Introduction
The p53 protein, a transcription factor which exerts
significant tumor-suppressive activity, regulates the expression of
various genes that induce different anti-carcinogenesis mechanisms
(1,2). These include cell cycle arrest,
apoptosis, senescence and DNA repair in response to genotoxic
stress, depending on the type and severity of stress and the
cellular context. Since the activation of p53 can lead to
irreversible cellular injury, p53 protein levels are tightly
regulated. Under basal conditions, the p53 protein has a short
half-life, due to its binding to the murine double minute (MDM)2
protein, an E3 ubiquitin ligase that targets it for 26S proteasomal
degradation (3). MDM2 binding also
hinders the interaction between p53 and transcriptional
co-activators, such as p300 and the CREB-binding protein (CBP),
thus rendering p53 inactive as a transcription factor (4). Due to its central role in cell death
and cell cycle arrest, the activation of p53 has become an
attractive therapeutic approach. In fact, currently used
anti-cancer treatments, including chemotherapy and radiotherapy,
induce p53-dependent apoptosis in cancer cells with an intact p53
gene. These therapeutic methods induce genomic DNA damage,
resulting in the activation of p53, which makes cancer cells with
rapid DNA replication more susceptible to these treatments compared
to non-transformed cells (5).
However, these treatments may cause DNA damage to normal cells and,
in fact, certain studies have reported the development of tumors,
such as leukemia, bladder, kidney and breast cancers, following
such anticancer treatments (6,7).
Therefore, small molecules such as MDM2 inhibitors, that induce p53
activation without causing DNA damage, may represent promising
anticancer drugs, since they reduce the risk of secondary tumor
development (8,9). Nutlin-3, a cis-imidazoline analog,
was the first selective MDM2 inhibitor to be discovered (10). The structure of nutlin-3 is similar
to that of amino acids in the binding domain between p53 and MDM2.
It binds to the p53-binding pocket of MDM2, thereby displacing p53
(from MDM2) and upregulating the p53 protein. As shown in previous
studies using the murine xenograft model, the treatment of cancer
cells with nutlin-3 triggers substantial p53-dependent cell cycle
arrest and apoptosis in vitro and exhibits antitumor
efficacy (10,11). It is noteworthy that MDM2
inhibitors, including nultin-3, are not toxic to normal cells
(12,13).
The Ras-Raf-MEK1/2-ERK1/2 pathway, which is
generally activated by growth factors, controls various cellular
responses, such as proliferation, migration, differentiation and
death (14,15). Upon growth factor stimuli, Ras is
activated via receptor tyrosine kinases (RTKs) and subsequently
activates Raf, MAPK/ERK kinase (MEK) and extracellular
signal-regulated kinase (ERK), leading to the activation of a
number of transcription factors that orchestrate various cellular
responses. The Ras-Raf-MEK-ERK signaling cascade has been
demonstrated to functionally interact with the p53 pathway. For
example, ERK1/2 is required for the phosphorylation of p53 and
apoptosis induced by cisplatin, colcemid, resveratrol and
benzo(a)pyrene in ovarian cancer, fibroblast, epidermal and
hepatocellular carcinoma cells, respectively (16–19).
However, it has also been reported that p53 is capable of
modulating ERK1/2 activation, although its effects on ERK1/2
activity do not appear to be consistent. In human bladder cancer,
osteosarcoma and colon cancer cells, p53 activates the
Ras-Raf-MEK1/2-ERK1/2 pathway via the transcriptional induction of
heparin-binding epidermal growth factor-like growth factor (HB-EGF)
and discoidin domain receptor 1 (DDR1), to protect cancer cells
against p53-dependent apoptosis (20–22).
By contrast, p53 suppresses ERK1/2 activity by activating
phosphatase genes, such as phosphatase of activated cells 1 (PAC1),
dual-specificity phosphatase 5 (DUSP1) and mitogen-activated
protein kinase (MAPK) phosphatase 1 (MKP1), which can
dephosphorylate ERK1/2 (23–25).
Since PAC1, DUSP1 and MKP1 can block the anti-apoptotic and cell
cycle progression effects of ERK1/2, p53 induces apoptosis and cell
cycle arrest via the upregulation of these phosphatase genes.
ERK1/2 thus appears to act as an upstream activator of p53
phosphorylation or a downstream effector of p53 that counteracts
p53-induced apoptosis, depending on the cell type and
p53-activating stimuli, although the mechanisms involved remain to
be elucidated.
Recent studies have reported that the combined
treatment with nutlin-3 and a MEK inhibitor synergistically induces
apoptosis in AML cells in which the MAPK pathway is aberrantly
activated, and which overexpress MDM2 (26,27).
This synergistic activation of apoptosis occurred, in part, via the
phosphorylation of FOXO3a, leading to an increase in pro-apoptotic
genes, such as p53 upregulated modulator of apoptosis (PUMA) and
BIM. These findings suggest that the growth-suppressive activity of
nutlin-3 may also be functionally counteracted by the MAPK pathway.
However, the correlation between nutlin-3-induced p53 and MAPK
activity remains unclear. To address this issue, we analyzed MAPK
activation and the underlying mechanisms in nutlin-3-treated U2OS
cells, a human osteosarcoma cell line with established
susceptibility to nutlin-3.
Materials and methods
Reagents
Nutlin-3 and U0126 were purchased from Tocris
(Bristol, UK). Pifithrin (PFT)-α and -μ,
2′,7′-dichlorodihydrofluorescein diacetate (H2DCF-DA) and
2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) were obtained from
Sigma-Aldrich Inc. (St. Louis, MO, USA). MitoSOX™ Red reagent was
obtained from Invitrogen (Carlsbad, CA, USA).
Cell culture
U2OS human osteosarcoma cells were cultured in DMEM
supplemented with 10% heat-inactivated fetal bovine serum (Hyclone,
Logan, UT, USA), 100 U/ml penicillin (Hyclone) and glutamate
(Invitrogen) at 37°C with 5% CO2.
Antibodies
Mouse antibodies against phospho-p42/44, p53 and
GAPDH were purchased from Santa Cruz Biotechnology (Santa Cruz, CA,
USA). Rabbit anti-p42/44 and mouse anti-phospho-MEK1/2 were
obtained from Cell Signaling Technology (Boston, MA, USA). Rabbit
anti-MEK1 and anti-MEK2 were obtained from Epitomics (Burlingame,
CA, USA). Secondary antibodies against rabbit or mouse IgG were
obtained from Sigma-Aldrich Inc.
Transfection of small interfering RNAs
(siRNAs)
p53 siRNA was obtained from Santa Cruz Biotechnology
and siRNAs against MEK1, MEK2, ERK1, ERK2 and epidermal growth
factor receptor (EGFR) were purchased from Sigma-Aldrich Inc. siRNA
transfection was carried out using Lipofectamine™ RNAiMAX
(Invitrogen), following the manufacturer’s instructions.
