Introduction
Ovarian cancer is one of the most common
gynecological cancers worldwide and the majority of patients are
diagnosed with late-stage disease. The five-year survival rates of
ovarian cancer are approximately 30% (1,2).
Without effective screening methods, women who are diagnosed with
ovarian cancer often present at an advanced stage associated with a
poor survival rate (1). Thus,
preventive strategies are required to reduce the mortality rate and
provide early detection of the tumor. Few risk factors for ovarian
cancer have been identified, such as age, family history of breast
and ovarian cancer, and genetic mutations (3).
Obesity is a well-established risk factor for
hormone-related cancers, such as breast, endometrial and prostate
cancer (4). Recently,
epidemiological studies have indicated that obesity is associated
with an increased risk of ovarian cancer (5). Obese pre-menopausal women have a
two-fold increased risk compared to individuals with a normal body
mass index (BMI) (1). A high BMI
strongly correlates with the occurrence of ovarian cancer (5,6). The
results of a meta-analysis showed that the risk of epithelial
ovarian cancer among obese women was 30% higher than women with a
normal BMI. Overweight women have a 16% increased risk compared to
those with a BMI within the healthy range (2). Obesity is not only positively
associated with the incidence of ovarian cancer but is also related
to a shorter time to recurrence and shorter overall survival.
Obesity is a poor prognostic factor for ovarian cancer survival
(7,8). An increasing BMI is an independent
negative predictor of disease-free and overall survival in ovarian
cancer (9).
Adipose tissue serves not only as energy storage but
also acts as endocrine tissue. Adiposity influences the synthesis
of endogenous sex hormones, such as estrogen, progesterone and
androgens. These sex hormones are believed to be involved in the
etiology of ovarian cancer (10,11).
Apart from sex hormones, another major hormone produced by
adipocytes that may mediate the correlation between obesity and
ovarian cancer is leptin. Leptin, encoded by the obesity gene (OB)
is a 16-kDa adipokine. Leptin has been identified as a growth
factor in certain hormone-related cancers, such as breast, prostate
and endometrial cancer (12–15).
Leptin exerts its activity through the membrane receptor, the
obesity receptor (OB-R). The overexpression of Ob-R has been
observed in 59.2% of ovarian cancers and significantly correlates
with poor progression-free survival (16). The growth factor-like functions of
leptin have been observed in many types of cancer cells (16–19).
However, the signaling pathways that directly underlie the
leptin-stimulated ovarian cell growth and inhibition of apoptosis
have not been extensively investigated.
Activated STAT3 had been shown to directly
contribute to oncogenesis through the upregulation of genes
encoding apoptosis inhibitors and cell-cycle regulators, such as
Bcl-xL, Mcl-1 and cyclin D1/D2, resulting in increased cell
proliferation and the prevention of apoptosis in a variety of human
cancer cells (20). During cancer
development and progression, anti-apoptotic proteins are usually
overexpressed and result in the cancer cells becoming resistant to
apoptosis (21). Mcl-1, a member
of the Bcl-2 family, was first cloned from the human myeloblastic
leukemia cell line, ML-1 (22).
Mcl-1 acts as an anti-apoptotic factor in various tumors, such as
human myeloid leukemia and hepatocellular carcinoma (23). Immunohistochemistry and
semi-quantitative PCR analyses of Mcl-1 expression in ovarian
cancer patients, have demonstrated that an increased Mcl-1
expression is associated with poor prognosis in ovarian cancer
patients. High Mcl-1 expression has been shown to significantly
correlate with advanced clinical stage, high histopathological
grade and poor survival (24).
Various growth factors and cytokines have been reported to induce
Mcl-1 expression, such as vascular endothelial growth factor
(VEGF), interleukin (IL)-3 and IL-6 (25–27).
In the current study, we investigated whether leptin
can stimulate ovarian cancer cell growth and prevent apoptosis
under serum-starvation conditions. We first observed that leptin
stimulated the expression of the anti-apoptotic protein, Mcl-1. Our
data demonstrate that leptin enhances cell growth by activating the
JAK2, MEK/ERK1/2 and PI3K/Akt pathways in ovarian cancer. Our
results indicate that leptin plays an important role between
obesity and the progression of ovarian cancer.
