Introduction
Breast cancer is functionally classified based on
molecular profiles. Estrogen receptor (ER), progesterone receptor
(PR) and ErbB-2/human epidermal growth factor receptor 2 (HER-2)
status are molecular markers used to determine breast cancer
subtypes as well as targets for treatment (1,2). In
contrast, triple-negative breast cancer (TNBC) is a breast cancer
subtype defined by the lack of expression of ER, PR and HER-2.
Treatment of TNBC, which often presents with a more aggressive
phenotype, is more difficult due to the paucity of potential target
molecules. Therefore, there is a critical need to enhance current
systemic treatments and/or identify new targets for the treatment
of TNBC (3–5).
Because of the strong correlation between tumor
development and specific mutations in the regulatory function of
certain cell cycle kinases and cyclin-dependent kinases (CDKs),
their potential as targets for anticancer drug design has
intensified, with the goal of overcoming the therapeutic challenges
presented by TNBC (6–9). Because the centrosome cycle is
regulated by protein phosphorylation and given the importance of
mitotic and centrosomal kinases, they are attractive targets for
anti-mitotic anticancer drugs (10–17).
NIMA-related kinase 2 (Nek2), a serine/threonine
centrosomal kinase that is highly expressed and activated during
the S and G2 phases, is such a target (18,19).
Overexpressed Nek2 results in premature centrosome splitting, while
centrosomal abnormalities, monopolar spindles and aneuploidy result
from overexpression of kinase-dead Nek2 (20,21).
Recently, studies have shown that Nek2 expression is elevated in
various cancer cell lines, including various breast tumors
(22–24). Tsunoda et al(23) demonstrated that Nek2 siRNA could
reduce the tumor volume in mouse xenografts. However, combinational
studies using Nek2 gene depletion with anticancer drugs have not
been reported. In spite of the increasing evidence of the
importance of Nek2 in cancer development, its role in cancer is
still far from clear.
In this study, we investigated whether Nek2
depletion by antisense oligonucleotides (ASO) or small interfering
RNA (siRNA) against Nek2, promoted drug sensitivity in the TNBC
cell lines MDA-MB-231 and MDA-MB-468. Doxorubicin and paclitaxel
treated cells were used as positive controls due to the reported
problem of low vulnerability (25)
with untreated cells as negative controls. Here, we show the effect
of Nek2 depletion using siRNA and ASO on two different TNBC cell
lines, alone and in combination with paclitaxel and doxorubicin.
Alone, siRNA and ASO showed significant reductions in cell
viability, mitotic spindle fiber formation and apoptosis. However,
in combination with paclitaxel or doxorubicin, Nek2 depletion
induced an increase in mitotic abnormalities and apoptosis above
either silencing alone or anticancer drug treatment alone. Given
the difficulty in effectively treating TNBC, our results suggest
that either siRNA or ASO targeted against Nek2 may increase TNBC
sensitivity to chemotherapy treatments.
Materials and methods
Cell culture and transfections
MDA-MB-231 and MDA-MB-468 breast cancer cells (ATCC,
Manassas, VA) were cultured in Dulbecco’s modified Eagle’s medium
(DMEM) supplemented with 10% fetal bovine serum (FBS) and
penicillin/streptomycin (100 IU/ml and 100 μg/ml, respectively)
under 5% CO2 in humid conditions at 37°C.
Oligonucleotide transfections were carried out using Lipofectamine
2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s
instructions. ASO and siRNA were synthesized by Integrated DNA
Technologies Inc., (Coralville, IA). The antisense sequence of
phosphorothioate-modified oligodeoxynucleotides against Nek2 was:
5′-GAGCCTGTGCCAATGGTG. The siRNA sequences targeted to Nek2 were
5′-CCAAGGAAAGGCAAUACUUUUdTdT-3′ (sense) and
5′-AAGUAUUGCCUUUCCUUGGUUdTdT-3′ (antisense). siRNA-A (Santa Cruz
Biotechnology, Santa Cruz, CA) was introduced into cells as
negative control. Null transfected cells were incubated with
Lipofectamine 2000 alone. Cells were collected 24 h after
transfection and analyzed for changes in transcript levels of Nek2.
Cells were analyzed 48 h after transfection, by fluorescence
activated cell sorting (FACS) and western blot analysis for protein
expression.
Semi quantitative reverse transcriptase
PCR (RT-PCR)
Total RNA from cells was isolated using RNeasy mini
kit (Qiagen, Valencia, CA) following supplier’s instructions and
treated with DNase I (Promega, Madison, WI) to remove DNA
contamination. To generate cDNA from purified total RNA, 1 μg of
total RNA was added to the SuperScript III first-strand synthesis
system reaction mixture according to the manufacturer’s
(Invitrogen) protocol. Equal amounts of synthesized cDNAs were used
to carry out the semi quantitative RT-PCR reactions using the
GeneAmp fast PCR Master mix (Applied Biosystems, Carlsbad, CA). The
Nek2 primer sequences were: forward, 5′-CCACAGACGAAGTGATGGTG-3′;
reverse, 5′-TGATTTTCCCAGCGAGTTCT-3′. Glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) was used as control. The primer sequences
were: forward, 5′-CACCACCATGGAGAAGGGTG-3′; reverse,
5′-GAGGCATTGCTGTAGATCTTGAGG-3′.
Immunoblotting analysis
Cells were lysed with 20 mmol/l Tris-HCl (pH 8.0),
137 mmol/l NaCl, 10% glycerol, 1% Triton X-100, 2 mmol/l EDTA
containing protease and phosphatase inhibitors at 4°C. A total of
20 μg of each lysate was separated by SDS-PAGE and transferred onto
a nitrocellulose membrane. Mouse monoclonal antibody against Nek2
(Santa Cruz Biotechnology, 1:1,000), mouse monoclonal
anti-γ-tubulin antibody (Sigma-Aldrich, St. Louis, MO, 1:10,000)
and peroxidase-conjugated goat anti-mouse IgG (AnaSpec, Freemont,
CA; 1:10,000) were used for immunoblot analyses.
Cell viability assay
The XTT Cell Proliferation Kit (Roche Applied
Science, Indianapolis, IN) was used to analyze cell viability.
Cells were seeded at 8,000 cells/well into a 96-well culture plate
in a final volume of 100 μl. The XTT mixture (50 μl) was added to
the wells and incubated for 10 h at 37°C. The absorbance of the
samples was measured using a Molecular Devices (Sunnyvale, CA)
microplate reader at 450 nm against the reference wavelength of 650
nm. Cell viability assays were carried out at least 3 times
independently.
Cell synchronization and FACS
Cells were synchronized by double thymidine.
Briefly, after 24 h incubation, 1×106 cells were exposed
to 2 mmole/l of thymidine for 18 h to presynchronize the cells in S
phase then released by refreshing the medium for 9 h. After
release, 2 mmole/l of thymidine was added to block cell cycle for
17 h followed by fresh DMEM to release cell cycle arrest allowing
cells to move forward synchronously throughout G2/M. Cells were
harvested 48 h later. Cell cycle distribution was analyzed by FACS.