Immunoblot analysis
Cells treated as indicated in figures were lysed
with RIPA buffer (Cell Signaling Technology) containing protease
inhibitor cocktail (Roche, Basel, Switzerland) or with 1X SDS
sample buffer. Equal amounts of protein were separated by
SDS-polyacrylamide gel electrophoresis and transferred onto
nitrocellulose membranes (Millipore, Billerica, MA, USA). The
membranes were incubated in 5% non-fat dried milk in TBS-T
(Tris-buffered saline with 0.05% Tween-20) containing primary
antibodies and then incubated with secondary antibodies, followed
by chemiluminescence (ECL, GE Healthcare, Buckinghamshire,
UK)-based detection.
Quantitative real-time RT-PCR
(qRT-PCR)
Total RNA was isolated using RNAiso Plus (Takara Bio
Inc., Shiga, Japan) and first-strand cDNA was generated using the
PrimeScript™ RT reagent kit (Takara Bio Inc.). Quantitative
real-time RT-PCR was performed using SYBR Premix Ex Taq (Takara Bio
Inc.) and the ABI 7300 Real-Time PCR system (Applied Biosystems,
Carlsbad, CA, USA). All reactions were performed following the
manufacturer’s instructions. Relative quantification was performed
using the ΔΔCt method with GAPDH as the endogenous control
(28).
Apoptosis assay
Apoptosis was assessed by measuring the number of
hypodiploidic cells and the translocation of phosphatidylserine
using flow cytometry (FACSCalibur; BD Biosciences, San Jose, CA,
USA). To measure the number of hypodiploidic cells, cells treated
as indicated in Fig. 5 were
harvested, fixed in ice-cold 70% ethanol for 2 h and then stained
with propidium iodide (PI, 0.2 mg/ml). The stained cells were
analyzed on a FACSCalibur and cell cycle distribution was
calculated by CellQuest and ModFit software (BD Biosciences). For
determination of phosphatidylserine translocation, cells were
stained with PI and Annexin V using the ApoScan kit (BioBud,
Gyunggido, Korea), followed by flow cytometry analysis as described
previously (29).
Isolation of cytosolic and mitochondrial
fractions
Cells treated as indicated in Fig. 4 were resuspended in ice-cold IBc
buffer (0.1 M Tris-MOPS, 0.5 M EGTA/Tris, 1 M sucrose, pH 7.4)
containing a protease inhibitor cocktail (Roche). The cells were
homogenized using dounce homogenizers and centrifuged at 6,000 × g
for 10 min at 4°C to remove unlysed cells and nuclei. The
supernatant was collected and centrifuged at 7,000 × g for 10 min
at 4°C. The clear fraction was saved as the cytosolic fraction and
the pellet was resuspended in IBc buffer, washed again and
centrifuged at 10,000 × g for 10 min at 4°C. The pellet was
resuspended in RIPA buffer and used as the mitochondrial
fraction.
Measurement of reactive oxygen species
(ROS)
To measure intracellular ROS levels and
mitochondrial ROS, cells were incubated in the presence of 20
μM H2DCF-DA dye and 5 μM MitoSOX Red reagent working
solution (Invitrogen), respectively, for 15 min. After the cells
were washed twice in pre-warmed PBS, they were examined under a
fluorescence inverted microscope (Olympus IX71, Tokyo, Japan) and
the intensity of the fluorescence was measured by flow
cytometry.
Results
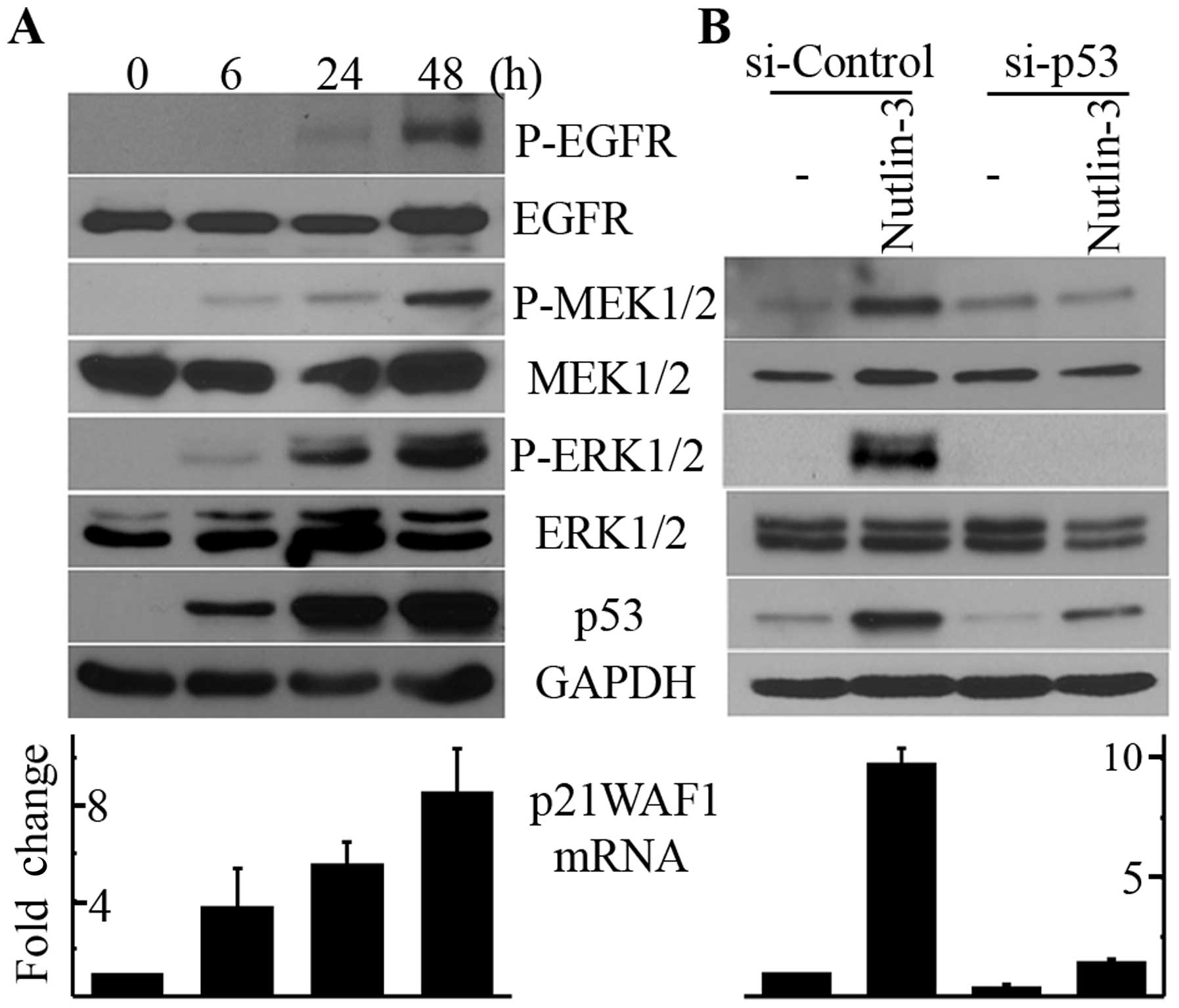
Nutlin-3 induces the phosphorylation of
MEK1/2-ERK1/2 in a p53-dependent manner
To determine whether the upregulation of p53 by
nutlin-3 induces the phosphorylation of ERK1/2, we treated U2OS
cells with 20 μM nutlin-3, treatment conditions that have
been reported to induce apoptosis in this cell line (10). As shown in Fig. 1A, treatment with nutlin-3 resulted
in the accumulation of p53 protein and p21WAF1 mRNA in a
time-dependent manner, indicating that p53 was activated by
nutlin-3. Along with p53 activation, the phosphorylation of ERK1/2
and MEK1/2 was induced by nutlin-3 as well, which was also
dependent on the incubation time (Fig.