Materials and methods
Reagents and antibodies
Human recombinant leptin, tyrphostin AG490, U0126,
PD98059, Ly294002 and wortmannin were purchased from Sigma Chemical
Co. (St. Louis, MO). Antibodies were obtained and included
monoclonal anti-β-actin antibody (Sigma Chemical Co.), polyclonal
antibodies against phospho-Akt or phospho-ERK1/2 (Cell Signaling
Technology, Beverly, MA), monoclonal antibody against Akt or cyclin
D1 (BD Pharmingen, Palo Alto, CA), and polyclonal antibodies
against ani-Mcl-1 or ERK1/2 (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA).
Cell culture
The OVCAR-3 human ovarian cancer cell line was
obtained from the American Type Culture Collection. The cells were
cultured in RPMI-1640 supplemented with 2 mM glutamine (HyClone),
15 mM HEPES (USB Corp.), 100 U/L penicillin G, 0.1 g/L
streptomycin, 25 mM D-glucose, 1 mM sodium pyruvate (Sigma) and 10%
fetal bovine serum (Biological Industries, Ltd., Kibbutz Beit
Haemek, Israel) in a 5% CO2 incubator at 37°C.
Cell number counting
Cells were seeded in 6-well plates, treated with the
vehicle, recombinant human leptin, or AG490, U0126, PD98059,
Ly294002 and wortmannin for the indicated periods of time, stained
with 0.5% trypan blue (Biological Industries, Ltd.) and then
counted.
MTT assay
The 3-(4,5-dimethylthiazolyl)-2,5-diphenyl
tetrazolium bromide (MTT) proliferation assay was carried out
according to a previous protocol (13). In brief, each cell treatment group
was incubated in medium containing 2 mg/ml MTT reagent (Sigma
Chemical Co.) at 37°C for 4 h, the formazan crystals converted from
tetrazolium salts by viable cells were dissolved in dimethyl
sulfoxide (150 μl/well) and their absorbance at 570 nm was
measured using a microplate spectrophotometer.
Western blot assay
Cells were extracted with lysis buffer (10 mM
Tris-HCl pH 7.5, 150 mM NaCl, 10% glycerol, 1% Triton X-100, 1 mM
DTT, 0.2 mM PMSF, 1 μg/ml aprotinin, 1 μg/ml