Collected cells were washed in ice-cold PBS, fixed in 70% ethanol
and stored at −20°C until analysis. For FACS, DNA was stained with
PBS containing 40 μg/ml of propidium iodide, 100 μg/ml of RNase A,
and 0.1% Triton X-100 for 30 min at 37°C. DNA from 10,000 cells was
evaluated with a FACSCalibur flow cytometer (Becton-Dickinson,
Franklin Lakes, NJ) and cell cycle phases were determined using FCS
Express 4.
Immunofluorescence analysis
Cells were fixed in cold methanol and processed for
immunocytochemistry. Primary antibodies: mouse monoclonal
anti-γ-tubulin (Sigma-Aldrich, 1:1,000) and mouse monoclonal
anti-α-tubulin (Abcam, Cambridge, MA; 1:1,000). Secondary
antibodies: FITC conjugated goat anti-mouse antibody (Abcam; 1:50)
and goat anti-mouse rhoda-mine conjugated antibody (Upstate,
Billerica; MA, 1:500). DNA was counterstained with
4′,6-diamidino-2-phenylindole (DAPI). Cells were visualized with an
Olympus IX71 inverted deconvolving epifluorescence microscope under
40X using SimplePCI software (Compix).
Apoptosis detection
Apoptosis of treated cells was detected using FITC
Annexin V/Dead cell apoptosis kit for flow cytometry (Invitrogen).
After staining, fluorescence emission at 530 nm for Annexin V (FL1)
and >575 nm for propidium iodide (FL3) was performed using a
FACSCalibur flow cytometer (Becton-Dickinson). Data were analyzed
using FCS Express 4. Each experiment was performed a minimum of
three times to generate statistically relevant results.
Results
Expression of Nek2 following transfection
with Nek2 siRNA or ASO
M231 cells transfected with siRNA and ASO showed
gradual loss of Nek2 mRNA expression (Fig. 1). In M468 cells, high
concentrations of siRNA demonstrated significantly decreased mRNA
expression (Fig. 1D). With ASO
transfection, Nek2 mRNA expression declined noticeably even at 5 nM
compared with 5 and 10 nM siRNA in M468 cells with higher ASO
concentrations suppressing Nek2 mRNA expression very effectively
(Fig. 1E).
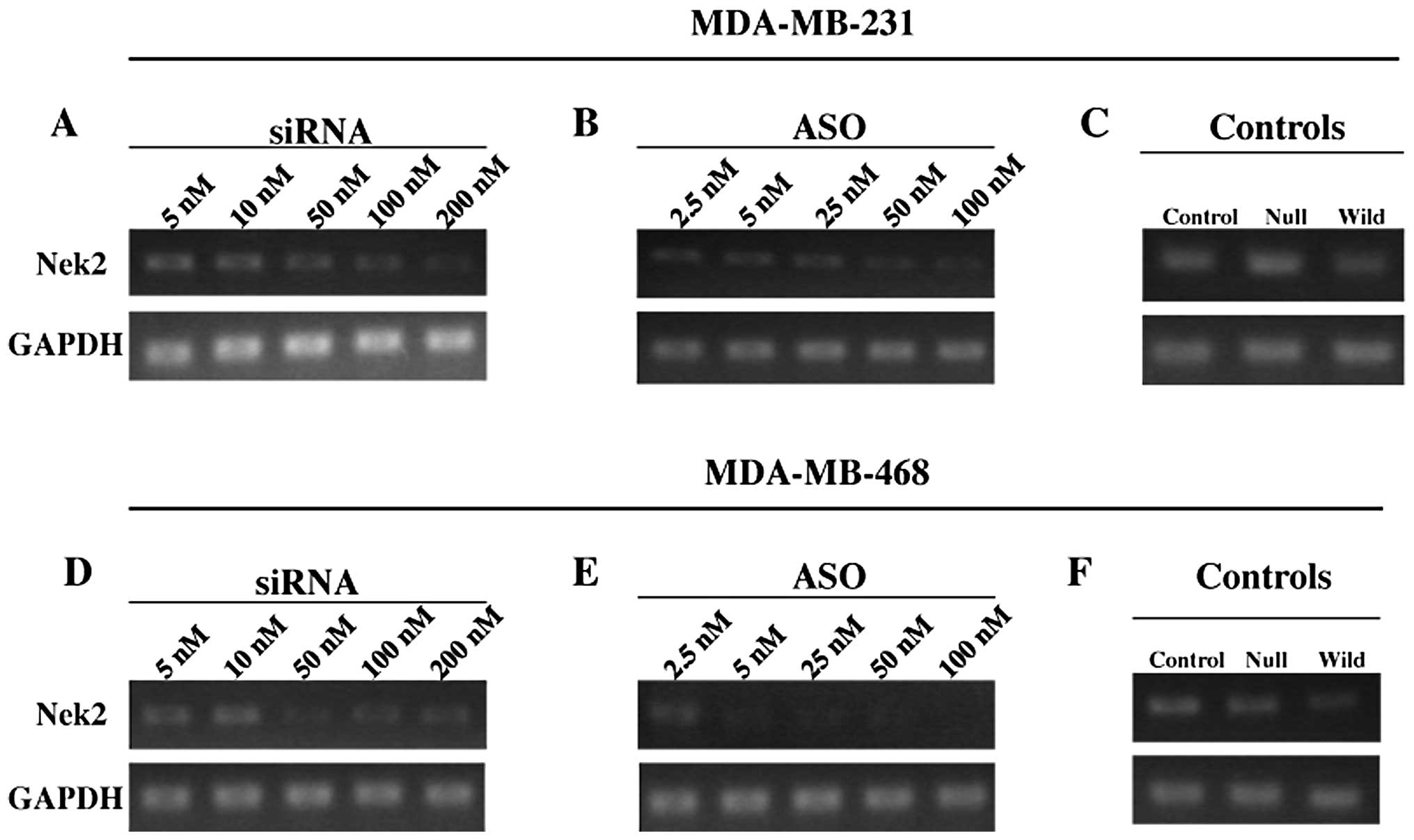 | Figure 1Representative semiquantitative RT-PCR
of siRNA or ASO transfection against Nek2 in M231 and M468 triple
negative breast cancer cells. RT-PCR analysis of M231 and M468
cells 24 h post-transfection with (A and D) 5, 10, 50, 100 or 200
nM siRNA against Nek2 and (B and E) 2.5, 5, 25, 50 and 100 nM ASO
against Nek2, respectively. Each representative gel shows the Nek2
gene product (upper) and GAPDH for control (below). Transfection
controls for (C) M231 and (F) M468 were analyzed using control
siRNA transfection reagents, null transfection with Lipofectamine
2000 alone, and wild-type, respectively. Each experiment was
conducted and analyzed from 3 independent trials. |
The next step was to determine whether Nek2
depletion attenuated protein production or if expression quickly
recovered. Therefore, effects of ASO or siRNA against Nek2 protein
expression were analyzed 48 h post-transfection. As shown in
Fig. 2, Nek2 protein expression
was significantly reduced at concentrations of 50 nM siRNA
(Fig. 2B). Similarly, higher
concentrations of ASO (25 to 100 nM) showed a significant decrease
in Nek2 expression (Fig. 2C).
