Introduction
Hepatocellular carcinoma (HCC) is a highly
aggressive cancer with multiple causes and no currently available
effective treatment (1,2). Understanding the molecular mechanism
of HCC carcinogenesis, its development and progression is
imperative for developing novel, effective and targeted therapies
for this fatal disease (3,4). Numerous investigations using
different improved approaches, such as insertional mutagenesis
screen (5), in vivo RNAi
screen (6) and omics-based methods
(7), are being conducted. Novel
tumor associated genes such as glypican-3 (GPC3), astrocyte
elevated gene-1 (AEG-1) and exportin 4 (XPO4) have
been discovered and studied extensively (8,9).
To search for new biomarkers for tumor diagnosis and
therapy, we previously detected tumor associated genes using
bioinformatic approaches on a large scale. HTA is a novel
tumor associated gene screened by cDNA xProfile, an in
silico identification tool based on the EST database provided
by the Cancer Genome Anatomy Project (CGAP) web site and
preliminarily confirmed by electronic-northern (E-northern). RT-PCR
revealed that HTA (NM 001101347) was specifically expressed
in certain types of tumor and had a high expression rate in HCC. To
further elucidate its mechanism in hepatoma carcinogenesis and its
potential role as a cancer biomarker, research were carried out and
the results showed that knockdown of endogenous HTA
expression in the malignant hepatic cell line HepG2 by small
interfering RNA (siRNA) attenuated cell growth, weakened its tumor
formation ability in nude mice (10) and changed the expression level of
some genes associated with apoptosis (Liu et al, unpublished
data), which suggests that HTA may be important in HCC
development and progression and its mechanism might be
apoptosis-related.
Since HTA is a gene screened from the EST
database, the full length sequences were essential for its
functional investigation. In this report, the full length cDNA of
the HTA gene was verified by rapid amplification of cDNA 3′-ends
[3′-rapid amplification of cDNA ends (RACE)] and amplification of
cDNA 5′-ends (5′-RACE) (11,12).
Northern blot assay was performed to determine the transcripts of
HTA mRNA expression in HCC and normal hepatic cell
lines.
Materials and methods
Cell lines
The HepG2, QGY-7703, HUVEC, L-02, QGY-7701 cell
lines were used in this study. The malignant hepatic cell line
HepG2 was stored by our laboratory. QGY-7703 was obtained from the
cell bank of the Chinese Academy of Sciences, Shanghai, China. The
benign hepatic cell lines QSG-7701, L-02 were stored by our
laboratory. Human umbilical vein endothelial cells (HUVEC) were
stored by our laboratory. These cell lines were cultured in
Gibco® RPMI-1640 medium, supplemented with 10% FBS. All
cells were maintained at 37°C in a 5% CO2 incubator.
Preparation of RNA
Total RNA from cell lines was extracted using TRIzol
RNA isolation reagents (Life Technologies) according to the
manufacturer’s instructions. The mRNA was prepared from total RNA
using the PolyATtract® mRNA Isolation Systems (Promega)
according to the manufacturer’s instructions. All the RNA samples
were tested for integrity and purity by an ultraviolet
spectrophotometer (OD260/OD280), RNA temperature incubation
experiment and formaldehyde degeneration electrophoresis.
Cloning and sequencing of full length HTA
cDNA
In accordance with the existing sequence of the HTA
gene obtained in a previous study (10), the gene specific primers for
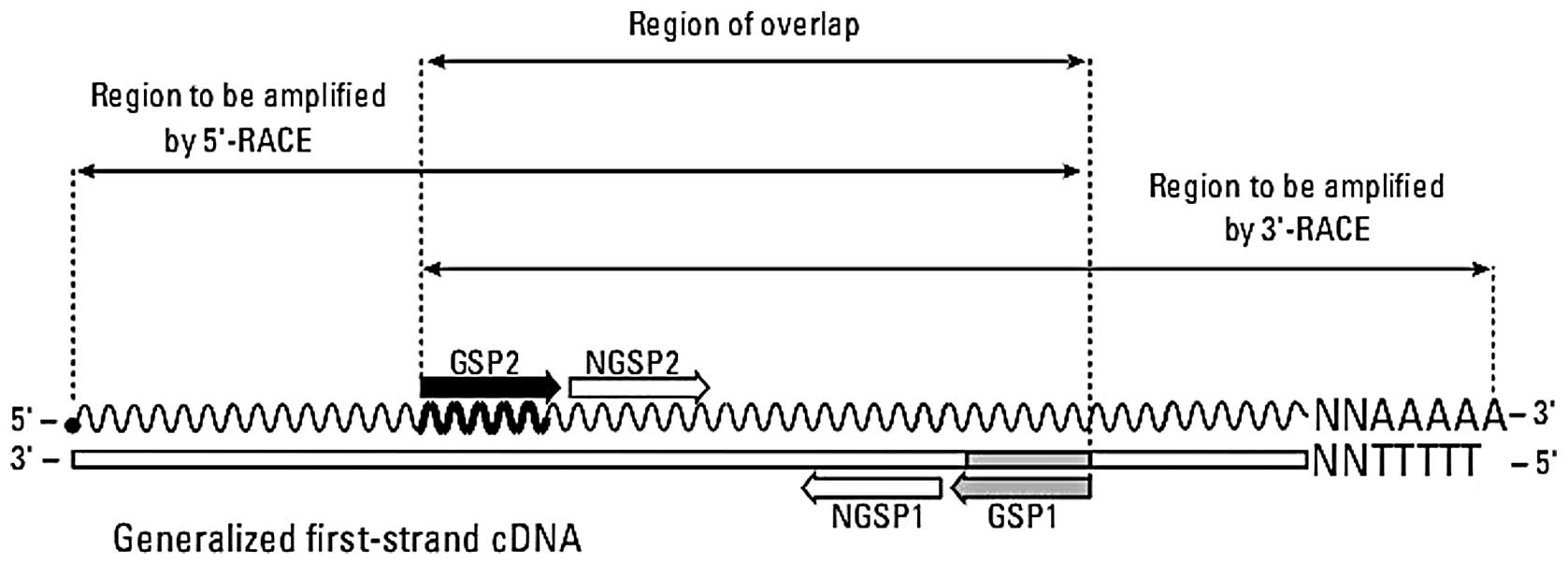
5′-RACE (GSP1), 3′-RACE (GSP2) and for nest PCR NGSP1 were designed
as shown in Fig. 1 and Table I. Then, the 5′-end sequence and
3′-end sequence of the cDNA encoding the HTA gene were
obtained from the HCC cell line HepG2 using Smarter™ RACE cDNA
Amplification kit 5′/3′ (Clontech) according to the manufacturer’s
instructions. Initially with 1 μg total RNA of HepG2 cells,
the 3′-RACE ready cDNA and 5′-RACE ready cDNA were synthesized
according to the protocol, respectively. GSP1 primer and Upper
primer mix were then used to amplify the 5′-end sequences of the
HTA gene. Upper primer mix and GSP2 primer were used to amplify the
3′-end sequence of the HTA gene. Then, a touch-down program was
performed as follows: 5 cycles of denaturing at 94°C for 30 sec,
72°C for 3 min, followed by 5 cycles of each 94°C for 30 sec, 70°C
for 30 sec and 72°C for 3 min, then followed by 35 cycles of 94°C
for 30 sec, 68°C for 30 sec and 72°C for 3 min. The RACE PCR
product was analyzed by 1% agarose gel electrophoresis and
photographed by Image Master VDS Gel Imaging system (Pharmacia).