1A). The activation of p53 and the phosphorylation of
MEK1/2-ERK1/2 commenced after 6 h of nutlin-3 treatment and were
still observed after 48 h of treatment. Therefore, to elucidate the
mechanism behind this phenomenon, we used 20 μM nutlin-3 and
a 24-h incubation time for the following experiments. We then
determined whether the nutlin-3-induced MEK1/2 and ERK1/2
phosphorylation was the direct effect of p53 activation. As shown
in Fig. 1B, the downregulation of
p53 by RNA interference almost completely inhibited the
nutlin-3-induced phosphorylation of MEK1/2 and ERK1/2 as well as
the increase in p21WAF1 mRNA levels. Therefore, it can be concluded
that nutlin-3 induces MEK1/2-ERK1/2 phosphorylation by activating
p53 in U2OS cells.
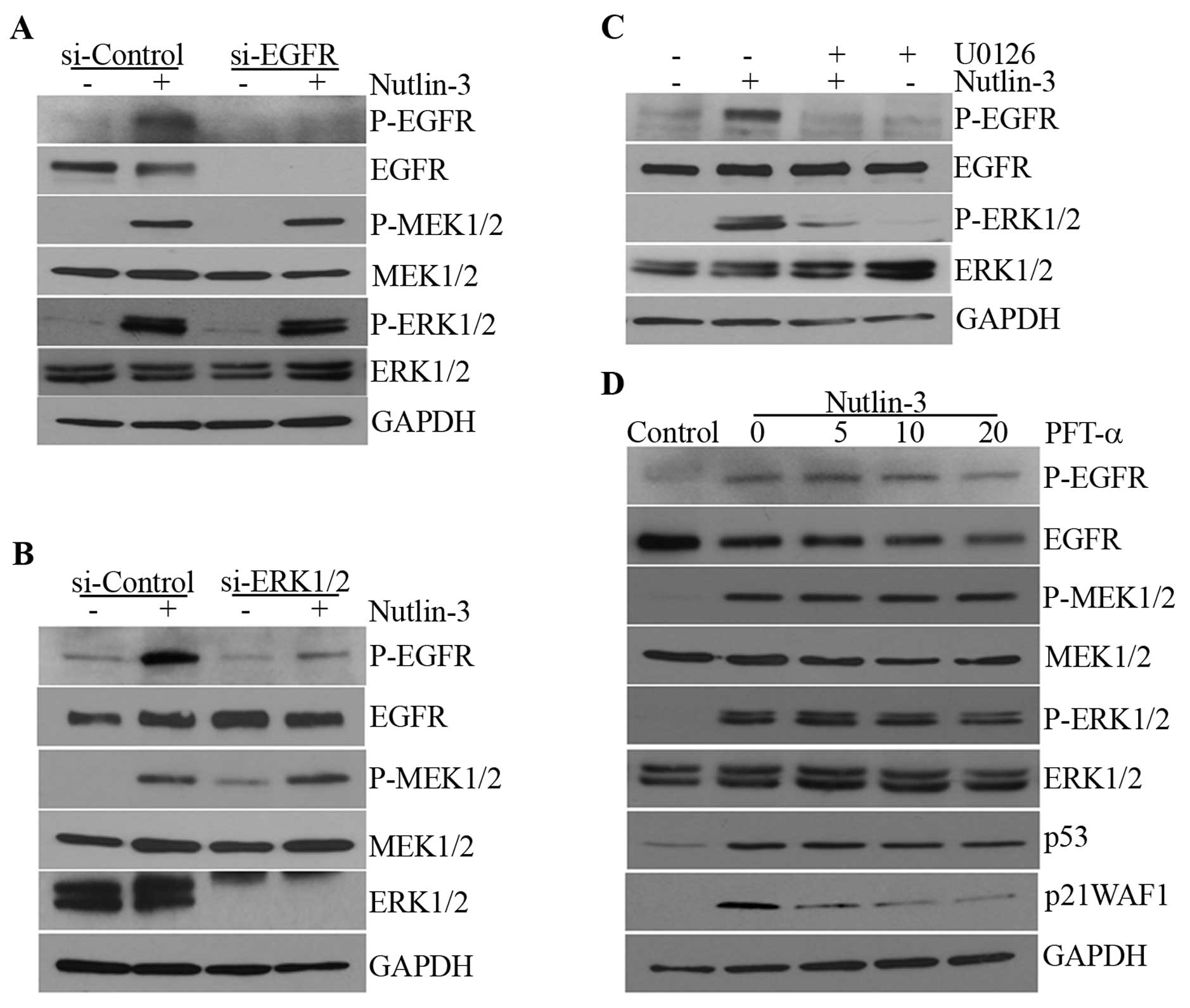
Nutlin-3-induced MEK1/2-ERK1/2 activation
is not dependent on EGFR activation or the transcriptional activity
of p53
Since p53 has been reported to induce EGFR
phosphorylation via the upregulation of EGFR ligands such as HB-EGF
(21) and it has also been argued
that EGFR ligands can be induced downstream of ERK1/2 via early
growth response-1 (EGR-1) (30),
we examined whether nutlin-3 induced the activation of EGFR and
whether this was responsible for the phosphorylation of
MEK1/2-ERK1/2. Nutlin-3 induced the phosphorylation of EGFR as well
as that of MEK1/2-ERK1/2 in a time-dependent manner (Fig. 1A). However, when EGFR
phosphorylation was prevented by EGFR siRNA and AG1478, an EGFR
kinase inhibitor, the phosphorylation of MEK1/2-ERK1/2 was not
affected (Fig. 2A; data not
shown). By contrast, EGFR phosphorylation was almost completely
prevented by ERK1/2 siRNA and U0126, a MEK inhibitor (Fig. 2B and C), suggesting that ERK1/2,
when activated by nutlin-3, induces the phosphorylation of EGFR and
that the transcriptional activity of p53 may not be involved in
this MEK1/2-ERK1/2 phosphorylation. Whereas an increase in p21WAF1
mRNA levels was almost completely inhibited by PFT-α, an inhibitor
of the transcriptional activity of p53, the phosphorylation of
MEK1/2-ERK1/2 was not altered (Fig.
2D). This suggests that nutlin-3 may induce MEK1/2-ERK1/2 and
subsequently, EGFR phosphorylation through a mechanism that is
independent of the transcriptional activity of p53.