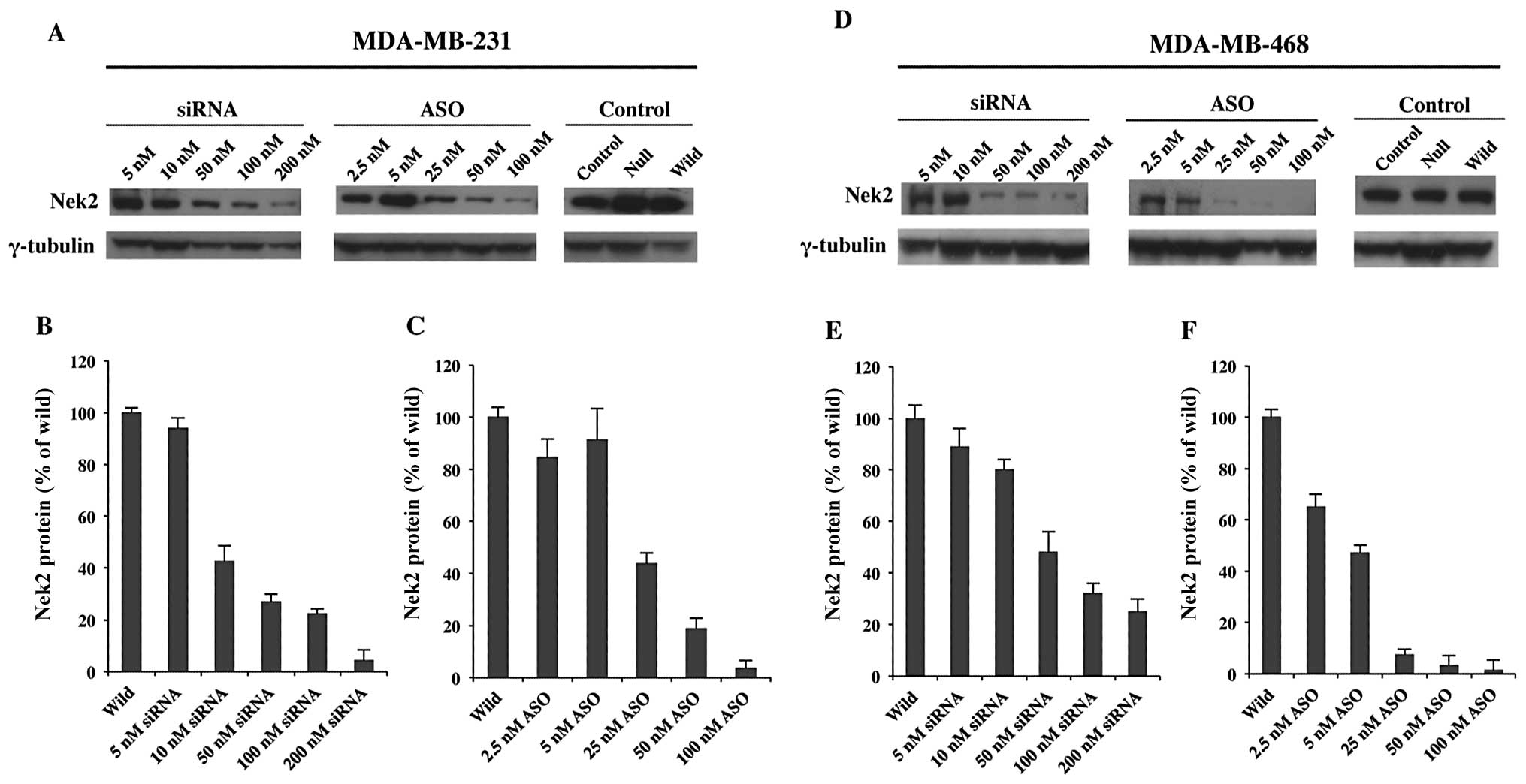 | Figure 2Protein expression levels of Nek2
siRNA or ASO transfected M231 and M468 TNBC cells analyzed by
western blot analysis. (A and D) Nek2 protein expression after 48-h
transfection in M231 and M468 cells, respectively, with 5, 10, 50,
100 and 200 nM of Nek2 siRNA (left) or 2.5, 5, 25, 50 and 100 nM of
Nek2 ASO (center). Control cells were treated with control siRNA,
Lipofectamine 2000 alone or untreated (right). γ-tubulin was used
as the loading control for each cell line. The bar graphs represent
Nek2 protein expression levels with different concentrations of (B
and E) siRNA or (C and F) ASO. Nek2 protein expression is given as
a percentage standardized against Nek2 expression levels in
wild-type cells. Average of three independent experiments and
standard deviations are shown (n=3; error bar, standard
deviations). |
M468 siRNA and ASO treatments (Fig. 2D–F) also demonstrated overall
downregulation of Nek2 protein expression. Higher siRNA
concentration results were not as dramatic as those observed for
ASO. However, protein expression was still markedly reduced. ASO
treatment in M486 cells exhibited a more profound effect when
administered at higher concentrations (Fig. 2F). Analyses confirmed that Nek2
transcription and translation was successfully depleted by both
siRNA and ASO and that the level of depletion was dependent upon
concentration.