Using 25-fold diluted RACE PCR product as template, nest PCR was
performed by nest primers and program as follows: 30 cycles of 94°C
for 30 sec, 68°C for 30 sec and 72°C for 3 min. The nest PCR
product was separated by 1% low-melting agarose gel electrophoresis
and Objective band was retrieved and ligated to pGEM-T easy vector,
then transformed into Escherichia coli (E. coli)
DH5α. Several positive clones were picked out and sequenced.
 | Table IPrimers used in this study. |
Table I
Primers used in this study.
| Primer | (5′-3′) |
|---|
| Universal primer
(long) |
CTAATACGACTCACTATAGGGCAAGCAGTGGTATCAACGCAGAGT |
| Universal primer
(short) |
CTAATACGACTCACTATAGGGC |
| Nested universal
primer |
AAGCAGTGGTATCAACGCAGAGT |
| GSP1 |
TCTGCTGGAGGTGAGGGCTCGTGGT |
| GSP2 |
ATGGGTTGCTTGGGGCATCCTGTG |
| NGSP1 |
GTCGGGGTTGTTCTTGTTTTCAG |
|
HTA-cDNA-F |
CGAAGATTTCTTATAGACAGGC |
|
HTA-cDNA-R |
GGAGGGCATTATGGTGAG |
|
HTA-probe-F |
CTATTTATGGGTTGCTTGG |
|
HTA-probe-R-T7 |
CTAATACGACTCACTATAGGGAGAGAGGGCATTATGGTGAGAT |
| GAPDH-probe-F |
AATCCCATCACCATCTTCC |
|
GAPDH-probe-R-T7 |
CTAATACGACTCACTATAGGGAGAGCTGTAGCCAAATTCGTTGT |
Analysis of HTA gene structure and
alternative splicing
The sequences of products of RACE PCR positive
clones were comprehensively analyzed by BLAST (http://www.ncbi.nlm.nih.gov/BLAST/) and online
software open reading frame (ORF) finder (http://www.ncbi.nlm.nih.gov/gorf/orfig.cgi) provided
by the National Center For Biotechnology Information (NCBI). The
full length sequence primers of HTA (HTA-cDNA-F and
HTA cDNA-R in Table I) were
designed and the full lengths of HTA transcripts were
amplified. The amplified product was sequenced and the alternative
splicing of the HTA gene was analyzed.
Northern blot analysis of HTA mRNA
expression
Probes for northern blotting were produced using a
DIG RNA labeling and detection kit (Roche Diagnostics). A 589-bp
HTA gene specific primer pair was designed for amplification of
northern bolt probe; at the same time, a 24-bp T7 promoter sequence
5′-CTAATACGACTCACTATAGGGAGA-3′ was added to the 5′-end of reverse
primer (Table I). Then, the
northern blot probe of HTA gene labeling with digoxin in UTP was
produced by in vitro transcription. GAPDH probe was produced
with the same procedure. The labeling efficiency of the probes was
determined according to the manufacturer’s instructions.
mRNA (1 μg) from four hepatic cell lines
(HUVEC, L-02, HepG2, QGY-7703) was separated on denaturing 0.667%
formaldehyde-2% agarose gel and was transferred onto positively
charged nylon membrane Biodyne Plus (Pall) by the capillary
transfer method. Following UV cross-linking (245 nm, 1.5
J/cm2 for 1 min 45 sec), prehybridization was carried
out at 68°C for 1 h using DIG Easy Hyb (Roche Diagnostics).
Hybridization was performed at 68°C for 12 h, 40 revolutions per
minute (rpm), using the DIG Easy Hyb containing 100 ng/ml of the
complementary RNA (cRNA) probe that was previously produced. The
membrane was then washed twice for 5 min in 2X SSC with 0.1% sodium
dodecyl sulfate (SDS) at room temperature, and was washed again
twice in 0.1X SSC with 0.1% SDS at 68°C for 15 min. After
incubating the membrane in alkaline phosphatase (AP) labeled
anti-digoxin antibody, the chemiluminescent signals were detected
using the DIG RNA labeling and detection kit according to the
manufacturer’s instructions. Signals were detected after exposure
on Pierce ECL Plus (Thermo Scientific). For standardization, the
HTA probe was stripped from the membrane and was reprobed using the
DIG-labeled GAPDH RNA probe under conditions similar to assessing
the RNA loading and transfer efficiency.
Construction of expression vectors of
HTA
Total RNA was extracted from HepG2 cells by
homogenization in TRIzol reagent (Invitrogen, USA) and first-strand
cDNAs were synthesized from 2 μg of DNase I-treated total
RNA using First Strand cDNA Synthesis kit (Fermentas, USA) by way
of a two-step system. Gene-specific polymerase chain reaction (PCR)
primers for HTA ORF amplification were:
5′-CGCGGATCCGCCGCCATGCTCTGTGTGCCCTTCCT-3′ (sense) and
5′-CCGGAATTCTTAGCCCAAGATGAAGACAAGGC-3′
(antisense) and introduced with BamHI site (sense) and an
EcoRI site (antisense) respectively (underlined). The italic
GCCGCC in the sense primer was a Kozak sequence (13) designed to improve gene expression
quantity. The bold ATG and TAA were initial and
terminator codon, respectively. The PCR products of the HTA
gene were purified by 1% agarose gel electrophoresis then double
digested and ligated into the eukaryotic expression vector
pcDNA3.1(+). The recombinant plasmid was transformed into E.
coli DH5α then identified by PCR, double endonuclease digestion
and DNA sequencing.
Transfection and selection of stable HTA
overexpression hepatic cell line
Plasmids pcDNA3.1(+)-HTA and pcDNA3.1(+) were
transfected into QSG-7701 cell lines using Lipofectamine™ 2000
(Invitrogen). Three days later, cells were selected in a medium
containing G418 (600 mg/ml) for 10 days, then the limiting
dilution method was used and G418-resistant colonies were cloned
and expanded. RT-PCR was performed to detect the HTA mRNA
expression level in empty vector and HTA overexpression clones. A
clone with the highest HTA expression was selected to perform
subsequent experiments.
Cell proliferation test in vitro
Growth experiments of QSG-7701-c [QSG-7701 cell line
stably transfected with pcDNA3.1 (+) empty vector as a control] and
QSG-7701-HTA-2 were performed using the Cell Counting kit-8 (CCK-8,
Beyotime) based on WST-8
[2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium].
Cell doubling time test and plate cloning formation assay were
reconfirmed by cell cycle analysis, which was performed by flow
cytometry.
In the CCK-8 assay, cells were seeded with
serum-free medium at a density of 103 cells/well in
96-well plates (n=6), grown overnight, washed with PBS, and
incubated in DMEM medium supplemented with 5% FBS. The samples were
tested every 24 h for 6 days. WST-8 (10 μl) each well was
added for 2 h and the optical density was measured at 450 nm in an
SM-3 automatic enzyme-linked immune-analyzer (TianShi, China).
In the cell doubling time test, cells were seeded at
a density of 104 cells/well in 24-well plates (n=3), grown with
serum-free medium overnight, washed with PBS, and incubated in DMEM
medium with 3% FBS. Cells were collected 6 days later and cell
densities were assessed by counting the cells in a hemocytometer.
Cell doubling time (TD) was calculated
with the following formula: TD=
t[lg2/(lgNt - lgNo)] [t, period of
culturing (hour). No, cell density when the cells were
seeded. Nt, cell density when the cells were cultured
t hours].