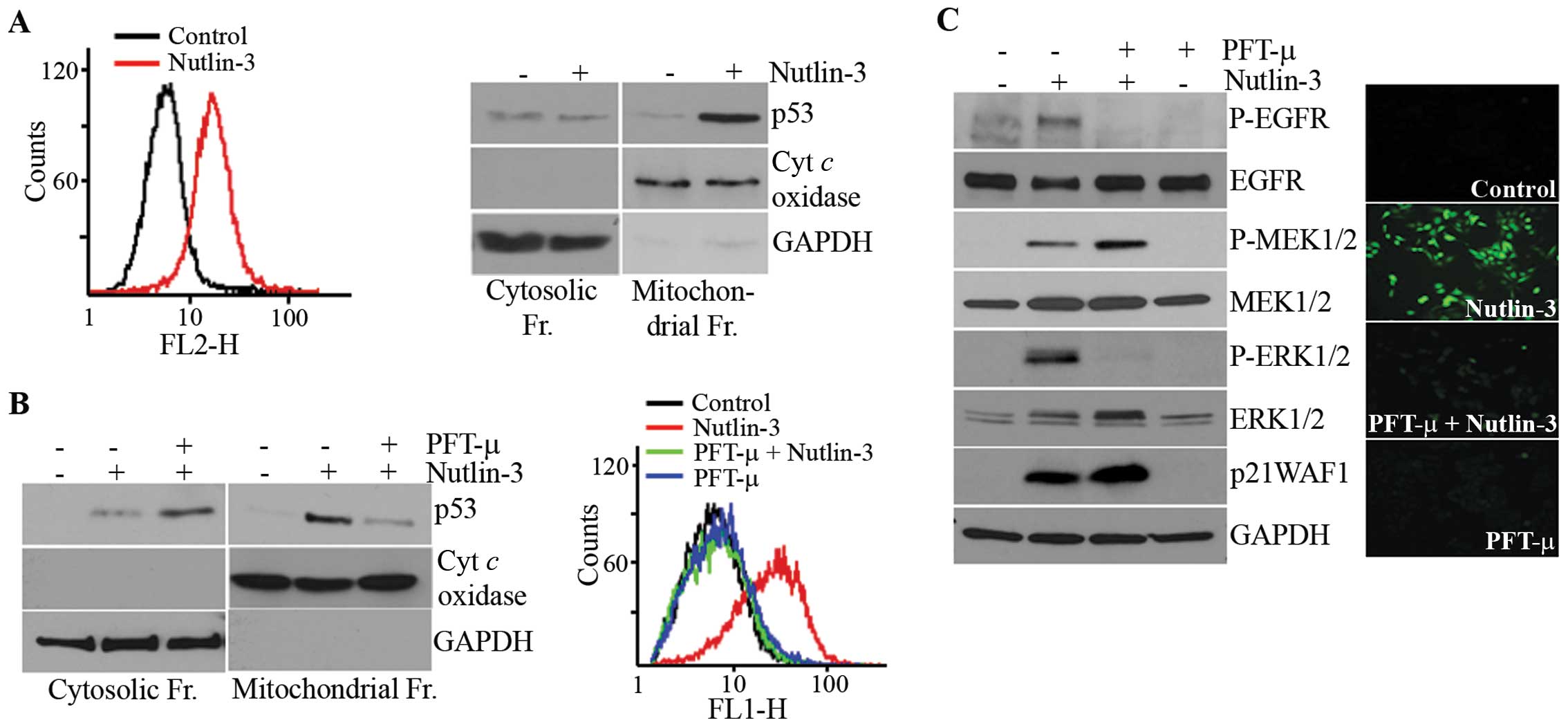
Involvement of ROS in the activation of
MEK1/2-ERK1/2 by nutlin-3
Since ROS have been reported to mediate the
activation of ERK1/2 in various cells (31,32)
and p53 modulates the production of ROS (33), we examined the involvement of ROS
in nutlin-3-induced ERK1/2 phosphorylation. As expected, ROS
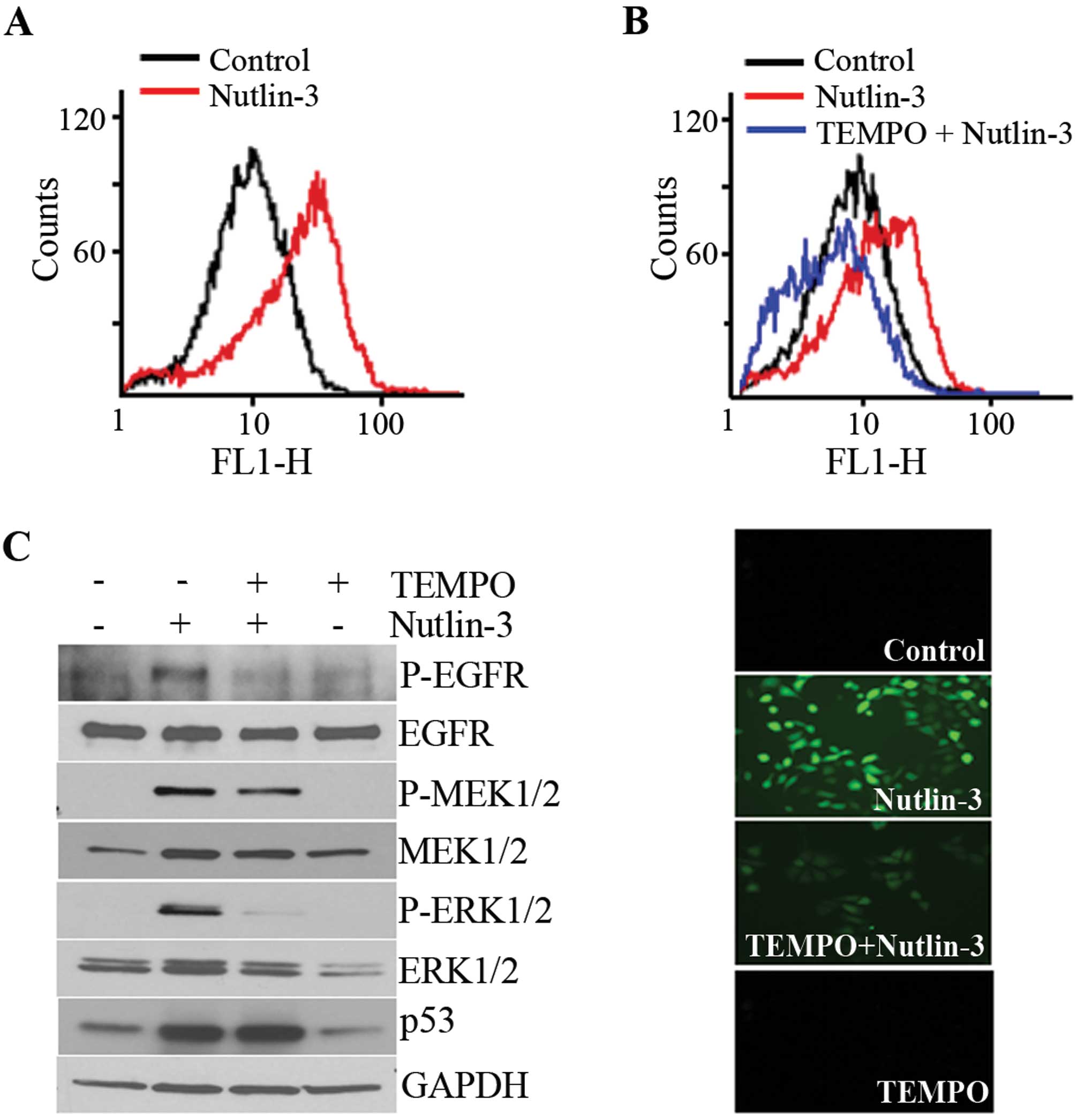
accumulated in the nutlin-3-treated cells (Fig. 3A) and this accumulation was
prevented by a pre-treatment with TEMPO, a ROS scavenger (Fig. 3B). Parallel to its effect on ROS
accumulation, TEMPO almost completely inhibited the phosphorylation
of ERK1/2 and EGFR, but the MEK1/2 phosphorylation induced by
nutlin-3 was only slightly inhibited (Fig. 3C). These findings demonstrated that
the nutlin-3 induced ROS generation and accumulation of ROS
contributed to the phosphorylation of ERK1/2.
