is induced by the combination of E2
and ionizing radiation in ER+ cells. MCF10A, MCF10A
stably expressing ER (10AER+), and human breast cancer
cell lines were transfected with the luciferase reporter construct
and treated with E2 (+E2), ionizing radiation (IR), combined E2 and
IR (E2+IR) or vehicle (con). Relative luciferase activity is shown.
(E) Mutation of the E box site in the NBS1 intron 1 inhibits
induction of activity by the combination of E2 and IR. MCF10A,
MCF10A stably expressing ER (10AER+), and human breast
cancer cell lines were transfected with the luciferase reporter
construct containing a mutation in the E box site and treated with
E2 (+E2), ionizing radiation (IR), combined E2 and IR (E2+IR) or
vehicle (con). Relative luciferase activity is shown. (F) Mutation
of the p53 binding site in the NBS1 intron 1 inhibits induction of
activity by the combination of E2 and IR. MCF10A, MCF10A stably
expressing ER (10AER+), and human breast cancer cell
lines were transfected with luciferase reporter construct
containing a mutation in the p53 binding site and treated with E2
(+E2), ionizing radiation (IR), combined E2 and IR (E2+IR) or
vehicle (con). Relative luciferase activity is shown. (G) c-myc and
p53 individually fail to activate NBS1 intron 1. MCF10A, MCF10A
stably expressing ER (10AER+), and human breast cancer
cell lines were transfected with the luciferase reporter construct
and expression vectors for c-myc, p53 or control vector (con).
Error bars indicate SEM of three independent experiments. |
We previously determined that the protective effects
of E2 on IR induced DNA damage was dependent on the epigenetic
coactivator CBP (16). To
determine if the coactivators CBP and SRC1 were recruited to the
myc and p53 binding sites of the NBS1 intron 1, we performed
chromatin immunoprecipitation. As shown in Fig. 4A and B, the combination of E2 and
IR recruited CBP and SRC1 to this region of intron 1 in
ER+ cells (2–3-fold increased occupancy; p<0.04).
Corresponding acetylation of histone H3 in this region was
increased by 2–3-fold which reduced nucleosomal occupancy by 90%.
E2 or IR alone did not increase coactivator occupancy of this
region and no increase in CBP or SRC1 binding was observed
following E2 and IR treatment of ER− cells. However,
transient overexpression of CBP induced NBS1 intron 1 activity by
2–5-fold and SRC1 overexpression induced intron activity by
2–4-fold in both ER+ and ER− cell lines
(p<0.01; Fig. 4C). Mutation of
the myc or p53 binding sites abolished the ability of CBP or SRC1
to induce NBS1 intron 1 activity (Fig.
4D and E). Constitutive overexpression of p53 induced apoptosis
in human breast cancer cell lines (data not shown), but stable
c-myc expression in combination with IR induced endogenous NBS1
expression by 3–5-fold in ER+ cell lines (Fig. 4F). CBP or SRC1 stable
overexpression was sufficient to induce NBS1 gene expression by
2–4-fold in ER+ human breast cancer cell lines (Fig. 4G). These results indicate that E2
and IR recruited coactivators to the myc and p53 binding sites of
the NBS1 intron 1. Coactivator induction of NBS1 gene expression
was dependent on these sites, and c-myc functionally substituted
for E2 treatment in ER+ cells. CBP and SRC1 functionally
substituted for E2 and IR induction of NBS1 gene expression,
indicating that the coactivators were sufficient to reproduce these
effects.