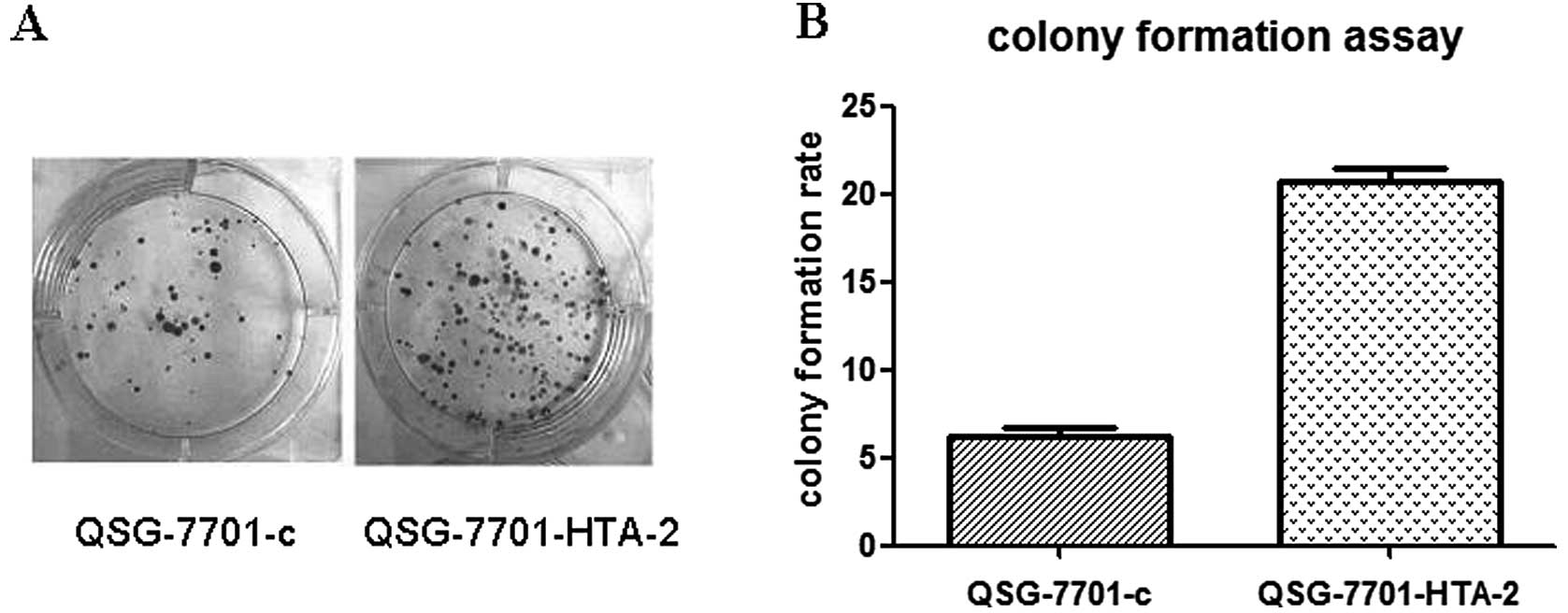
Plate colony formation assay was used to assess the
colony formation ability of cells. The cells were incubated in
serum-free medium overnight. A total of 1,000 single-cell
suspension cells resuspended in 2 ml growth media were plated in
triplicate on 6-well plates. Colonies >50 cells were counted 14
days after plating. Colony formation ratio was calculated with the
following formula: Colony formation rate = (number of colonies
counted /numbers of cells seeded) x 100%.
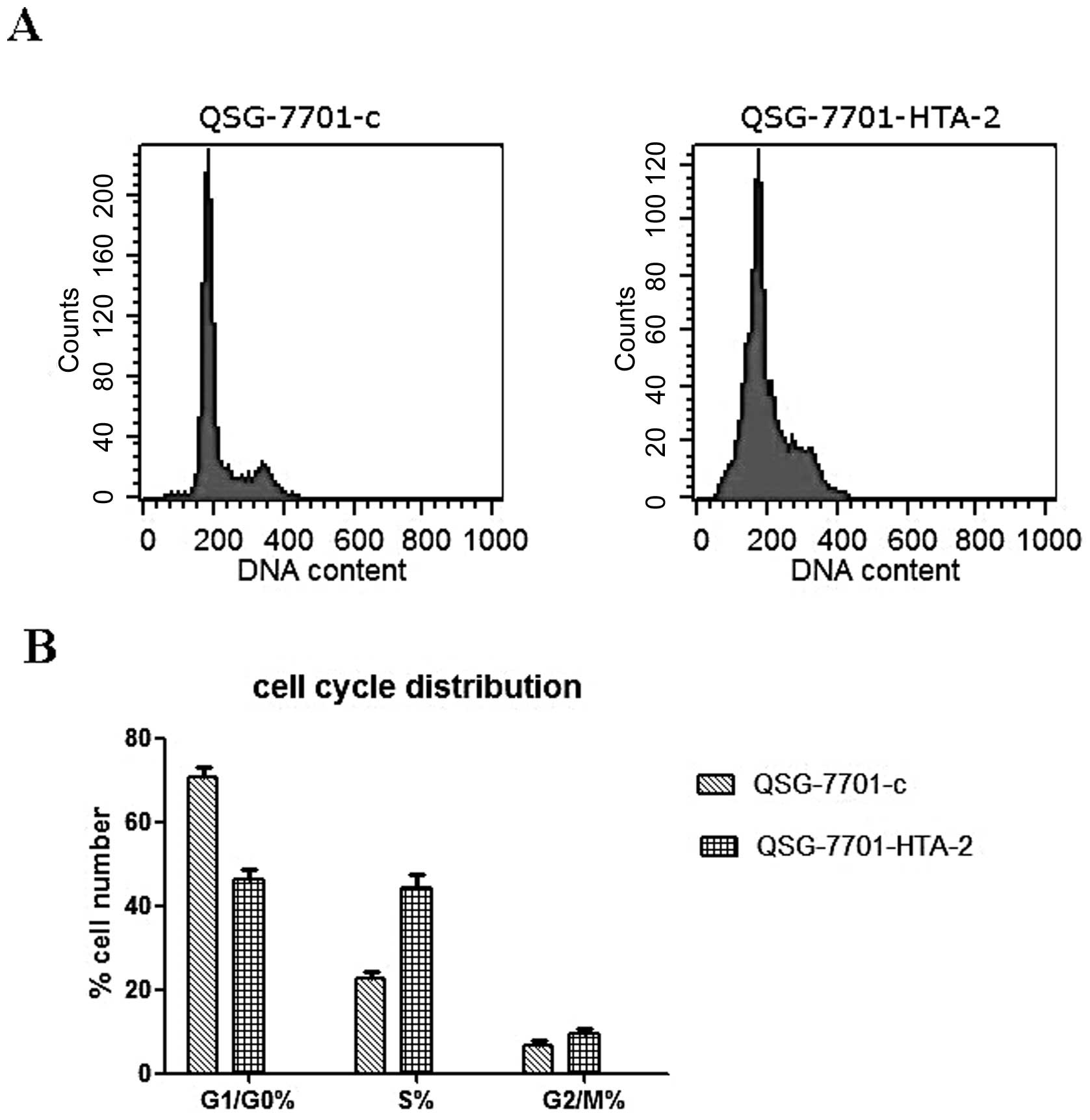
For flow cytometry, cells were incubated with
serum-free medium for 24 h, and then cultured in DMEM with 10% FBS
for 48 h (n=3). Cells were harvested and resuspended in fixation
fluid at a density of 106/ml, 1,500 μl propidium
iodide (PI) solution was added, and the cell cycle was detected by
FACSCalibur (Becton-Dickinson).