Nutlin-3-induced ROS generation and
MEK1/2-ERK1/2 activation are dependent on the mitochondrial
translocation of p53
Since PFT-α had no effect on the nutlin-3-induced
MEK1/2-ERK1/2 phosphorylation (Fig.
2D), ROS accumulation caused by nutlin-3 may be independent of
the transcriptional activity of p53. p53 has been reported to
translocate to the mitochondria (34,35),
the main source of ROS, prompting us to examine the correlation
between ROS accumulation and the mitochondrial translocation of p53
in nutlin-3-treated cells. For this purpose, we used MitoSOX, a
reagent that captures mitochondrial ROS and immunoblot analyses of
subcellular fractions. As shown in Fig. 4A, the nutlin-3-treated cells were
stained with MitoSOX. The results showed that accumulating ROS were
generated from the mitochondria and that nutlin-3 caused the
mitochondrial translocation of p53. Both this mitochondrial p53
translocation and ROS generation were prevented by PFT-μ, an
inhibitor of the mitochondrial translocation of p53 (Fig. 4B). The staining of
PFT-μ-pre-treated cells with H2DCF-DA demonstrated that PFT-μ
completely prevented ROS accumulation, suggesting that
nutlin-3-induced ROS originated solely in mitochondria (Fig. 4B, right panel). It should be noted
that, while the phosphorylation of ERK1/2 and EGFR by nutlin-3 was
almost completely inhibited by PFT-μ pre-treatment, the
phosphorylation of MEK1/2 was not inhibited, but rather potentiated
by PFT-μ. These results demonstrate that nutlin-3 induces the
mitochondrial translocation of p53, which generates ROS in the
mitochondria and that mitochondrial ROS are involved in the
phosphorylation of ERK1/2 but not that of MEK1/2.
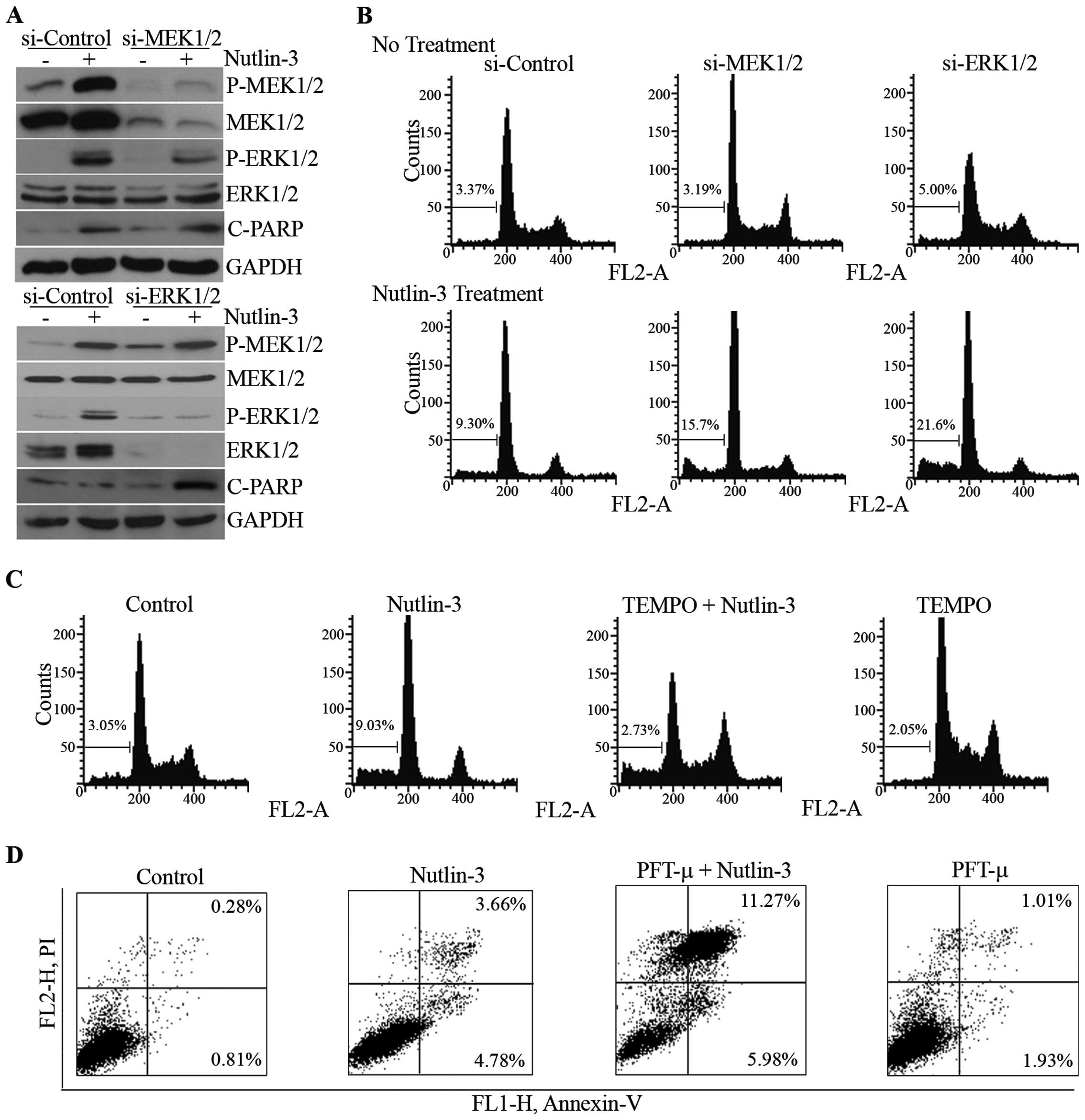
Effect of ERK1/2 inhibition on
nutlin-3-induced cell death
We then attempted to determine the role of
MEK1/2-ERK1/2 phosphorylation on nutlin-3-induced cell death. When
the phosphorylation of MEK1/2 and ERK1/2 was reduced by the
knockdown of MEK1/2 and ERK1/2 mediated by the respective siRNAs
(Fig. 5A), nutlin-3-induced
apoptotic features, such as the cleavage of poly(ADP-ribose)
polymerase-1 (PARP-1) and the accumulation of hypodiploidic cells,
were augmented (Fig. 5A and B).