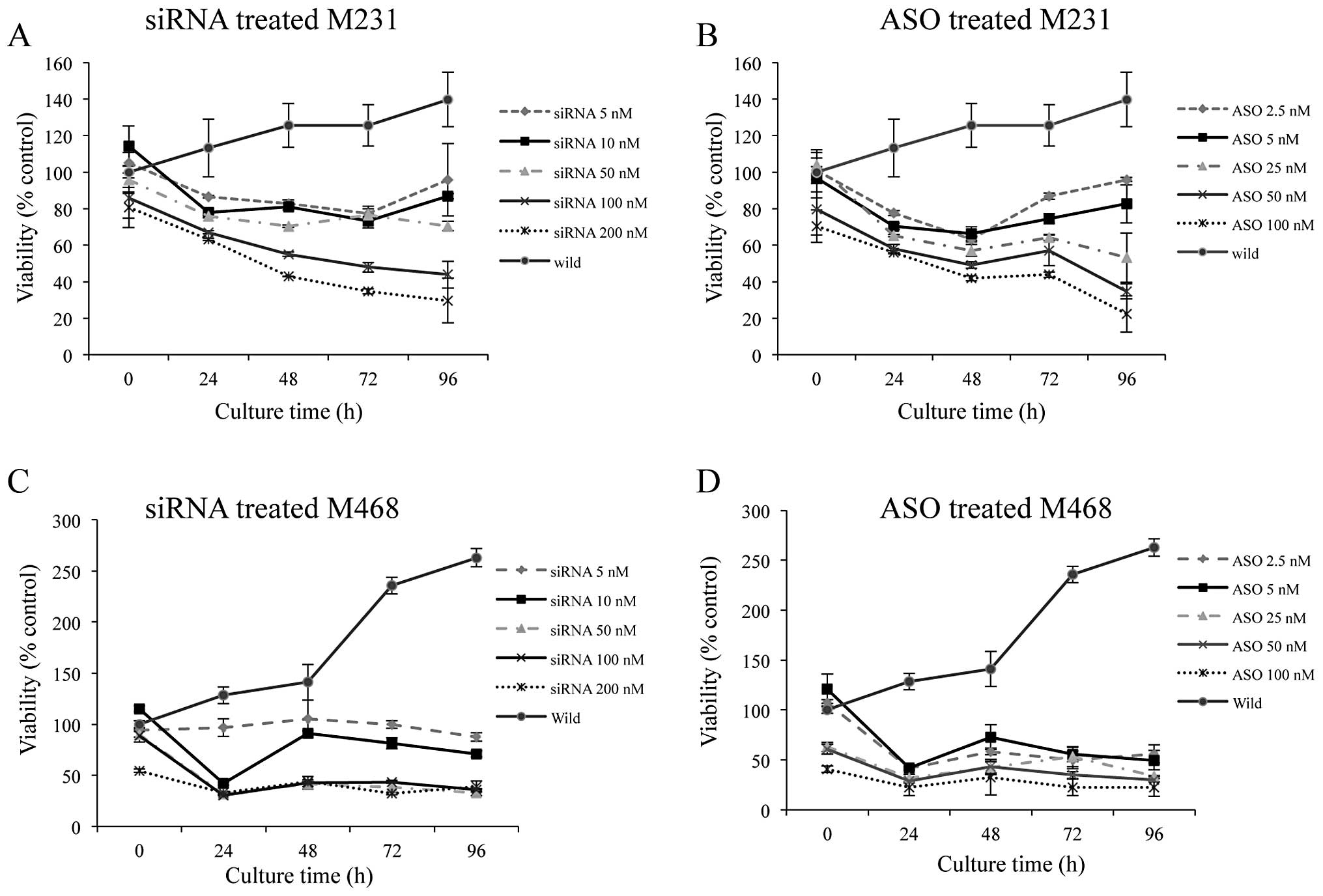
Cell viability of Nek2 depleted
cells
Viability of siRNA transfected M231 cells decreased
continuously from 24 to 72 h. After 96 h post-transfection,
viability levels plateaued except in 5 and 10 nM siRNA transfected
cells (Fig. 3A). Similarly, cell
viability for ASO-transfected M231 cells demonstrated continuous
decrease to 72 h with concentrations of 25 nM and greater showing
no significant changes at 96 h (Fig.
3B).
Transfection of M468 cells with increasing
concentrations of siRNA or ASO reduced cell viabilities more
effectively (Fig. 3C and D)
compared to M231 cells (Fig. 3A and
B). Cell viability of M468 cells transfected with 5 nM siRNA
showed no change in Nek2 expression. However, with the addition of
50–200 nM siRNA, cell viability significantly decreased after 24 h
(down to 32%) and remained low (33%) through 96 h (Fig. 3C).
M468 cells transfected with Nek2-ASO demonstrated
significantly reduced viabilities (between 55 to 22%) for all
concentrations (Fig. 3D). Based on
these results, 50 nM siRNA and 25 nM ASO were chosen as the optimal
concentrations for both cell lines in the subsequent combinatorial
studies of siRNA or ASO pretreatment before antitumor agent
application. Cell viability of the wild-type (untreated cells) at
time 0 h was normalized as 100%. Cell viability fluctuations for
treated samples at time 0 h were most likely due to the effects of
Nek2 depletion on cell proliferation during the 10-h XTT reaction
time. Cell viability observed over 100% in the wild-type control
was attributed to continued cell proliferation and metabolism
through subsequent time points.
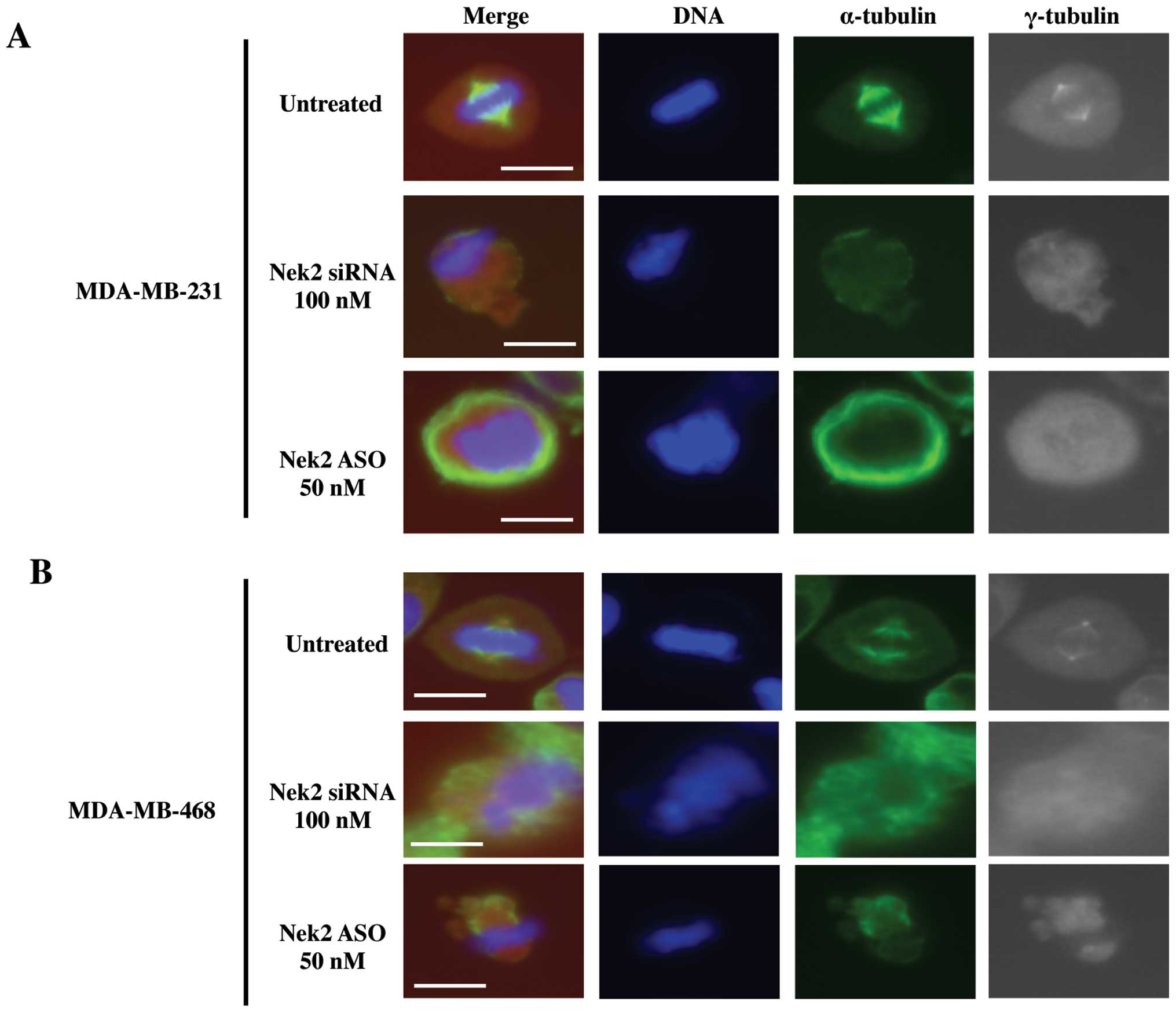
Nek2 is crucial for mitotic spindle
formation
In order to determine the effects of silenced Nek2
on microtubules and spindle pole formation, immunofluorescence
microscopy was used to visualize mitotic spindle structural
changes. Upon release from cell cycle arrest, disrupted mitoses
were observed in both siRNA and ASO transfected cells (Fig. 4A and B) whereas untreated cells
exhibited no mitotic deformation. Both cell lines with silenced
Nek2 demonstrated diffuse spindle poles (Fig. 4A and B, gray images) and malformed
microtubules (Fig. 4A and B, green
images). In addition, α-tubulin signal intensities in the
spindle microtubules for transfected cells were substantially
reduced compared with controls. Similar spindle structures in both
Nek2 depleted cell lines were observed. These abnormal mitotic
structures were classified into several types. First, Nek2
downregulated cells retained fewer microtubules (Fig. 4A and B, green images), and the
γ-tubulin centrosome-associated signal was weak (Fig. 4A and B, gray images) compared to
untreated cells. Lack of clear centrosomal staining against
γ-tubulin suggests it is lost due to Nek2 depletion. Alternatively,
duplicated centrosomes were not separated properly, or microtubule
formation from the recently separated centrosomes was defective.