Statistical analysis
The values are presented as the means ± SD for three
or more individual experiments. Data were tested with SPSS software
(version 13.0, SPSS Inc.) for significance. The analysis of
variance and t-test were applied in comparing the intergroup
difference of measurement data. p<0.05 was considered to
indicate a statistically significant difference. The diagrams were
drawn by GraphPad Prism 5.
Results
Molecular cloning of the full-length HTA
cDNA
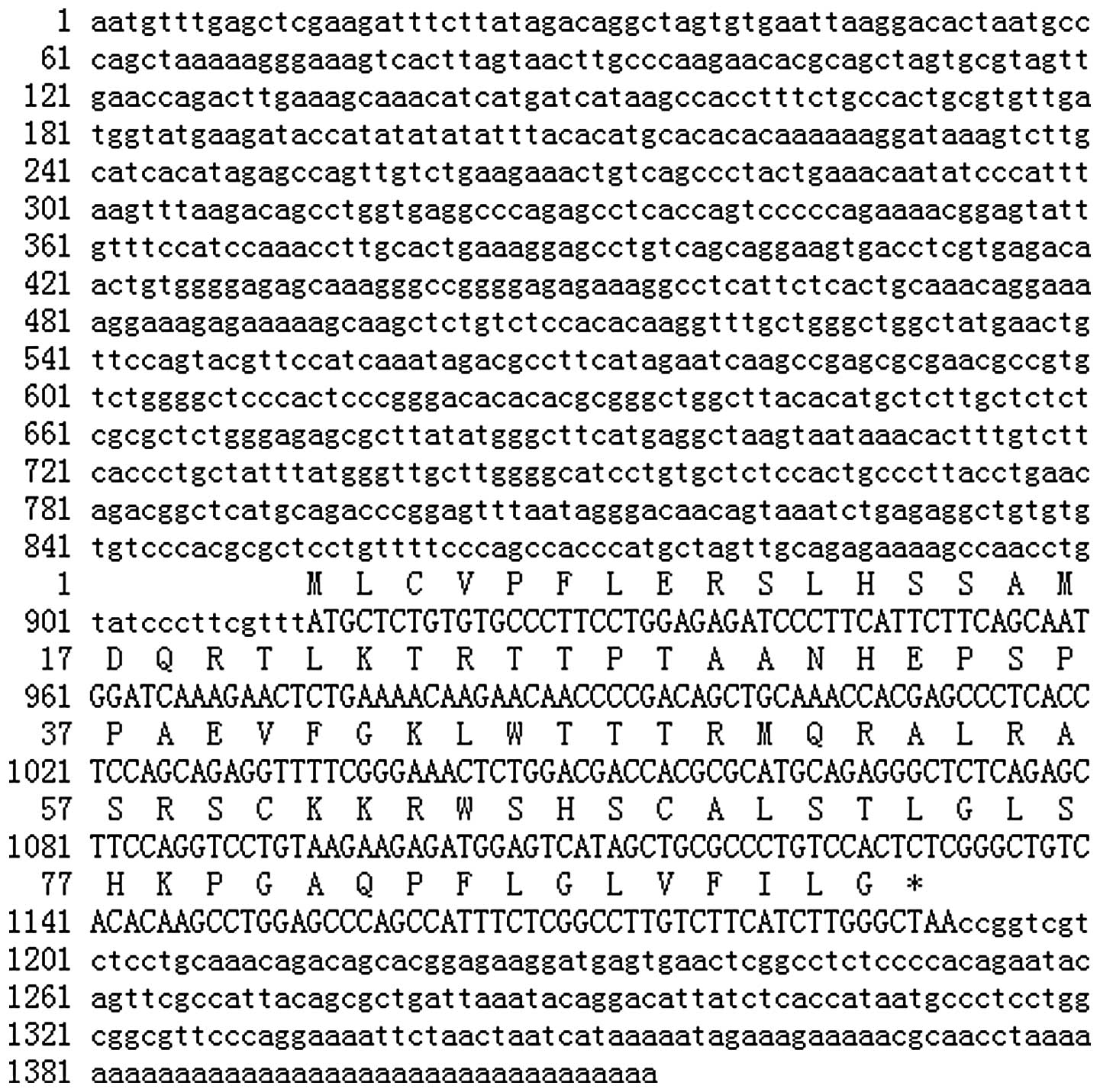
We cloned the 5′-RACE and 3′-RACE product and
determined the sequence. The products using this method were partly
consistent with a previous known sequence of HTA. Finally, the
1414-bp obtained HTA cDNA was reconfirmed to be full length by
northern blot analysis. Fig. 2
shows the nucleotide sequence and the deduced amino acid sequence
using 5′-RACE and 3′-RACE. The 3′-end of the product was in line
with the sequence previously obtained. The ORF of the HTA gene
encoded a protein of 92 amino acids with an estimated molecular