The effect of the inhibition of ERK1/2 phosphorylation by U0126, a
chemical inhibitor of MEK1/2, was consistent with that of siRNAs
against MEK1/2 and ERK1/2 (data not shown). Furthermore,
pre-treatment with TEMPO and PFT-μ, decreased ERK1/2
phosphorylation, and increased nutlin-3-induced apoptosis (Fig. 5C and D). These results demonstrate
that the MEK1/2-ERK1/2 activation suppresses nutlin-3-induced
apoptosis, suggesting that it may constitute a negative feedback
loop against nutlin-3-induced apoptosis in U2OS cells.
Discussion
In this study, we report that the p53 protein, when
stabilized by nutlin-3 treatment, causes the activation of the
MEK1/2-ERK1/2 pathway that functions as a survival pathway, thereby
constituting a negative feedback loop against p53-induced
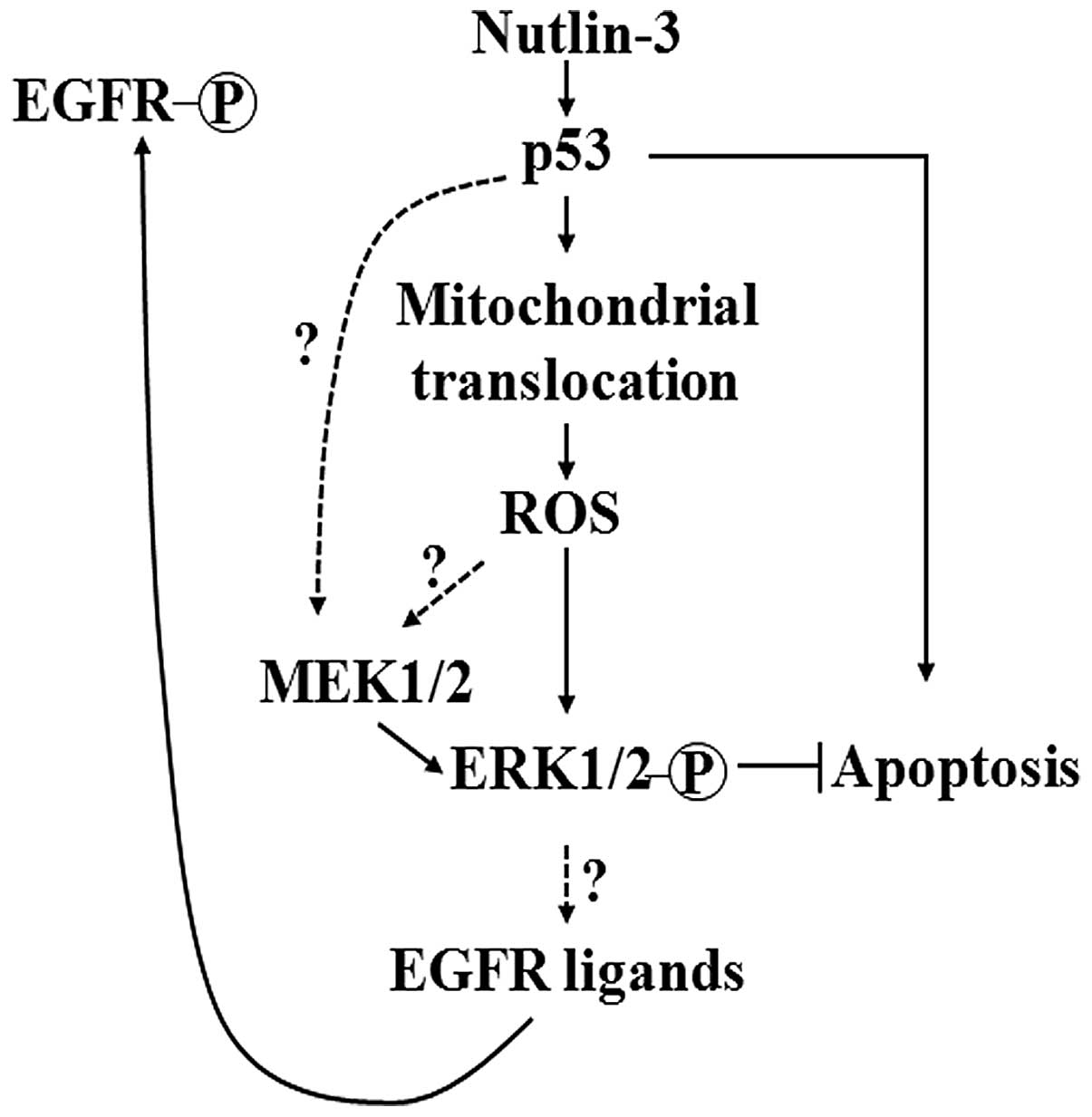
apoptosis. The findings of the present study are summarized in the
form of a schematic diagram, presented in Fig. 6. Since MEK1/2-ERK1/2
phosphorylation occurs downstream of EGFR activation and p53
activates EGFR by increasing the expression of HB-EGF at the
transcription level (21), we
hypothesized that the sequence of events in the nutlin-3-treated
cells would be an increase of p53, followed by an increase of EGFR
ligands, EGFR activation and finally MEK1/2-ERK1/2 phosphorylation.
However, while the phosphorylation of MEK1/2-ERK1/2 was unaffected
by the EGFR inhibition and PFT-α, the phosphorylation of EGFR was
completely prevented by the inhibition of ERK1/2, indicating that
ERK1/2 activation occurs upstream of EGFR activation (Fig. 2). In accordance with this
hypothesis, nutlin-3 induced the transcription of EGFR ligands such
as amphiregulin, epiregulin and HB-EGF and
this induction was suppressed by ERK1/2 inhibitors but not by PFT-α
(data not shown), leading to the assumption that the MEK1/2-ERK1/2
pathway may be activated by nutlin-3-induced p53 in a
transcription-independent manner prior to the phosphorylation of
EGFR. Mitochondrial ROS generation was increased during the
nutlin-3 treatment and ROS accumulation closely correlated with the
mitochondrial translocation of p53. While MEK1/2 activation was not
inhibited or slightly inhibited by PFT-μ or ROS scavengers,
respectively, ERK1/2 appeared to be almost completely inhibited by
these compounds as well as by MEK inhibition (Fig. 3). Collectively, these findings led
us to conjecture that the mitochondrial translocation of p53 and
MEK1/2 activation are central events in nutlin-3-induced ERK1/2
activation and that the nutlin-3-induced phosphorylation of ERK1/2
may be induced by two pathways which act independently or mutually
cooperatively: the sequence of events of one pathway is the
increase of p53, the mitochondrial translocation of p53, followed
by the mitochondrial generation of ROS and ERK1/2 phosphorylation,
while that of the other pathway is the activation of MEK1/2 by p53,
followed by ERK1/2 phosphorylation (Fig. 6).