Also, because Nek2 regulates chromosome alignment and signaling of
the spindle assembly checkpoint (26), inhibition of Nek2 by siRNA or ASO
lead to misaligned chromosomes at meta-phase (Fig. 4A and B, DAPI staining). This data
suggests that Nek2 is required for proper mitotic spindle formation
since cell death ensued as a result of abnormal microtubule
generation or centrosome duplication/separation upon Nek2 silencing
with either siRNA or ASO.
The effects of Nek2 siRNA or ASO
transfection on cell cycle distribution and apoptosis
Before studying combinatorial treatment efficacy for
siRNA and ASO with anticancer drugs, it was important to establish
whether cell cycle distribution and apoptosis levels would be
changed with Nek2 depletion.
siRNA or ASO treated M231 cell lines showed little
mitotic activity (Fig. 5). While a
small increase in the percent of cells in 4n was observed
regardless of treatment, significant changes in cell cycle profiles
between control and siRNA or ASO transfected cells was not evident.
Treatment with 100 nM siRNA increased cells in 4n by 10%. Cells
treated with 50 nM ASO demonstrated a 6% increase in 4n compared to
controls (Fig. 5A).
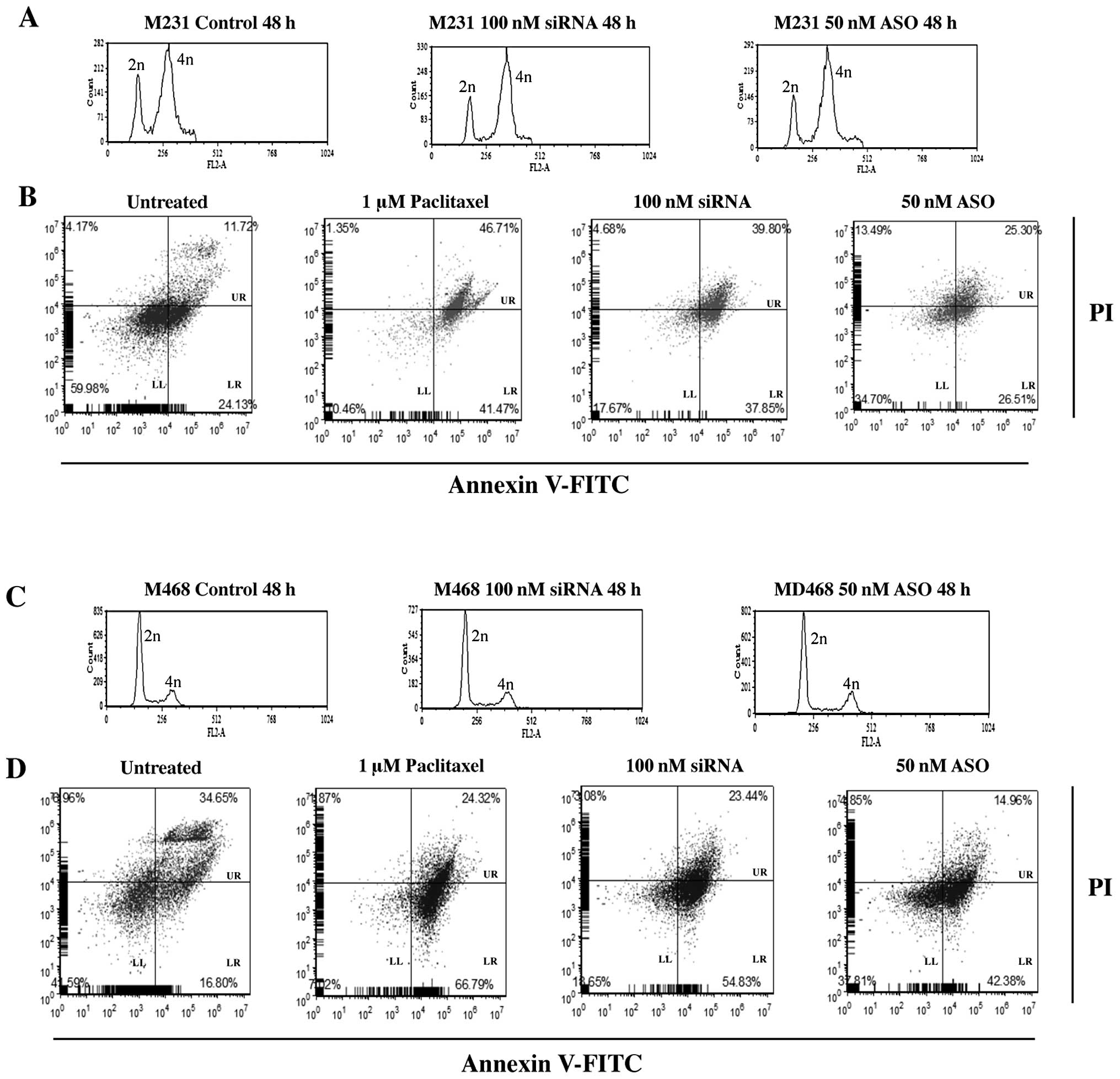 | Figure 5Representative results of cell cycle
distribution and apoptosis for 100 nM Nek2 siRNA or 50 nM Nek2 ASO
in M468 and M231 TNBC cells. (A) M231 and (C) M468 cell cycle
distribution of non-transfected (left), 100 nM siRNA (middle), or
50 nM ASO (right). Cell cycle was synchronized using a double
thymidine block. Time indicates released time after thymidine
treatment. After cell cycle synchronization, cells were transfected
with siRNA or ASO. Cells were harvested 48 h later. Cells were
fixed in ethanol and the DNA content was analyzed by flow cytometry
following propidium iodide (PI) staining. In A and C, the x-axis
demonstrates fluorescence intensity based on DNA content, and the
y-axis corresponds to the number of fluorescent cells. B and D
represent parallel cell cultures stained using FITC-conjugated
Annexin V and PI to analyze apoptosis using flow cytometry (LL,
live cells; LR, apoptotic cells; UR, dead, necrotic and late
apoptotic cells). Cells were treated with 1 μM paclitaxel, 100 nM
siRNA or 50 nM ASO, respectively. Data are representative results
from at least 3 independent experiments demonstrating
reproducibility. |
To determine whether Nek2 gene silencing alone
caused apoptosis and how apoptosis induction compared to a commonly
used anticancer drug for TNBC, cells were analyzed by FACS 24 h
after siRNA/ASO transfection and paclitaxel treatment. M231 cells
treated with 1 μM paclitaxel alone, showed 41% increase in cell
death. Similarly, 37% increase in cell death was observed with 100
nM siRNA alone. Transfection of 50 nM ASO showed no appreciable
difference in cell death (26%), over untreated cells (24%)
(Fig. 5B).