weight of 10.2 kDa. A comparison of the HTA cDNA sequence with the
GenBank database showed no significant homology with other genes
except its homologous sequences in Macaca fascicularis and
Macaca fascicularis genome. Furthermore, a comparison of the
deduced amino acid sequence with a protein sequence database showed
no overall significant homology with other proteins whose function
is known. It also showed no conserved domains and no valid domain
hit for architecture searched by NCBI.
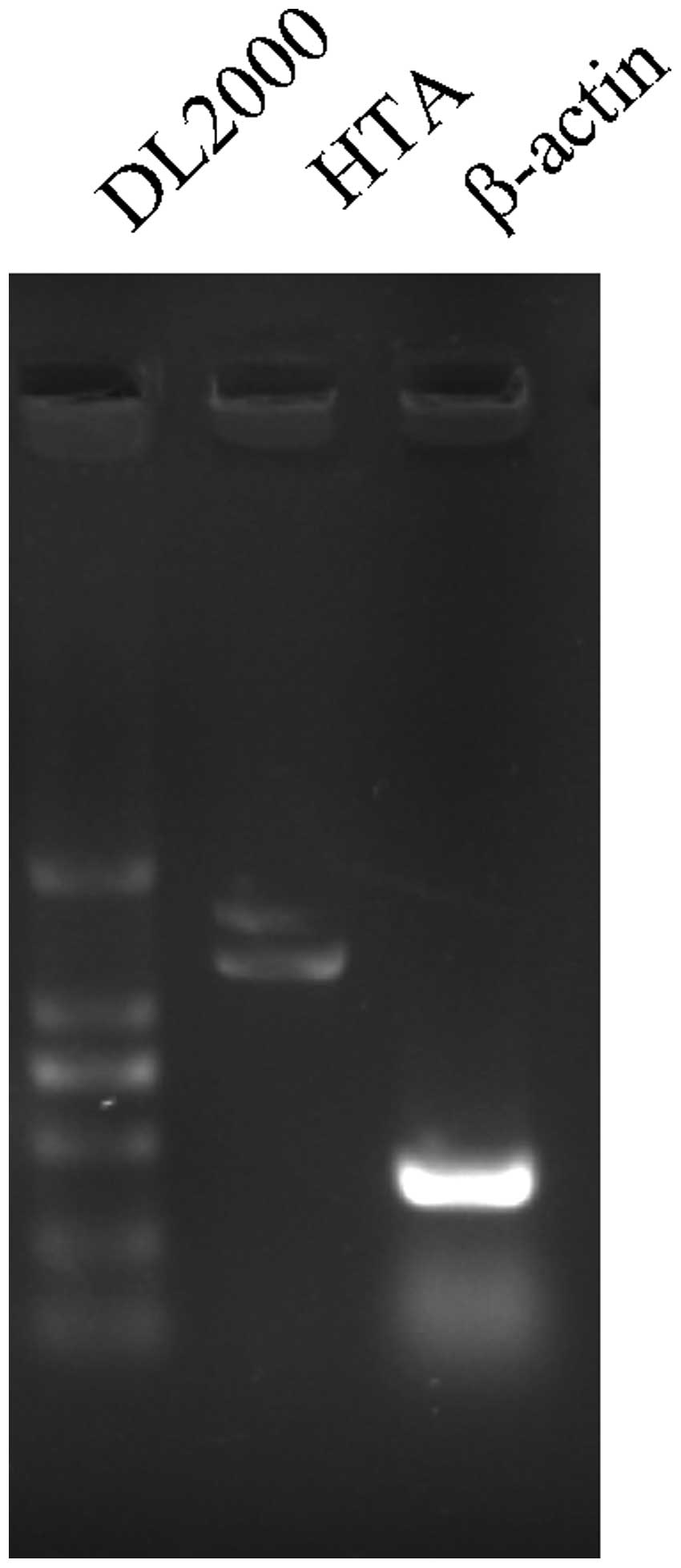
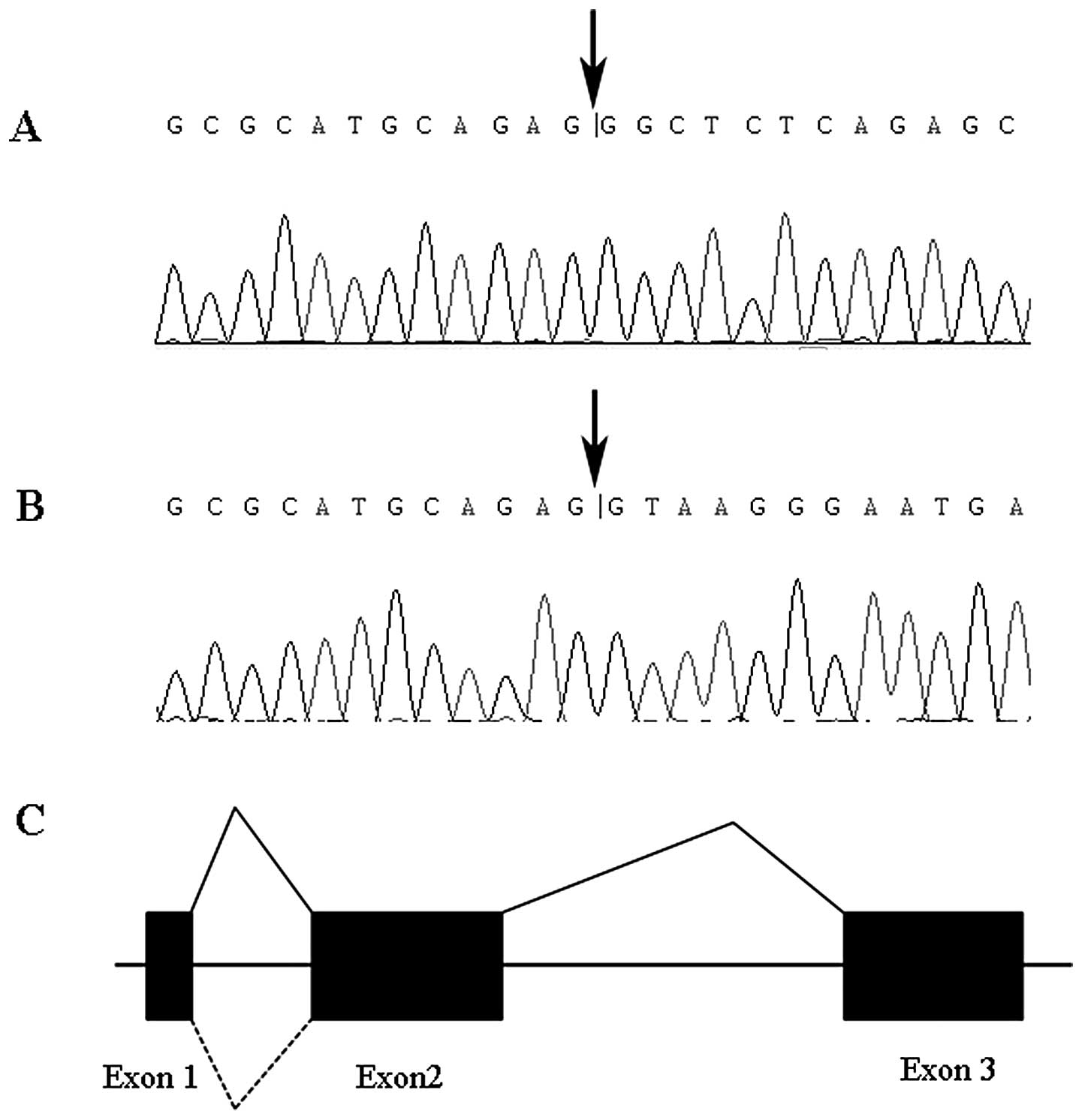
Alternative splicing analysis of the HTA
gene
Online prediction software ORF finder provided by
NCBI revealed that the HTA gene was constituted with 3 exons and 2
introns. RT-PCR with HTA full length primer pair HTA-cDNA-F and
HTA-cDNA-R showed that the HTA gene has two transcripts which were
1.4 and 1.7 kb, respectively (Fig.
3). Sequencing of RT-PCR product suggested an alternative
splicing of the HTA gene. The 252-bp second introns of the HTA gene
can be spliced, or retained as a part of mRNA (Fig. 4).