leupeptin, 1 mM Na3VO4 and 1 mM NaF) and
separated on 8–15% SDS-PAGE gel. The proteins were
electrotransferred onto a nitrocellulose membrane (Perkin-Elmer
Life Sciences, Boston, MA). The membrane was blocked with 5% skim
milk in TBST (20 mM Tris-HCl pH 7.6, 137 mM NaCl and 0.1%
Tween-20), probed with the appropriate primary antibodies at 4°C
overnight and incubated with HRP-conjugated secondary antibodies
(Jackson Immuno Research Laboratories Inc., West Grove, PA) at room
temperature for 1 h. The blots were incubated with a
chemiluminescence substrate (Amersham Biosciences, Little Chalfont,
UK) and exposed to X-ray film (Fuji Photo Film Co., Tokyo,
Japan).
Statistical analysis
Statistical analyses were calculated using a
two-sided Student’s t-test. All data are presented as the means ±
SD and the statistical differences are shown in the figure
legends.
Results
Leptin stimulates ovarian cancer cell
growth
Leptin exerts its functional effects through binding
to the leptin receptors, OB-Rb and OB-Ra expressed in ovarian
cancer cells (19). In the present
study, we examined the effect of leptin on ovarian cancer cell
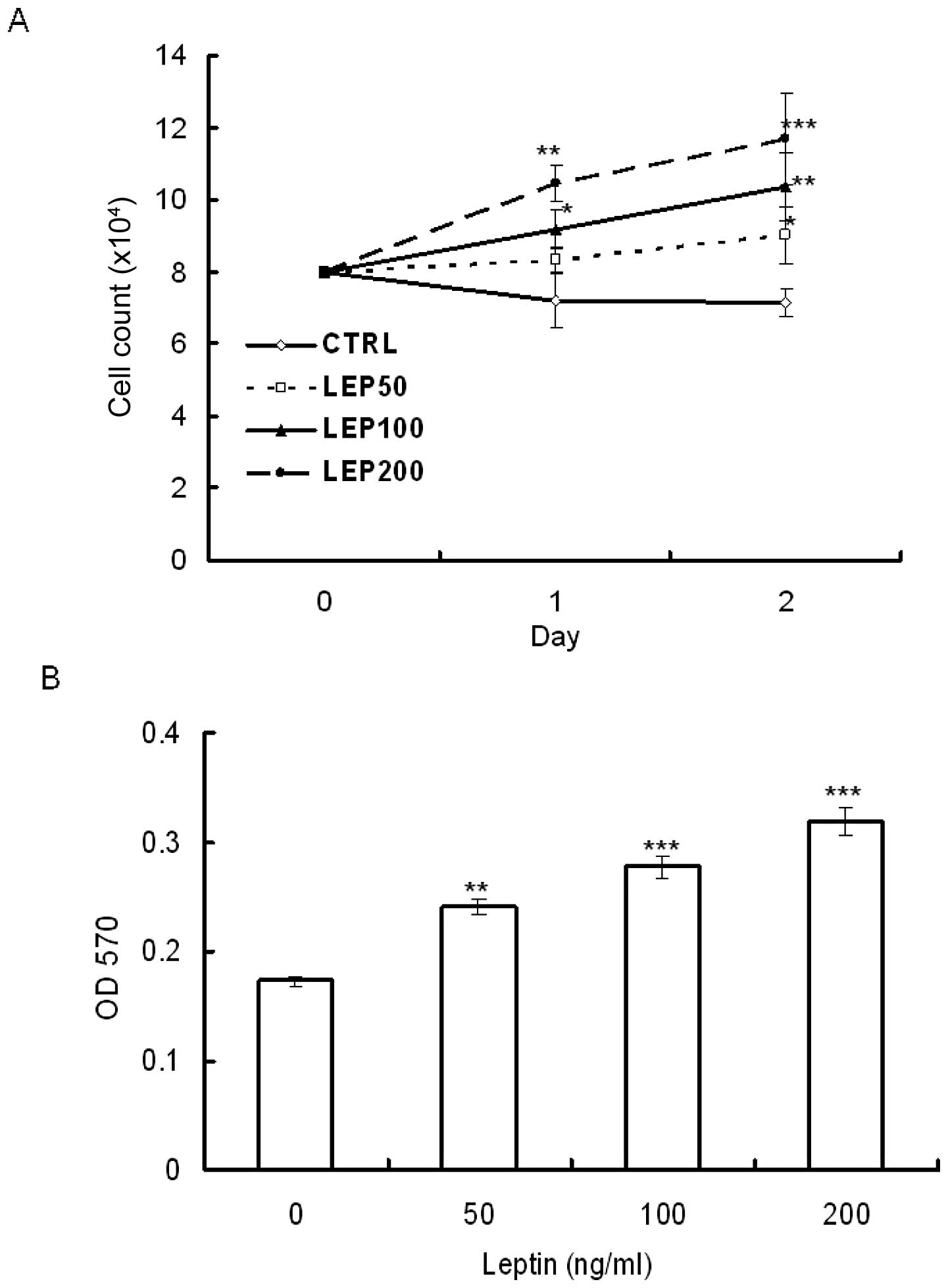
growth using trypan blue exclusion staining and MTT assay. OVCAR-3
cells were serum-deprived for 48 h and then treated with various
concentrations of leptin for 48 h. MTT assay showed that leptin
stimulated OVCAR-3 cell growth in a dose-dependent manner. The
stimulation was observed at a dose as low as 50 ng/ml (Fig. 1B). Using the trypan blue exclusion
assay to estimate the amount of viable cells, the results showed
that leptin enhanced cell growth not only in a dose-dependent but
also in a time-dependent manner (Fig.
1A).
Leptin inhibits serum starvation-induced
apoptosis
In addition to the stimulation of cancer cell
growth, leptin treatment inhibited apoptosis either induced by
sodium butyrate in colon cancer cells or by TGFβ-1 in
hepatocellular carcinoma cells (18,28).