M468 cell treatments demonstrated similar cell cycle
distribution results. There was a 12% accumulation in 4n DNA
content for control cells but only 17% for siRNA and 15% for
ASO-transfected cells (Fig. 5C).
However, M468 cells were more sensitive to apoptosis induction.
Paclitaxel (1 μM) induced apoptosis in 66% of the treated cell
population. siRNA (100 nM) increased apoptosis to 54%.
Interestingly, 50 nM ASO treatment increased apoptosis to 42%
compared to the 16% observed for controls.
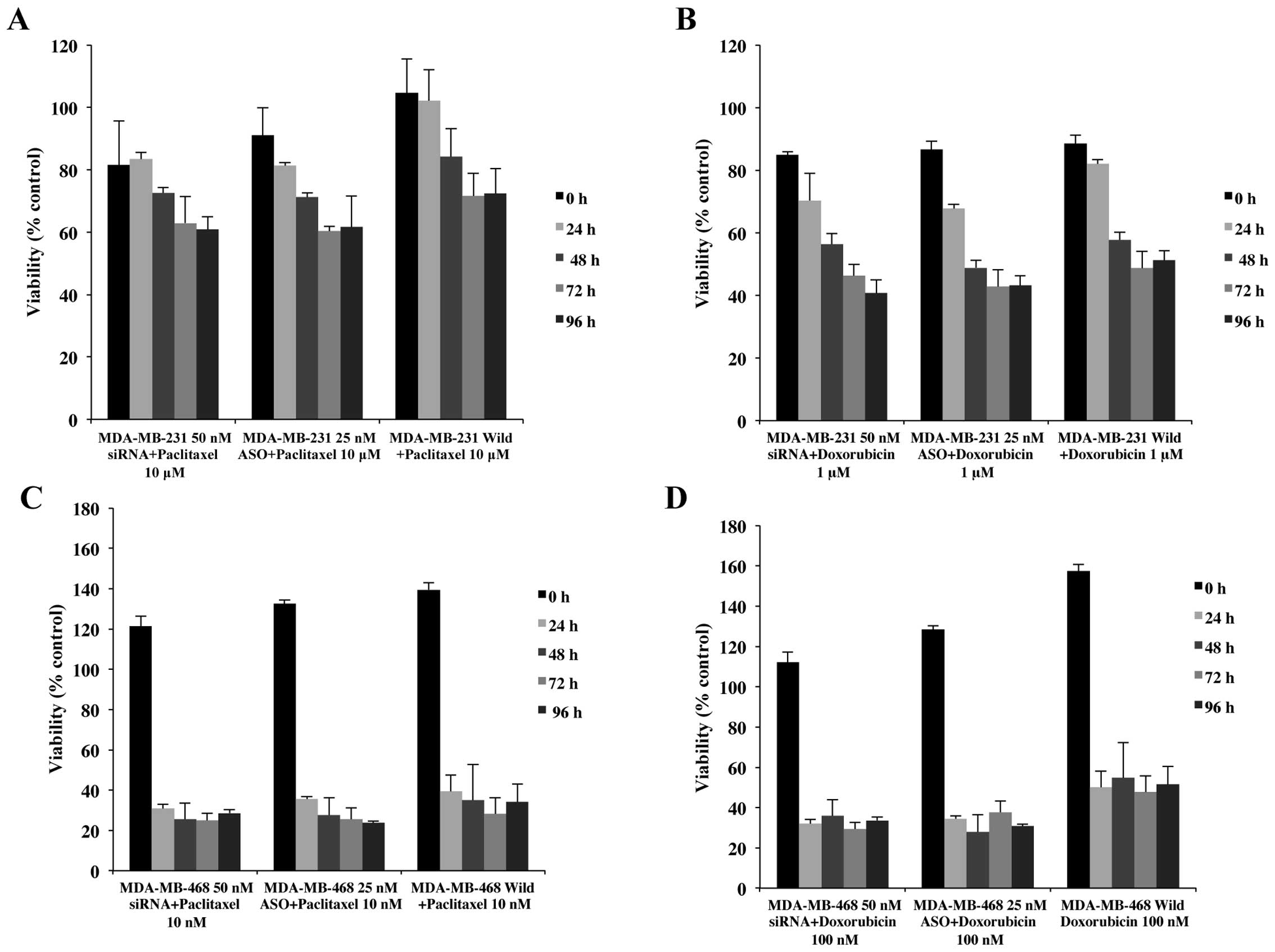
Combinatorial anticancer drug treatment
effects on triple negative breast cancer cells
In order to address this question, paclitaxel and
doxorubicin were chosen because they work through different
mechanisms of action. Paclitaxel binds to the tubulin heterodimer,
effectively stabilizing microtubules by inhibiting
depolymerization, resulting in transient arrest in mitosis,
development of a multinucleated interphase, followed by apoptosis
(27,28). Doxorubicin inhibits release of DNA
torsion during replication and transcription by immobilizing the
topoisomerase II-DNA complex (29–31).
The effects of combinatorial treatment on
M231 cells
To determine whether M231 cell sensitivity to
paclitaxel was augmented with this combinatorial approach,
concentrations from 10 nM to 10 μM of paclitaxel were added to M231
cells pretreated with 50 nM siRNA or 25 nM ASO (Fig. 6A). Although cell viability with 50
nM siRNA + paclitaxel treated cells showed levels below paclitaxel
only treated cells, there was no significant difference between
siRNA combinatorial treatment and paclitaxel concentrations up to 1
μM (data not shown). However, as demonstrated in Fig. 6A, significant decreases in cell
viability (60% vs. 72%) were observed in combination with 10 μM
paclitaxel. Additionally, this combination of siRNA and paclitaxel
generated a consistent decrease through 96 h whereas other
combinations showed cell recovery at 72 or 96 h.
Interestingly, 25 nM ASO transfected M231 cells, in
combination with low concentrations of paclitaxel (10, 100 nM and 1
μM paclitaxel), demonstrated decreases in cell viability from 13 to
30% (data not shown), comparable to 50 nM siRNA + 10 μM paclitaxel
(Fig. 6A). Cell viability for 25
nM ASO + 10 μM paclitaxel was reduced by 19 to 29% (Fig. 6A) compared to controls. These data
confirmed that Nek2 silenced by siRNA or ASO, increased M231 cell
sensitivity to paclitaxel resulting in overall decreased cell
viability.