Expression of HTA mRNA in hepatic cell
lines
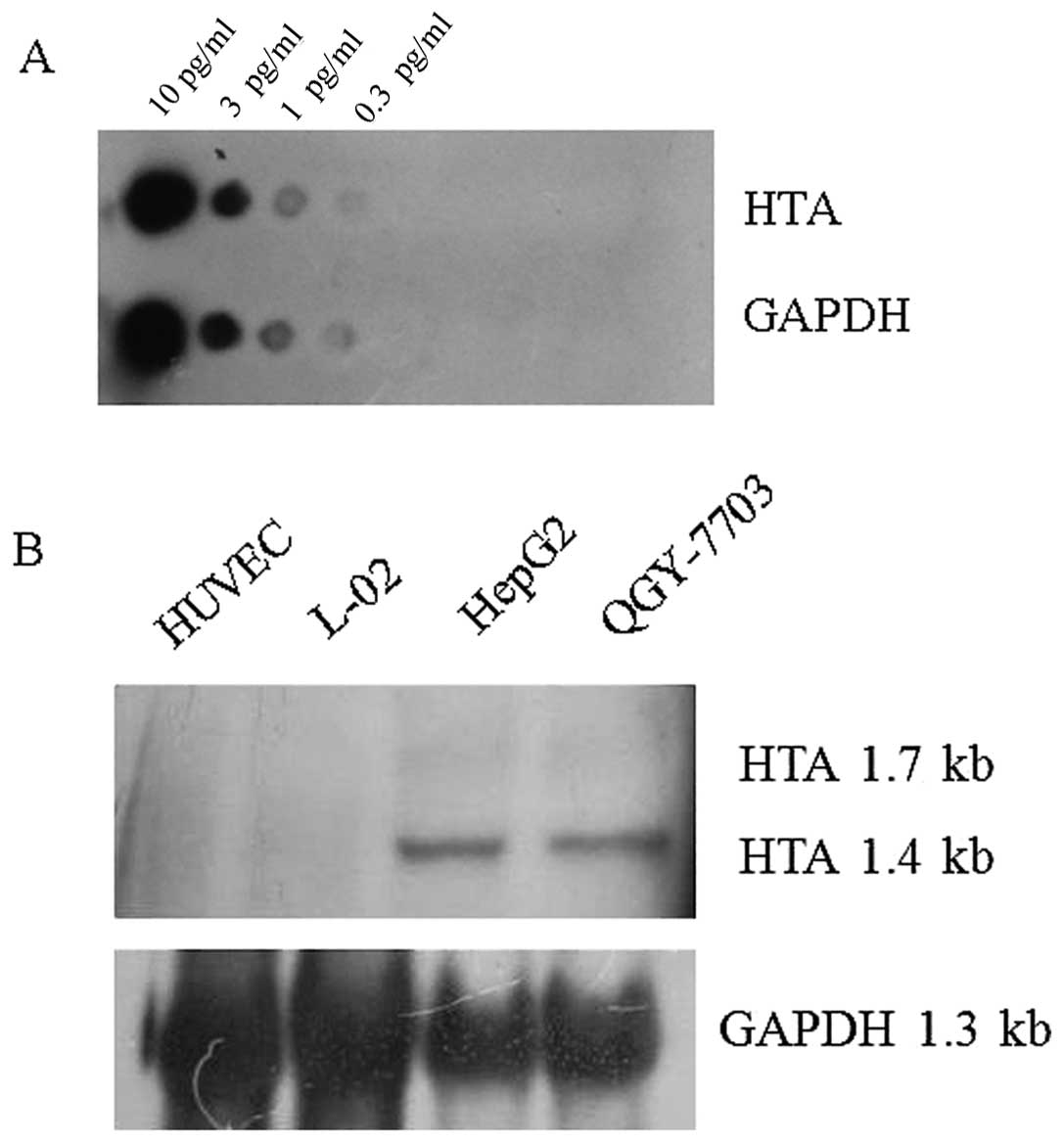
The labeling efficiency of HTA and GAPDH digoxin
probes were defined. When the samples were diluted to the
concentration of 0.3 pg/μl, the signal could still be
detected, which means that adequate amounts of labeled RNA probe
for northern blot analysis were obtained (Fig. 5A). Northern blot analysis showed
that the 1.4-kb HTA mRNA and 1.7-kb HTA mRNA transcripts were
present in HCC cell lines HepG2 and QGY-7703. The expression amount
of the 1.4-kb transcript was much higher than the 1.7-kb
transcript. In the normal hepatic cell line L-02 and the normal
cell line HUVEC, no HTA transcript was detected (Fig. 5B).
Establishment of the overexpression of
the HTA hepatic cell line
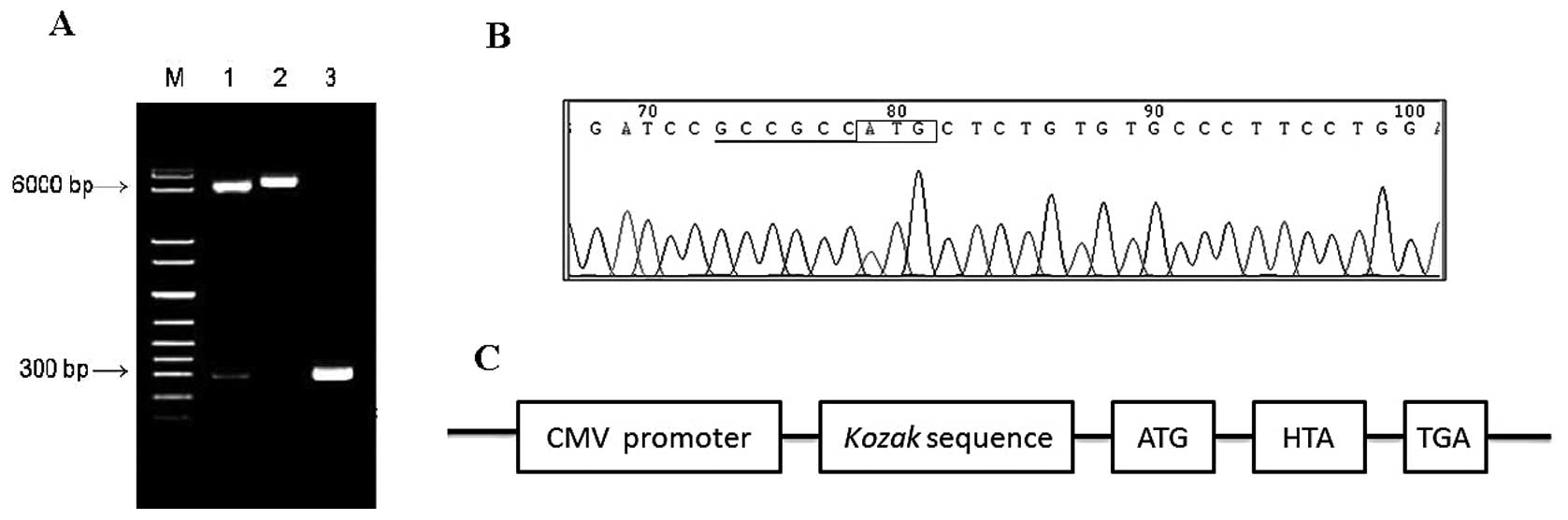
The recombinant eukaryotic expression plasmid
pcDNA3.1(+)-HTA was successfully constructed and verified by
bacterial colony PCR, restriction enzyme digestions (Fig. 6A) and complete sequencing (Fig. 6B). The recombinant HTA protein
obtained from this plasmid had a Kozak sequence for high potency of
expressing heterologous protein (Fig.
6C).