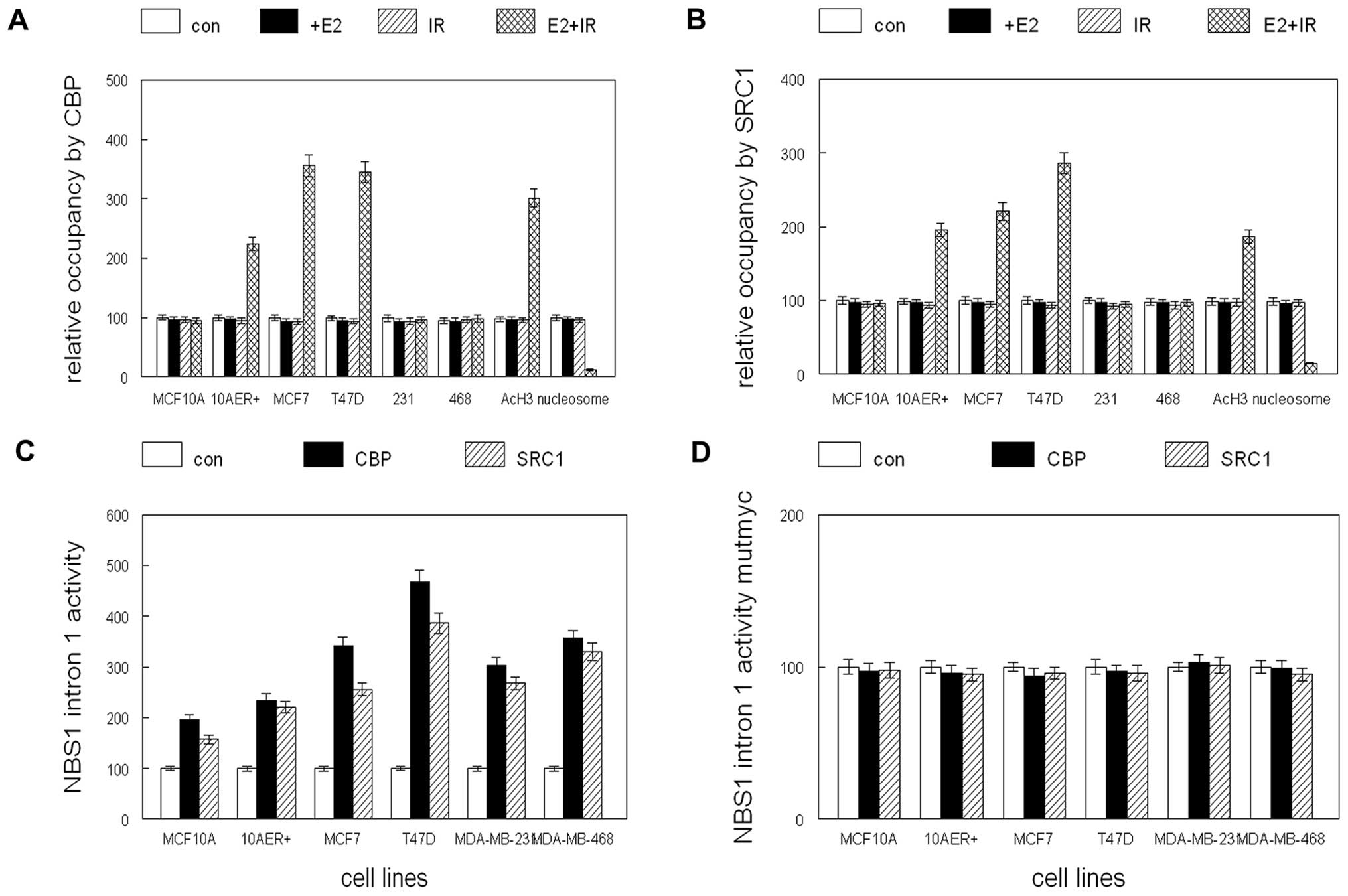 | Figure 4CBP and SRC1 coactivators are
recruited to the NBS1 intron 1 by the combination of E2 and
ionizing radiation. (A) MCF10A, MCF10A stably expressing ER
(10AER+), and human breast cancer cell lines were
treated with E2 (+E2), ionizing radiation (IR), combined E2 and IR
(E2+IR) or vehicle (con) and subjected to chromatin
immunoprecipitation. Relative occupancy of intron 1 by CBP, AcH3,
or nucleosomes is shown. (B) MCF10A, MCF10A stably expressing ER
(10AER+), and human breast cancer cell lines were
treated with E2 (+E2), ionizing radiation (IR), combined E2 and IR
(E2+IR) or vehicle (con) and subjected to chromatin
immunoprecipitation. Relative occupancy of intron 1 by SRC1, AcH3,
or nucleosomes is shown. (C) CBP and SRC1 activate NBS1 intron 1
activity. MCF10A, MCF10A stably expressing ER (10AER+)
and human breast cancer cell lines were transiently transfected
with luciferase reporter construct and expression vectors for CBP,
SRC1 or control vector (con). (D) Mutation of the E box site
inhibits coactivator mediated induction of NBS1 intron 1 activity.