Cells treated with doxorubicin in combination with
siRNA or ASO against Nek2 demonstrated variable results. M231 cells
treated with 10 nM doxorubicin alone showed no significant changes
in cell viability until 96 h at which time, cell viability
increased significantly. Cells treated with 50 nM siRNA or 25 nM
ASO in combination with 10 nM doxorubicin showed significant
decreased cell viability until 72 h. At 96 h cells treated with
siRNA + doxorubicin recovered slightly, whereas ASO-doxorubicin
mirrored the cell viability increase observed in doxorubicin alone.
Similar trends were observed for combinations of siRNA or ASO with
100 nM doxorubicin.
Comparable cell viability effects were observed with
50 nM siRNA and 25 nM ASO transfected cells + 1 μM doxorubicin
(Fig. 6B) and 10 μM of doxorubicin
(data not shown). Cell viabilities continued to decrease through
the end-point of the experiment. In summary, although 10 and 100 nM
doxorubicin + siRNA or ASO appeared to increase cell viability
during the culture period, the effects of 1 and 10 μM doxorubicin
on M231 cells were significantly enhanced with the addition of Nek2
silencing by siRNA or ASO.
The effects of combinatorial treatment on
M468 cells
Control cells treated with 10 nM to 10 μM of
paclitaxel, all showed increased viabilities at 96 h of treatment.
However, even though the viability levels of transfected M468 cells
were not significantly different between siRNA and ASO transfected
cells with various concentrations of paclitaxel treatment, all of
the cell viabilities of transfected cells were lower than
non-transfected cells and remained at similar levels or showed
minor increases at 96 h (Fig. 6C).
In general, paclitaxel treatment in combination with 50 nM siRNA or
25 nM ASO did not significantly decrease viability compared with
paclitaxel alone.
Doxorubicin treatment in combination with Nek2
inhibition had a more profound effect in M468 cells than M231
cells. No significant increase in cell viability was observed for
control cells treated with 100 nM, 1 or 10 μM doxorubicin. M468
cells transfected with siRNA or ASO resulted in significantly
decreased cell viability (Fig.
6D). Cells transfected with 50 nM siRNA or with 25 nM ASO plus
100 nM doxorubicin showed >34% viability. In contrast, cells in
doxorubicin alone showed ∼50% viability (Fig. 6D). These results indicate that a
combinatorial approach of Nek2 gene silencing with siRNA or ASO and
an anticancer drug increased M468 cell sensitivity compared to
anticancer drug administration alone.
Interestingly, we observed fluctuations in cell
viability with 10 nM doxorubicin + 50 nM siRNA or 25 nM ASO. After
24 h, cell viability for Nek2 silenced cells + doxorubicin was 36
and 43%, respectively. In 10 nM doxorubicin treatment alone, cell
viability was 53%. After 24 h cell viability increased by 20% and
after 72 h an additional 40% increase was observed. Although siRNA
and ASO + doxorubicin showed slight increases in cell viability,
the overall increases compared to non-transfected, at 96 h,
doxorubicin controls were ∼70% lower than controls.
Discussion
In this study, we investigated Nek2 as a potential
drug target in cancer treatment. Additionally, we were interested
in whether pre-conditioning the cells through Nek2 silencing would
augment the effectiveness of current anticancer drugs and in
determining whether a combinatorial approach would maintain
treatment effectiveness while decreasing anticancer drug
concentrations, potentially decreasing associated side-effects. In
order to address these questions, we utilized two
well-characterized TNBC cell lines (MDA-MB-231 and MDA-MB-468).
When comparing the effects of Nek2 depletion by
siRNA or ASO on cell viability, we observed that overall, ASO
treated cells exhibited lower cell viability than siRNA treated
cells. Indeed, the efficiency of siRNA and ASO is controversial.
Some studies reported that when compared, siRNA was more efficient
and its effect was longer than ASO (32,33).
In contrast, Tsui et al(2005) described that the efficiency
of siRNA was comparable to ASO (34). Our cell viability results suggest
that the effectiveness of siRNA and ASO may be cell type dependent.
Alternatively, efficiency may depend upon the target gene.
Before depleting Nek2 using siRNA and ASO, we
predicted that cell cycle would be arrested in the G2/M phase based
on other cell cycle kinase studies (33). Although it has been previously
shown that Nek2 is one of the cell cycle kinases (18–24),
our results showed that Nek2 gene silencing did not induce strong
mitotic arrest at G2/M phase, but instead induced apoptosis,
indicating that the role of Nek2 may be different from other cell
cycle-related kinases in its regulation of cell cycle.
Additionally, inactive Nek2A in human cells did not block cell
cycle progression (21). It may be
possible that other Nek2 family members (i.e., Nek1 to Nek11), or
other cell cycle kinases such as Plk1 or Aurora A compensate for
the loss of Nek2 function. However, Nek2 depleted cells have been
reported to have abnormal mitotic characteristics induced through
several different categories. First, Nek2 depletion interferes with
centrosome duplication or maturation and arrangement of proteins
including γ-tubulin, Plk1 and nucleophosmin/B23 onto the mitotic
spindle poles. Secondly, Nek2 silencing induces abnormal chromosome
segregation in human cells (35).
Thirdly, Nek2 depletion interferes with the regulation of
centrosome separation. Finally, depletion of Nek2 causes arrest of
cell proliferation and increases apoptosis as a result of mitotic
errors (36). As we suspected,
Nek2 depletion affected the kinetochores-microtubule attachment,
inducing a spindle checkpoint imbalance and abnormal clustering of
kinetochore components, resulting in increased sensitivity of cells
to the microtubule targeting anticancer drug paclitaxel.
The increase in cell sensitivity of Nek2-depleted
TNBC cells in combination with doxorubicin may be due to the
down-regulation of TRF1 and the checkpoint kinase, Chk2, inducing
chromosomal abnormalities and altering the cell cycle (31). Prime et al demonstrated
interactions between Trf1 with Mad1 and Nek2 (37). However, more study is needed to
elucidate their relationship with Nek2 and the implications for
cancer development.
Our results suggest that combinational
administration of Nek2 depletion and these chemotherapy agents
increase TNBC cell sensitivity to anticancer treatment. We observed
that the effects of siRNA and ASO + anticancer agent was comparable
for both cells and showed significant decreases from control
non-transfected cells treated with agent alone. By pretreating the
patient with siRNA or ASO therapies, the potential exists for
equivalent or higher sensitization with lower dosages anticancer
drugs. While achieving the same overall effect, the side-effects to
the patient may be reduced.
Acknowledgements
We wish to thank the TTU Imaging
Center, the TTU Biotechnology Core Facilities as well as Dr Dmitri
Pappas (Department of Chemistry and Biochemistry) for access to the
FACSCalibur cell sorter.
References
|
1
|
Baselga J, Norton L, Albanell J, Kim YM
and Mendelsohn J: Recombinant humanized anti-HER2 antibody
(Herceptin) enhances the antitumor activity of paclitaxel and
doxorubicin against HER2/neu overexpressing human breast cancer
xenografts. Cancer Res. 58:2825–2831. 1998.
|
|
2
|
Cleator S, Heller W and Coombes RC:
Triple-negative breast cancer: therapeutic options. Lancet Oncol.