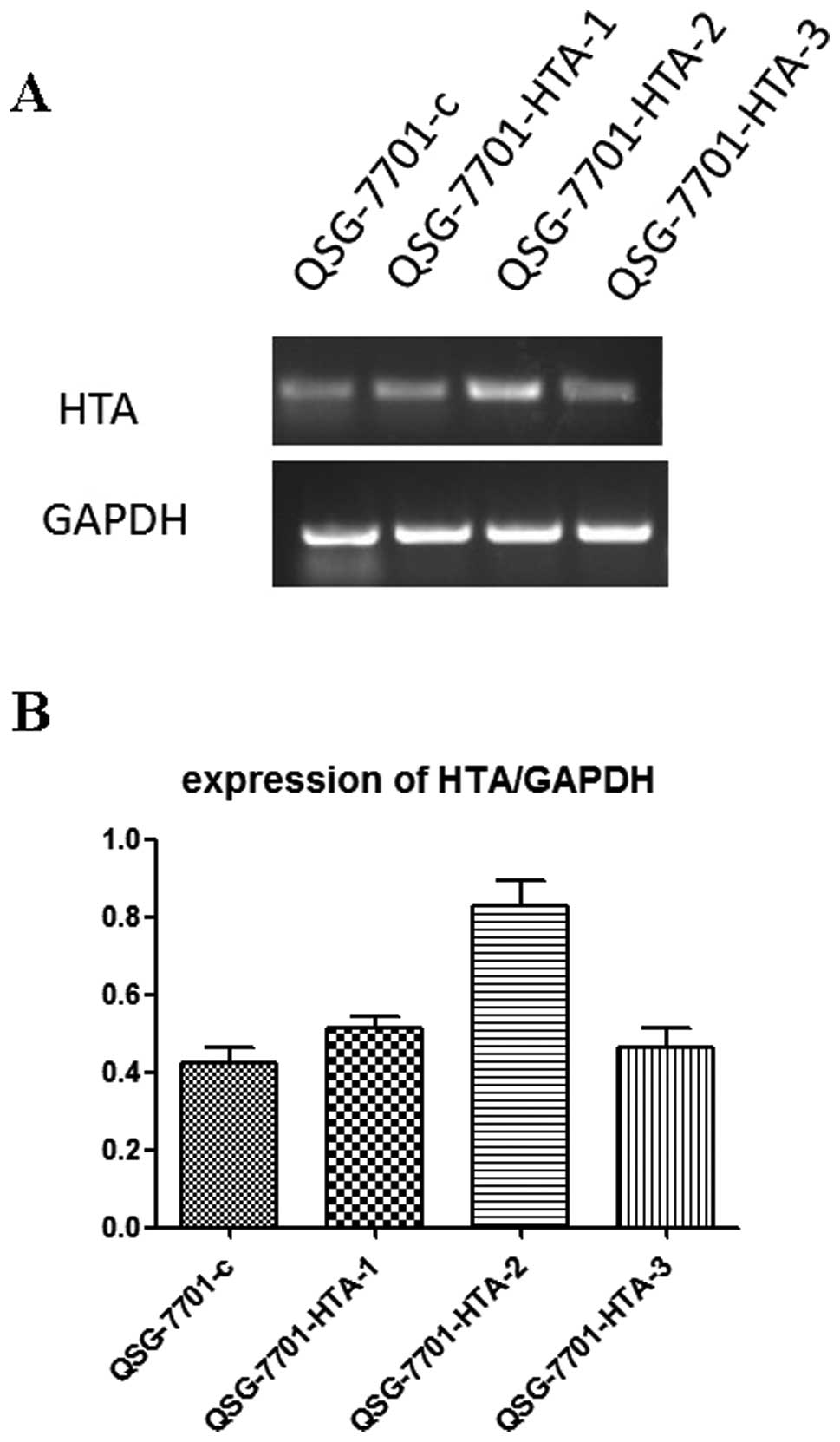
After G418-resistant stable cell lines were
picked up, a semi-quantitative RT-PCR analysis was performed to
determine the HTA mRNA expression in different clones.
QSG-7701-HTA-2, the clone with the highest HTA expression,
was chosen to perform subsequent in vitro experiments
(Fig. 7).
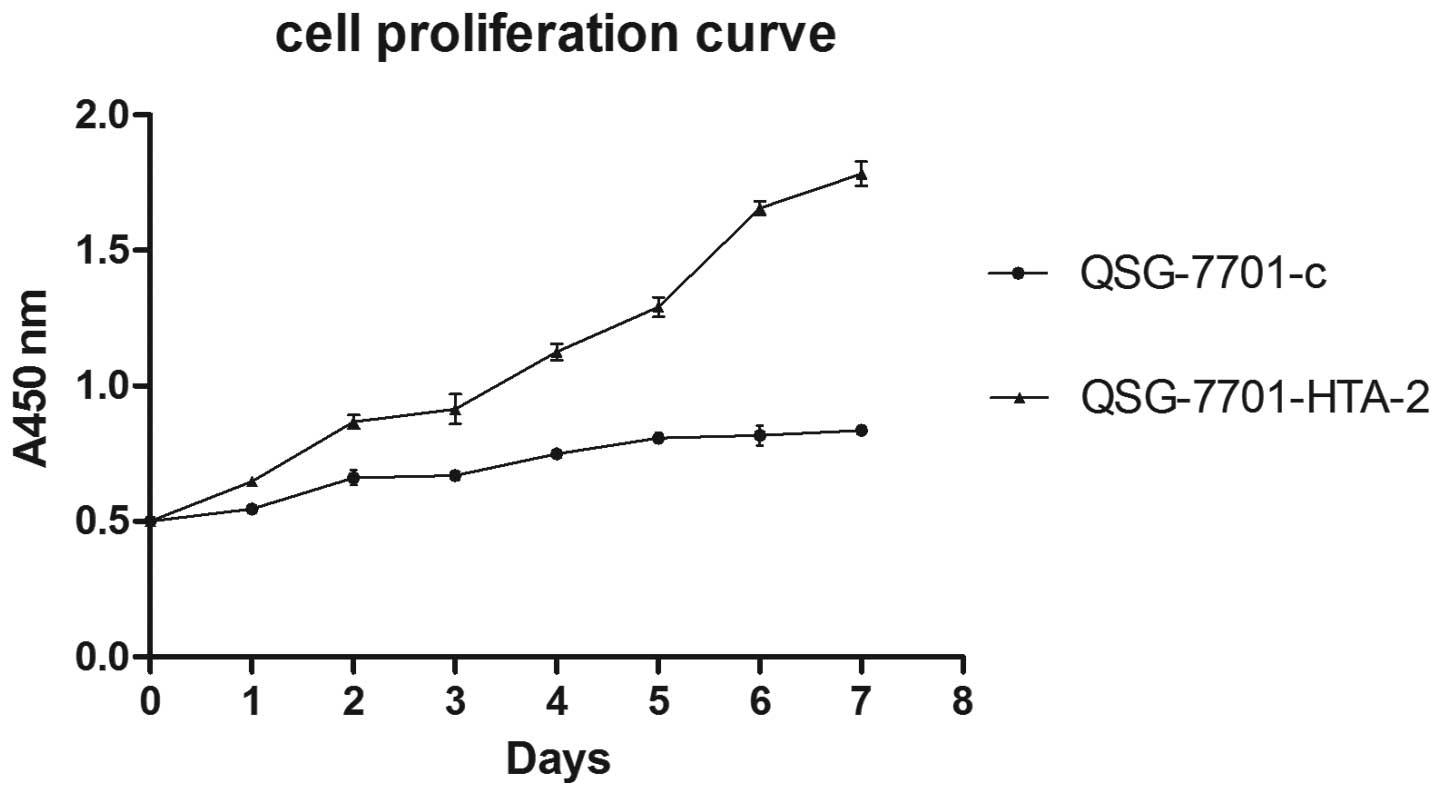
Overexpression of HTA promotes cell
growth in vitro
The rate of the cell proliferation in the clone
QSG-7701-HTA-2 was significantly increased compared to QSG-7701-c.
Cell proliferation curve detected by CCK-8 assay (Fig. 8) and colony formation assay
(Fig. 9) showed that the
overexpression of HTA upregulated the cell proliferation and
colony formation ability of QSG-7701 cells. The cell doubling time
of QSG-7701-HTA-2 was 3.87±0.187 days, while QSG-7701-c was
9.14 ±0.143 days. Cell cycle distribution detected by flow
cytometry is shown in Fig. 10.
This result is in line with the above results.
Discussion
HTA is a novel tumor associated gene screened
by our research group. To date, only a few studies have focused on
the HTA gene. Specific expression characteristics and the
cancer-promoting effect revealed by knockdown experiment suggested
that HTA may play a role in HCC development (10). In order to thoroughly explore the
role of HTA, the full length sequence of HTA is imperative
for the further investigations such as overexpression and
functional clarification. In this study, the full length cDNA of
HTA was obtained by 3′-RACE and 5′-RACE; HTA mRNA expression
in different hepatic cell lines and normal cell lines was analyzed
by northern blot assay, and the over expression of HTA cDNA in the
hepatic cell line was performed to clarify the biological function
of the HTA gene.