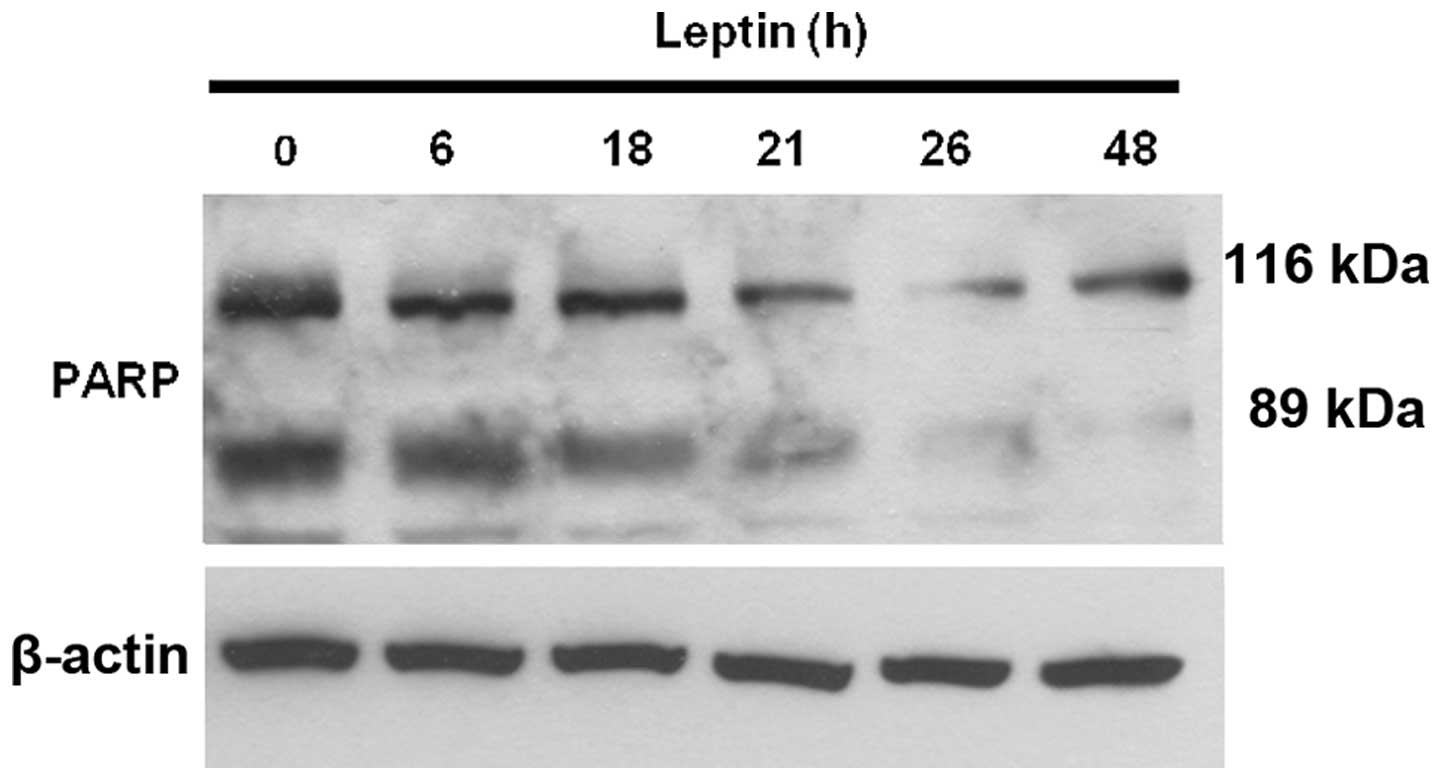
Therefore, we further examined whether leptin exerts anti-apoptotic
effects on OVCAR-3 cells. We measured the level of cleaved
poly(ADP-ribose) polymerase (PARP) using a western blot assay. In
apoptotic mammalian cells, PARP, known as a death substrate, is
degraded to an 89-kDa signature fragment. PARP cleavage by
caspase-3 is a well-characterized event in the apoptotic pathway in
mammalian cells. PARP is cleaved by caspase-3 early during
apoptosis in many different cell lines (29,30).
Our results showed that the 116-kDa active form of PARP was readily
cleaved into 89- and 28-kDa fragments during serum-deprivation,
showing that serum deprivation induces apoptosis in OVCAR-3 cells.
The addition of leptin into the serum-free medium for 0, 6, 18, 21,
26 and 48 h during serum starvation blocked the cleavage of PARP
(Fig. 2). These results suggest
that leptin exerts an anti-apoptotic effect on ovarian cancer
cells.