MCF10A, MCF10A stably expressing ER (10AER+) and human
breast cancer cell lines were transfected with luciferase reporter
construct containing a mutation in the E box site and expression
vectors for CBP, SRC1 or control vector (con). (E) Mutation of the
p53 binding site inhibits coactivator mediated induction of NBS1
intron 1 activity. MCF10A, MCF10A stably expressing ER
(10AER+), and human breast cancer cell lines were
transfected with luciferase reporter construct containing a
mutation in the p53 binding site and expression vectors for CBP,
SRC1 or control vector (con). (F) Stable overexpression of c-myc
substitutes for E2 treatment in IR mediated induction of NBS1 gene
expression. The human breast cancer cell lines MCF7 and T47D were
stably transfected with c-myc or control (con) expression vector
and exposed to ionizing radiation (IR). c-myc, NBS1, and β-actin
expression was determined by western blot analysis. (G) Stable
overexpression of CBP or SRC1 substitutes for E2 and IR mediated
induction of NBS1 expression. The human breast cancer cell lines
MCF7 and T47D were stably transfected with CBP, SRC1, or control
(con) expression vectors. CBP, SRC1, NBS1, and β-actin expression
was determined by western blot analysis. |
Discussion
Our previously published studies indicated that E2
treatment decreased DNA damage and improved survival of
ER+ human breast cancer cell lines following IR
treatment (16). We now
demonstrate that the combination of E2 and IR treatment induces
NBS1 expression in ER+ but not ER− human
breast cancer cell lines. While inhibition of gene products in the
MRN complex inhibited DNA repair, NBS1 was responsible for
mediating the anti-apoptotic effects of E2 in irradiated
ER+ breast cancer cell lines. A previous study
demonstrated that cells from mice expressing a C-terminal deleted
NBS1 exhibited decreased apoptosis (18). Additionally E2 was previously shown
to sustain the growth of irradiated breast cancer cell lines
(19). This effect was due to
inactivation of p21 which sustained Rb hyperphosphorylation
allowing increased cell cycle progression in irradiated cells.
These studies demonstrate important control of cell cycle and
apoptosis by E2 and NBS1 in human breast cancer cells.
Our results demonstrated that induction of c-myc by
E2 and p53 by IR was required for increased NBS1 expression in
ER+ human breast cancer cell lines. The lack of NBS1
induction by E2 or IR alone may be due to the proximity of the myc
and p53 response elements in the NBS1 intron 1 (20). p53 has been shown to bind to half
sites in target gene promoters (21). Myc or p53 overexpression alone was
not sufficient to induce NBS1 expression, but myc expression could
substitute for E2 in irradiated cells. A previous report
demonstrated that ER could bind directly to p53 and repress the
function of the tumor suppressor (22). This interaction may provide an
additional mechanism by which activated ER may inactivate p53 to
facilitate cell cycle progression and inhibit apoptosis.
Our results demonstrated that CBP and SRC1
coactivators were recruited to the myc and p53 response elements in
the NBS1 intron 1 and were sufficient to activate gene expression.
Previous studies demonstrated that SRC1 physically interacts with
p53 and potentiated p53 mediated transactivation (23). The coactivators CBP/p300 associate
with and acetylate p53, and results in acetylation of histones in
p53 target gene promoters (24–26).
These studies demonstrate the importance of coactivator function in
mediating the effects of DNA damage response in human breast cancer
cells.
Mutations in the NBS1 gene have been associated with
increased risk of breast cancer (9,10,27,28).