In a number of studies, p53 has been shown to
migrate to the mitochondria where it interacts with Bcl-xL, thereby
activating pro-apoptotic BAX and BAK proteins to induce apoptosis
(34,35). Palacios et al and Talos
et al induced apoptosis using p53-overexpressing constructs
that target the mitochondria, such as Lp53WT and Lp53CBT,
emphasizing the importance of the mitochondrial translocation of
p53 in p53-induced apoptosis (36,37).
Moreover, it was recently reported that nutlin-3 also induced
apoptosis via the mitochondrial translocation of p53 (38). Based on these reports, we
hypothesized that the targeting of mitochondrial p53 using these
constructs might induce ROS generation in the U2OS cell line.
However, although these constructs appeared to move to the
mitochondria, they failed to induce ROS generation and ERK1/2
phosphorylation (data not shown). It was therefore concluded that
the mitochondrial translocation of p53 is necessary but not
sufficient for ROS accumulation and ERK1/2 activation in U2OS
cells. This conclusion led to the conjecture that two central
pathways involving ROS accumulation and MEK1/2 activation may
cooperate rather than act independently to activate ERK1/2 in
nutlin-3-treated U2OS cells. The discrepancy between the effects of
nutlin-3-induced p53 mitochondrial translocation and the
p53-overexpressing constructs that target the mitochondria on ROS
generation, suggests that there are other critical factors, such as
the precise location and binding partners of p53 in mitochondria,
that determine the effect of the mitochondrial translocation of p53
on apoptosis, ROS generation and MAPK activation.
The exact mechanism by which the nutlin-3-induced
mitochondrial translocation of p53 regulates ROS accumulation
remains to be clarified. Similar to the mechanism of apoptosis
induction, p53 has been reported to induce ROS accumulation by both
transcription-dependent and -independent mechanisms. Elevated p53
levels caused by genotoxic stress induce the transcription of
pro-oxidant genes, such as NQO1, POX, BAX and PUMA, and suppress
anti-oxidant genes, including MnSOD, thereby shifting the balance
between ROS-generating and ROS-scavenging capacities in favor of
ROS accumulation (33).
Furthermore, when in the mitochondria, p53 increases ROS by binding
to MnSOD and inhibiting its ROS-scavenging activity (39). ROS accumulated by the upregulation
of p53 eventually trigger the induction of apoptosis. However, in
this study, we did not observe any significant transcriptional
changes in pro- and anti-oxidant genes as a result of nutlin-3
treatment, which is consistent with the absence of an effect of
PFT-α on p53-induced MEK1/2-ERK1/2 phosphorylation and the
interaction between p53 and MnSOD (data not shown). In addition,
the overexpression of MnSOD did not prevent nutlin-3-induced ROS
accumulation. Therefore, it can be postulated that binding targets
of p53 other than MnSOD may be present in the mitochondria that are
responsible for ROS accumulation in nutlin-3-treated cells. It is
noteworthy that ROS accumulation induced by nutlin-3 treatment
resulted in the survival rather than the apoptosis of U2OS cells,
as has been reported thus far, indicating the peculiarity of the
mechanism and the role of ROS accumulation as a result of nutlin-3
treatment in this cell line.
Although MEK1/2 and ERK1/2 are independent of the
transcriptional activity of p53, as evidenced by the effect of
PFT-α on the activation of these kinases, the mechanisms for their
activation appear to vary regarding the dependency on ROS. Whereas
ERK1/2 activation was suppressed by the prevention of ROS induction
and the mitochondrial translocation of p53, this prevention had no
effect on MEK1/2 activation. We were also unable to observe the
dependency of MEK1/2 activation on Raf kinases (data not shown). Of
note, PFT-μ augmented the nutlin-3-induced phosphorylation of
MEK1/2 as well as the expression of p21WAF1 protein (Fig. 4C). PFT-μ has been reported to have
no effect on the transcriptional activity of p53 (40) and, moreover, the nutlin-3-induced
increase of p21 mRNA and the level of MEK1/2 protein were not
affected by PFT-μ-pre-treatment (Fig.
4C; data not shown), suggesting that the potentiating effect of
PFT-μ on the phosphorylation of MEK1/2 and the expression of
p21WAF1 protein occurred at the post-transcriptional level. In
addition, on the immunoblot analysis of subcellular fractions, p53
accumulated in the cytosol of PFT-μ-pre-treated cells. Therefore,
it may be inferred that p53 activates MEK1/2 at the
post-transcriptional level and through cytosolic events that need
to be identified hereafter to elucidate the mechanism behind the
p53-induced MEK1/2 activation.
Nutlin-3 is a promising anticancer drug since it
does not induce genomic DNA damage that can contribute to
carcinogenesis secondary to anticancer therapy. In the present
study, we propose a mechanism that explains the manner by which
nultin-3 treatment activates the cell survival pathway
simultaneously with apoptosis via ROS accumulation and
MEK1/2-ERK1/2 activation induced by the mitochondrial translocation
of p53 in human cancer cells. For the optimal use of nutlin-3,
interfering with the nutlin-3-induced survival pathway is critical.
In order to characterize the nutlin-3-induced survival pathway in
detail, proteins interacting with p53 in the mitochondria, which
are critical for ROS accumulation and the major determinants
between ROS generation and apoptosis induced by mitochondrial p53,
should be identified in the future and their distribution or
expression characteristics among cancer cells, as well as their
effect on nutlin-3-induced apoptosis should be elucidated for the
optimal use of nutlin-3 against cancer.
Acknowledgements
We are deeply grateful to Professor
Ute M. Moll at Stony Brook University and her colleagues for
providing us with the Lp53WT and Lp53CBT plasmids and helpful
advice. This study was supported by the Basic Science Research
Program through the National Research Foundation of Korea (NRF)
funded by the Ministry of Education, Science and Technology
(2011-0027115).
References
|
1
|
Menendez D, Inga A and Resnick MA: The
expanding universe of p53 targets. Nat Rev Cancer. 9:724–737. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Riley T, Sontag E, Chen P and Levine A:
Transcriptional control of human p53-regulated genes. Nat Rev Mol
Cell Biol. 9:402–412. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Honda R, Tanaka H and Yasuda H:
Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53.
FEBS Lett. 420:25–27. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oliner JD, Pietenpol JA, Thiagalingam S,
Gyuris J, Kinzler KW and Vogelstein B: Oncoprotein MDM2 conceals
the activation domain of tumor suppressor p53. Nature. 362:857–860.
1993. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reinhardt HC and Schumacher B: The p53
network: cellular and systemic DNA damage responses in aging and
cancer. Trends Genet. 28:128–136. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sill H, Olipitz W, Zebisch A, Schulz E and
Wölfler A: Therapy-related myeloid neoplasms: pathobiology and
clinical characteristics. Br J Pharmacol. 162:792–805. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Travis LB, Ng AK, Allan JM, et al: Second
malignant neoplasms and cardiovascular disease following
radiotherapy. J Natl Cancer Inst. 104:357–370. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shangary S and Wang S: Targeting the
MDM2-p53 interaction for cancer therapy. Clin Cancer Res.