8:235–244. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rakha EA and Ellis IO:
Triple-negative/basal-like breast cancer: review. Pathology.
41:40–47. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gluz O, Liedtke C, Gottschalk N, Pusztai
L, Nitz U and Harbeck N: Triple-negative breast cancer - current
status and future directions. Ann Oncol. 20:1913–1927. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Carey L, Winer E, Viale G, Cameron D and
Gianni L: Triple-negative breast cancer: disease entity or title of
convenience? Nat Rev Clin Oncol. 7:683–692. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pérez de Castro I, de Cárcer G, Montoya G
and Malumbres M: Emerging cancer therapeutic opportunities by
inhibiting mitotic kinases. Curr Opin Pharmacol. 8:375–383.
2008.PubMed/NCBI
|
|
7
|
Chi YH and Jeang KT: Aneuploidy and
cancer. J Cell Biochem. 102:531–538. 2007. View Article : Google Scholar
|
|
8
|
Kops GJ, Weaver BA and Cleveland DW: On
the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev
Cancer. 5:773–785. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yuen KW, Montpetit B and Hieter P: The
kinetochore and cancer: what’s the connection? Curr Opin Cell Biol.
17:576–582. 2005.
|
|
10
|
Fry AM, Mayor T and Nigg EA: Regulating
centrosomes by protein phosphorylation. Curr Top Dev Biol.
49:291–312. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nigg EA: Mitotic kinases as regulators of
cell division and its checkpoints. Nat Rev Mol Cell Biol. 2:21–32.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Takai N, Miyazaki T, Fujisawa K, Nasu K,
Hamanaka R and Miyakawa I: Polo-like kinase (PLK) expression in
endometrial carcinoma. Cancer Lett. 169:41–49. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Holtrich U, Wolf G, Bräuninger A, et al:
Induction and down-regulation of PLK, a human serine/threonine
kinase expressed in proliferating cells and tumors. Proc Natl Acad
Sci USA. 91:1736–1740. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li D, Zhu J, Firozi PF, et al:
Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human
pancreatic cancer. Clin Cancer Res. 9:991–997. 2003.PubMed/NCBI
|
|
15
|
Bettencourt-Dias M and Glover DM:
Centrosome biogenesis and function: centrosomics brings new
understanding. Nat Rev Mol Cell Biol. 8:451–463. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Krämer A, Neben K and Ho AD: Centrosome
aberrations in hematological malignancies. Cell Biol Int.
29:375–383. 2005.
|
|
17
|
Nigg EA: Centrosome aberrations: cause or
consequence of cancer progression? Nat Rev Cancer. 2:815–825. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schultz SJ, Fry AM, Sütterlin C, Ried T
and Nigg EA: Cell cycle-dependent expression of Nek2, a novel human
protein kinase related to the NIMA mitotic regulator of
Aspergillus nidulans. Cell Growth Differ. 5:625–635.
1994.PubMed/NCBI
|
|
19
|
Fry AM, Schultz SJ, Bartek J and Nigg EA:
Substrate specificity and cell cycle regulation of the Nek2 protein
kinase, a potential human homolog of the mitotic regulator NIMA of
Aspergillus nidulans. J Biol Chem. 270:12899–12905. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fry AM, Meraldi P and Nigg EA: A
centrosomal function for the human Nek2 protein kinase, a member of
the NIMA family of cell cycle regulators. EMBO J. 17:470–481. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Faragher AJ and Fry AM: Nek2A kinase
stimulates centrosome disjunction and is required for formation of
bipolar mitotic spindles. Mol Biol Cell. 14:2876–2889. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hayward DG, Clarke RB, Faragher AJ, Pillai
MR, Hagan IM and Fry AM: The centrosomal kinase Nek2 displays
elevated levels of protein expression in human breast cancer.
Cancer Res. 64:7370–7376. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tsunoda N, Kokuryo T, Oda K, et al: Nek2
as a novel molecular target for the treatment of breast carcinoma.
Cancer Sci. 100:111–116. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kokuryo T, Senga T, Yokoyama Y, Nagino M,
Nimura Y and Hamaguchi M: Nek2 as an effective target for
inhibition of tumorigenic growth and peritoneal dissemination of
cholangio-carcinoma. Cancer Res. 67:9637–9642. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Crown J and Pegram M: Platinum-taxane
combinations in metastatic breast cancer: an evolving role in the
era of molecularly targeted therapy. Breast Cancer Res Treat.
79(Suppl 1): S11–S18. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wei R, Ngo B, Wu G and Lee WH:
Phosphorylation of the Ndc80 complex protein, HEC1, by Nek2 kinase
modulates chromosome alignment and signaling of the spindle
assembly checkpoint. Mol Biol Cell. 22:3584–3594. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jordan MA and Wilson L: Microtubules as a
target for anti-cancer drugs. Nat Rev Cancer. 4:253–265. 2004.
View Article : Google Scholar
|
|
28
|
Blajeski AL, Kottke TJ and Kaufmann SH: A
multistep model for paclitaxel-induced apoptosis in human breast
cancer cell lines. Exp Cell Res. 270:277–288. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Isaacs RJ, Davies SL, Sandri MI, Redwood
C, Wells NJ and Hickson ID: Physiological regulation of eukaryotic
topoisomerase II. Biochim Biophys Acta. 1400:121–137. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nitiss JL and Beck WT: Antitopoisomerase
drug action and resistance. Eur J Cancer. 32A:958–966. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Spallarossa P, Altieri P, Aloi C, et al:
Doxorubicin induces senescence or apoptosis in rat neonatal
cardiomyocytes by regulating the expression levels of the telomere
binding factors 1 and 2. Am J Physiol Heart Circ Physiol.
297:H2169–H2181. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bertrand JR, Pottier M, Vekris A, Opolon
P, Maksimenko A and Malvy C: Comparison of antisense
oligonucleotides and siRNAs in cell culture and in vivo. Biochem
Biophys Res Commun. 296:1000–1004. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Spänkuch-Schmitt B, Bereiter-Hahn J,
Kaufmann M and Strebhardt K: Effect of RNA silencing of polo-like
kinase-1 (PLK1) on apoptosis and spindle formation in human cancer
cells. J Natl Cancer Inst. 94:1863–1877. 2002.PubMed/NCBI
|
|
34
|
Tsui P, Rubenstein M and Guinan P: siRNA
is not more effective than a first generation antisense
oligonucleotide when directed against EGFR in the treatment of PC-3
prostate cancer. In Vivo. 19:653–656. 2005.PubMed/NCBI
|
|
35
|
Lou Y, Yao J, Zereshki A, et al: NEK2A
interacts with MAD1 and possibly functions as a novel integrator of
the spindle checkpoint signaling. J Biol Chem. 279:20049–20057.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fletcher L, Cerniglia GJ, Yen TJ and
Muschel RJ: Live cell imaging reveals distinct roles in cell cycle
regulation for Nek2A and Nek2B. Biochim Biophys Acta. 1744:89–92.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Prime G and Markie D: The telomere repeat
binding protein Trf1 interacts with the spindle checkpoint protein
Mad1 and Nek2 mitotic kinase. Cell Cycle. 4:121–124. 2005.
View Article : Google Scholar : PubMed/NCBI
|