There were certain difficulties in obtaining the
full length cDNA sequence of the HTA gene, due to its relatively
low expression even in the HepG2 cell line, which is considered to
have the highest expression of the HTA gene. The smart technology,
which is considered the most advanced approach to obtain the full
length cDNA of a certain gene with a partial known sequence, was
chosen for the full length cDNA of the HTA gene (11). At the same time, hot start PCR
polymerase was applied to avoid non-specific amplification in lower
temperatures (14) and a
touch-down PCR program was applied to avoid non-specific
amplification in early cycles and to improve the efficiency of
specific amplification in later cycles of the program (15).
The accuracy and integrity of the cDNA sequence
obtained by 3′/5′-RACE require further confirmation. Following
3′/5′-RACE, RT-PCR was performed. The sequence of HTA cDNA was
amplified by full length primer. As the HTA gene is unique in
quadrumana and exclusively found in the genome of humans and
anthropoids, we could not confirm its full length cDNA sequence by
homologous alignment (16).
Therefore, the full length of the HTA gene according to the
disciplines as following: the ready frame of the gene was integral:
there was a terminal codon in the same frame in the upstream of the
ORF’s initial codon ATG and there was polyA tail. At the same time,
it was proved that the length of HTA transcripts defined by
northern blot analysis was consistent with the sequence obtained by
RACE.
Alternative splicing occurs as a normal phenomenon
in eukaryotes, where it greatly increases the biodiversity of
proteins that can be encoded by the genome (17,18).
In humans, approximately 95% of multi-exonic genes are
alternatively spliced (18). There
are numerous modes of alternative splicing observed, of which the
most common is exon skipping (17). It was reported that alternative
splicing plays a crucial role in receptor diversity generation and
growth and development regulation (19,20),
particularly reflected in the nervous (21,22)
and the immune system (23,24),
which was closely related to their function diversity and reaction
sensibility. It was estimated that 15% of disease occurred
mutations would affect the splicing of pre-mRNA (25,26).
We clarified that the second introns of the HTA gene were
selectively retained in mRNA and produced a small quantity of
longer transcript. Whether this alternative splicing plays a role
in carcinogenesis requires further elucidation.
In vitro experiments revealed that
overexpression of the HTA gene in the hepatic cell line QSG-7701
promotes the proliferation and colony formation ability of the
cells, which proved the carcinogenesis by the HTA gene, and it was
consistent with the interference experiment in a previous study
(10). The result confirmed that
the HTA gene plays an important role in HCC development.
In conclusion, this study obtained and identified
the full length cDNA sequence of the HTA gene and analyzed its
alternative splicing form, further elucidating the structure of the
HTA gene and providing the foundations for HTA gene function
studies. Primary function study of the HTA gene by overexpression
in vitro proved its role in HCC carcinogenesis.
Acknowledgements
This study was supported by the
National Natural Science Foundation of China (81201903 and
2010CB833605), the Science and Technology Department Research
Foundation of Hunan Province (2010SK3132), and the PhD Candidates’
Research Innovation Project of Hunan Province (CX2011B050),
China.
References
|
1
|
El-Serag HB: Hepatocellular carcinoma. N
Engl J Med. 365:1118–1127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
3
|
DuBray BJ, Chapman WC and Anderson CD:
Hepatocellular carcinoma: a review of the surgical approaches to
management. Mo Med. 108:195–198. 2011.PubMed/NCBI
|
|
4
|
Jia HL, Ye QH, Qin LX, et al: Gene
expression profiling reveals potential biomarkers of human
hepatocellular carcinoma. Clin Cancer Res. 13:1133–1139. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Keng VW, Villanueva A, Chiang DY, et al: A
conditional transposon-based insertional mutagenesis screen for
genes associated with mouse hepatocellular carcinoma. Nat
Biotechnol. 27:264–274. 2009. View
Article : Google Scholar
|
|
6
|
Zender L, Xue W, Zuber J, et al: An
oncogenomics-based in vivo RNAi screen identifies tumor suppressors
in liver cancer. Cell. 135:852–864. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pei Y, Zhang T, Renault V and Zhang X: An
overview of hepatocellular carcinoma study by omics-based methods.
Acta Biochim Biophys Sin (Shanghai). 41:1–15. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Villanueva A, Minguez B, Forner A, Reig M
and Llovet JM: Hepatocellular carcinoma: novel molecular approaches
for diagnosis, prognosis, and therapy. Annu Rev Med. 61:317–328.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zender L, Villanueva A, Tovar V, Sia D,
Chiang DY and Llovet JM: Cancer gene discovery in hepatocellular
carcinoma. J Hepatol. 52:921–929. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Y, Li Y, Guo F, et al: Identification
of HTA as a novel-specific marker for human hepatocellular
carcinoma. J Cancer Res Clin Oncol. 136:1187–1192. 2010.
|
|
11
|
Scotto-Lavino E, Du G and Frohman MA: 5′
end cDNA amplification using classic RACE. Nat Protoc. 1:2555–2562.
2006.
|
|
12
|
Scotto-Lavino E, Du G and Frohman MA: 3′
end cDNA amplification using classic RACE. Nat Protoc. 1:2742–2745.
2006.
|
|
13
|
Kozak M: Structural features in eukaryotic
mRNAs that modulate the initiation of translation. J Biol Chem.
266:19867–19870. 1991.PubMed/NCBI
|
|
14
|
Paul N, Shum J and Le T: Hot start PCR.
Methods Mol Biol. 630:301–318. 2010. View Article : Google Scholar
|
|
15
|
Pratyush DD, Tiwari S, Kumar A and Singh
SK: A new approach to touch down method using betaine as co-solvent
for increased specificity and intensity of GC rich gene
amplification. Gene. 497:269–272. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Loytynoja A: Alignment methods:
strategies, challenges, benchmarking, and comparative overview.
Methods Mol Biol. 855:203–235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Black DL: Mechanisms of alternative
pre-messenger RNA splicing. Annu Rev Biochem. 72:291–336. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pan Q, Shai O, Lee LJ, Frey BJ and
Blencowe BJ: Deep surveying of alternative splicing complexity in
the human transcriptome by high-throughput sequencing. Nat Genet.
40:1413–1415. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roberts GC and Smith CW: Alternative
splicing: combinatorial output from the genome. Curr Opin Chem
Biol. 6:375–383. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kelemen O, Convertini P, Zhang Z, et al:
Function of alternative splicing. Gene. Aug 15–2012.(Epub ahead of
print).
|
|
21
|
Norris AD and Calarco JA: Emerging roles
of alternative pre-mRNA splicing regulation in neuronal development
and function. Front Neurosci. 6:1222012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dever SM, Xu R, Fitting S, Knapp PE and
Hauser KF: Differential expression and HIV-1 regulation of
μ-opioid receptor splice variants across human central
nervous system cell types. J Neurovirol. 18:181–190.
2012.PubMed/NCBI
|
|
23
|
Magg T, Mannert J, Ellwart JW, Schmid I
and Albert MH: Subcellular localization of FOXP3 in human
regulatory and nonregulatory T cells. Eur J Immunol. 42:1627–1638.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Martinez NM, Pan Q, Cole BS, et al:
Alternative splicing networks regulated by signaling in human T
cells. RNA. 18:1029–1040. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu J, Lee W, Jiang Z, et al: Genome and
transcriptome sequencing of lung cancers reveal diverse mutational
and splicing events. Genome Res. Nov 1–2012.(Epub ahead of
print).
|
|
26
|
Singh RK and Cooper TA: Pre-mRNA splicing
in disease and therapeutics. Trends Mol Med. 18:472–482. 2012.
View Article : Google Scholar : PubMed/NCBI
|