14:5318–5324. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dickens MP, Fitzgerald R and Fischer PM:
Small-molecule inhibitors of MDM2 as new anticancer therapeutics.
Semin Cancer Biol. 20:10–18. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vassilev LT, Vu BT, Graves B, et al: In
vivo activation of the p53 pathway by small-molecule antagonists of
MDM2. Science. 303:844–848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Secchiero P, Bosco R, Celeghini C and
Zauli G: Recent advances in the therapeutic perspectives of
nutlin-3. Curr Pharm Des. 17:569–577. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tovar C, Rosinski J, Filipovic Z, et al:
Small-molecule MDM2 antagonists reveal aberrant p53 signaling in
cancer: implications for therapy. Proc Natl Acad Sci USA.
103:1888–1893. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mendrysa SM, O’Leary KA, McElwee MK, et
al: Tumor suppression and normal aging in mice with constitutively
high p53 activity. Genes Dev. 20:16–21. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mebratu Y and Tesfaigzi Y: How ERK1/2
activation controls cell proliferation and cell death: Is
subcellular localization the answer? Cell Cycle. 8:1168–1175. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McCubrey JA, Steelman LS, Chappell WH, et
al: Roles of the Raf/MEK/ERK pathway in cell growth, malignant
transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Persons DL, Yazlovitskaya EM and Pelling
JC: Effect of extracellular signal-regulated kinase on p53
accumulation in response to cisplatin. J Biol Chem.
275:35778–35785. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sablina AA, Chumakov PM, Levine AJ and
Kopnin BP: p53 activation in response to microtubule disruption is
mediated by integrin-Erk signaling. Oncogene. 20:899–909. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
She QB, Bode AM, Ma WY, Chen NY and Dong
Z: Resveratrol-induced activation of p53 and apoptosis is mediated
by extracellular-signal-regulated protein kinases and p38 kinase.
Cancer Res. 61:1604–1610. 2001.PubMed/NCBI
|
|
19
|
Lin T, Mak NK and Yang MS: MAPK regulate
p53-dependent cell death induced by benzo[a]pyrene: involvement of
p53 phosphorylation and acetylation. Toxicology. 247:145–153.
2008.PubMed/NCBI
|
|
20
|
Lee SW, Fang L, Igarashi M, Ouchi T, Lu KP
and Aaronson SA: Sustained activation of Ras/Raf/mitogen-activated
protein kinase cascade by the tumor suppressor p53. Proc Natl Acad
Sci USA. 97:8302–8305. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fang L, Li G, Liu G, Lee SW and Aaronson
SA: p53 induction of heparin-binding EGF-like growth factor
counteracts p53 growth suppression through activation of MAPK and
PI3K/Akt signaling cascades. EMBO J. 20:1931–1939. 2001. View Article : Google Scholar
|
|
22
|
Ongusaha PP, Kim JI, Fang L, et al: p53
induction and activation of DDR1 kinase counteracts p53-mediated
apoptosis and influence p53 regulation through a positive feedback
loop. EMBO J. 22:1289–1301. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yin Y, Liu YX, Jin YJ, Hall EJ and Barrett
JC: PAC1 phosphatase is a transcription target of p53 in signaling
apoptosis and growth suppression. Nature. 422:527–531. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ueda K, Arakawa H and Nakamura Y:
Dual-specificity phosphatase 5 (DUSP5) as a direct transcriptional
target of tumor suppressor p53. Oncogene. 22:5586–5591. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li M, Zhou JY, Ge Y, Matherly LH and Wu
GS: The phosphatase MKP1 is a transcriptional target of p53
involved in cell cycle regulation. J Biol Chem. 278:41059–41068.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kojima K, Konopleva M, Samudio IJ, Ruvolo
V and Andreeff M: Mitogen-activated protein kinase kinase
inhibition enhances nuclear proapoptotic function of p53 in acute
myelogenous leukemia cells. Cancer Res. 67:3210–3219. 2007.
View Article : Google Scholar
|
|
27
|
Zhang W, Konopleva M, Burks JK, et al:
Blockade of mitogen-activated protein kinase/extracellular
signal-regulated kinase kinase and murine double minute
synergistically induces apoptosis in acute myeloid leukemia via
BH3-only proteins Puma and Bim. Cancer Res. 70:2424–2434. 2010.
View Article : Google Scholar
|
|
28
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jang JY, Kim MK, Jeon YK, et al:
Adenovirus adenine nucleotide translocator-2 shRNA effectively
induces apoptosis and enhances chemosensitivity by the
down-regulation of ABCG2 in breast cancer stem-like cells. Exp Mol
Med. 44:251–259. 2012. View Article : Google Scholar
|
|
30
|
Sauer L, Gitenay D, Vo C and Baron VT:
Mutant p53 initiates a feedback loop that involves Egr-1/EGF
receptor/ERK in prostate cancer cells. Oncogene. 29:2628–2637.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
McCubrey JA, Lahair MM and Franklin RA:
Reactive oxygen species-induced activation of the MAP kinase
signaling pathways. Antioxid Redox Signal. 8:1775–1789. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liu CM, Sun YZ, Sun JM, Ma JQ and Cheng C:
Protective role of quercetin against lead-induced inflammatory
response in rat kidney through the ROS-mediated MAPKs and NF-κB
pathway. Biochim Biophys Acta. 1820:1693–1703. 2012.PubMed/NCBI
|
|
33
|
Liu B, Chen Y and St Clair DK: ROS and
p53: a versatile partnership. Free Radic Biol Med. 44:1529–1535.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sansome C, Zaika A, Marchenko ND and Moll
UM: Hypoxia death stimulus induces translocation of p53 protein to
mitochondria. Detection by immunofluorescence on whole cells. FEBS
Lett. 488:110–115. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mihara M, Erster S, Zaika A, et al: p53
has a direct apoptogenic role at the mitochondria. Mol Cell.
11:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Palacios G and Moll UM: Mitochondrially
targeted wild-type p53 suppresses growth of mutant p53 lymphomas in
vivo. Oncogene. 25:6133–6139. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Talos F, Petrenko O, Mena P and Moll UM:
Mitochondrially targeted p53 has tumor suppressor activities in
vivo. Cancer Res. 65:9971–9981. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Vaseva AV, Marchenko ND and Moll UM: The
transcription-independent mitochondrial p53 program is a major
contributor to nutlin-induced apoptosis in tumor cells. Cell Cycle.
8:1711–1719. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhao Y, Chaiswing L, Velez JM, et al: p53
translocation to mitochondria precedes its nuclear translocation
and targets mitochondrial oxidative defense protein-manganese
superoxide dismutase. Cancer Res. 65:3745–3750. 2005. View Article : Google Scholar
|
|
40
|
Strom E, Sathe S, Komarov PG, et al:
Small-molecule inhibitor of p53 binding to mitochondria protects
mice from gamma radiation. Nat Chem Biol. 2:474–479. 2006.
View Article : Google Scholar : PubMed/NCBI
|