Persistent radiation induced NBS1 foci has been associated with
chromosomal instability and increased breast cancer risk (13). In mice, NBS1 null mutation is
embryonic lethal but heterozygosity renders mice susceptible to
tumor formation (5). However,
mammary tumors are uncommon in mouse strains with reduced NBS1
function (6). Defects in cellular
proliferation were noted in the cells of NBS1 deficient mice in
previous studies (7). Loss of p53
has been shown to greatly increase tumorigenesis in NBS1 mutant
mice, suggesting that p53 mediated DNA damage response may be
responsible for apoptosis and increased tumor latency (29). A previous study demonstrated
nuclear export of NBS1 following ionizing radiation as a mechanism
of downregulating the DNA damage response (30). Loss of NBS1 has been shown to
induce supernumerary centrosomes similar to those observed in BRCA1
deficient cells, leading to increased chromosomal instability
(31). These studies demonstrate
that impaired NBS1 function can result in cellular proliferation
defects leading to increased tumor latency. It is interesting to
speculate that tumorigenic clones that escape defective
proliferation may be more aggressive and metastatic due to
chromosomal aberrations induced by diminished NBS1 function.
Acknowledgements
This study was supported by Susan G.
Komen for the Cure award BCTR0504295 and Department of Defense
Breast Cancer Research Program award W81XWH-10-1-0081 to DLC.
References
|
1
|
Hakem R: DNA damage repair; the good, the
bad, and the ugly. EMBO J. 27:589–605. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Niida H and Nakanishi M: DNA damage
checkpoints in mammals. Mutagenesis. 21:3–9. 2006. View Article : Google Scholar
|
|
3
|
Thoms KM, Kuschal C and Emmert S: Lessons
learned from DNA repair defective syndromes. Exp Dermatol.
16:532–544. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sung P and Klein H: Mechanism of
homologous recombination: mediators and helicases take on
regulatory functions. Nature Rev Mol Cell Biol. 7:739–750. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kang J, Bronson RT and Xu Y: Targeted
disruption of NBS1 reveals its roles in mouse development and DNA
repair. EMBO J. 21:1447–1455. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dumon-Jones V, Frappart PO, Tong WM, et
al: Nbn heterozygosity renders mice susceptible to tumor formation
and ionizing radiation induced tumorigenesis. Cancer Res.
63:7263–7269. 2003.PubMed/NCBI
|
|
7
|
Frappart PO, Tong WM, Demuth I, et al: An
essential function for NBS1 in the prevention of ataxia and
cerebellar defects. Nat Med. 11:538–544. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bartkova J, Tommiska J, Oplustilova L, et
al: Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex
in human breast cancer: MRE11 as a candidate familial cancer
predisposing gene. Mol Oncol. 2:296–316. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bogdanova N, Feschchenko S, Schurmann P,
et al: Nijmegen breakage syndrome mutations and risk of breast
cancer. Int J Cancer. 122:802–806. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lu J, Wei Q, Bondy ML, et al:
Polymorphisms and haplotypes of the NBS1 gene are associated with
risk of sporadic breast cancer in non-Hispanic white women less
than or equal to 55 years. Carcinogenesis. 27:2209–2216. 2006.
|
|
11
|
Hsu HM, Wang HC, Chen ST, Hsu GC, Shen CY
and Yu JC: Breast cancer risk is associated with the genes encoding
the DNA double strand break repair Mre11/Rad50/Nbs1 complex. Cancer
Epidemiol Biomarkers Prev. 16:2024–2032. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Soderlund K, Stal O, Skoog L, Rutqvist LE,
Nordenskjold B and Askmalm MS: Intact Mre11/Rad50/Nbs1 complex
predicts good response to radiotherapy in early breast cancer. Int
J Rad Oncol Biol Physics. 68:50–58. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Someya M, Sakata K, Tauchi H, Matsumoto Y,
Nakamura A, Komatsu K and Hareyama M: Association of ionizing
radiation induced foci of NBS1 with chromosomal instability and
breast cancer susceptibility. Rad Res. 166:575–582. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Russo IH and Russo J: Role of hormones in
cancer initiation and progression. J Mammary Gland Biol Neoplasia.
3:49–61. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Goodman RH and Smolik S: CBP/p300 in cell
growth, transformation, and development. Genes Dev. 14:1553–1577.
2000.PubMed/NCBI
|
|
16
|
Crowe DL and Lee MK: New role for nuclear
hormone receptors and coactivators in regulation of BRCA1 mediated
DNA repair in breast cancer cell lines. Breast Cancer Res. 8:1–12.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Eggleston P and Zhao Y: A sensitive and
rapid assay for homologous recombination in mosquito cells: impact
of vector topology and implications for gene targeting. BMC Genet.
2:21–29. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stracker TH, Morales M, Couto SS, Hussein
H and Petrini JHJ: The carboxy terminus of NBS1 is required for
induction of apoptosis by the MRE11 complex. Nature. 447:218–222.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Toillon RA, Magne N, Laios I, et al:
Estrogens decrease gamma ray induced senescence and maintain cell
cycle progression in breast cancer cells independently of p53. Int
J Rad Oncol Biol Physics. 67:1187–1200. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chiang YC, Teng SC, Su YN, Hsieh FJ and Wu
KJ: c-myc directly regulates the transcription of the NBS1 gene
involved in DNA double strand break repair. J Biol Chem.
278:19286–19291. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cai BH, Chen JY, Lu MH, Chang LT, Lin HC,
Chang YM and Chao CF: Functional four base A/T gap core sequence
CATTAG of p53 response elements specifically bound tetrameric p53
differently than two base A/T gap core sequence CATG bound both
dimeric and tetrameric p53. Nuc Acids Res. 37:1984–1990. 2009.
View Article : Google Scholar
|
|
22
|
Liu W, Konduri SD, Bansai S, et al:
Estrogen receptor alpha binds p53 tumor suppressor protein directly
and represses its function. J Biol Chem. 281:9837–9840. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee SK, Kim HJ, Kim JW and Lee JW: Steroid
receptor coactivator 1 and its family members differentially
regulate transactivation by the tumor suppressor protein p53. Mol
Endocrinol. 13:1924–1933. 1999. View Article : Google Scholar
|
|
24
|
Barlev NA, Liu L, Chehab NH, Mansfield K,
Harris KG, Halazonetis TD and Berger SL: Acetylation of p53
activates transcription through recruitment of coactivators/histone
acetyltransferases. Mol Cell. 8:1243–1254. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mujtaba S, He Y, Zeng L, et al: Structural
mechanism of the bromodomain of the coactivator CBP in p53
transcriptional activation. Mol Cell. 13:251–263. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Watts GS, Oshiro MM, Junk DJ, Wozniak RJ,
Watterson S, Domann FE and Futscher BW: The acetyltransferase
p300/CBP associated factor is a p53 target gene in breast tumor
cells. Neoplasia. 6:187–194. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Walsh T and King MC: Ten genes for
inherited breast cancer. Cancer Cell. 11:103–105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nowak J, Mosor M, Ziolkowska I, et al:
Heterozygous carriers of the I171V mutation of the NBS1 gene have a
significantly increased risk of solid malignant tumors. Eur J
Cancer. 44:627–630. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kang J, Ferguson D, Song H, Bassing C,
Eckersdorff M, Alt FW and Xu Y: Functional interaction of H2AX,
NBS1, and p53 in ATM dependent DNA damage responses and tumor
suppression. Mol Cell Biol. 25:661–670. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vissinga CS, Yeo TC, Warren S, Brawley JV,
Phillips J, Cerosaletti K and Concannon P: Nuclear export of NBN is
required for normal cellular responses to radiation. Mol Cell Biol.
29:1000–1006. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shimada M, Sagae R, Kobayashi J, Habu T
and Komatsu K: Inactivation of the Nijmegen breakage syndrome gene
leads to excess centrosome duplication via the ATR/BRCA1 pathway.
Cancer Res. 69:1768–1775. 2009. View Article : Google Scholar : PubMed/NCBI
|