Leptin stimulates the expression of
cyclin D1 and Mcl-1
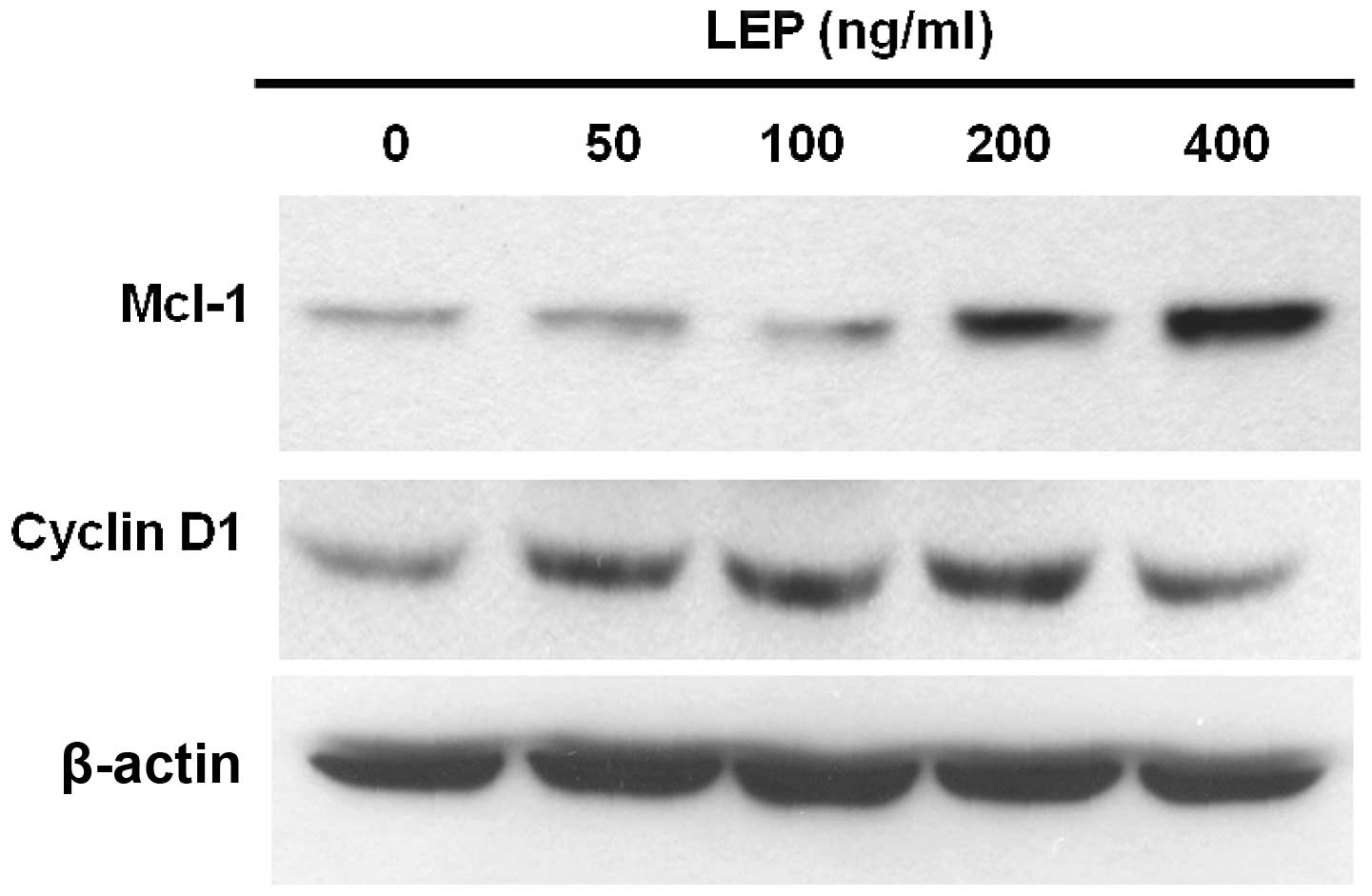
Cyclin D1 is a growth sensor induced by a variety of
growth factors and mitogens to trigger cell cycle progression
(31). Therefore, we used western
blot analysis to measure the expression of cyclin D1 in OVCAR-3
cells. At a low concentration (50 ng/ml), leptin increased the
expression of cyclin D1 and the effect of leptin on cyclin D1
expression reached a maximum at the concentration of 200 ng/ml
(Fig. 3). Since leptin exerts
anti-apoptotic effects on OVCAR-3 cells, we sought to determine the
mechanisms responsible for this response. Usually, the
overexpression of Mcl-1 delays apoptosis induced by growth factor
withdrawal (32). Increased Mcl-1
expression has been associated with poor prognosis in ovarian
cancer (24). OVCAR-3 cells
treated with leptin showed a dose-dependent increase in the amount
of Mcl-1 protein. The effect was maximal at 400 ng/ml leptin
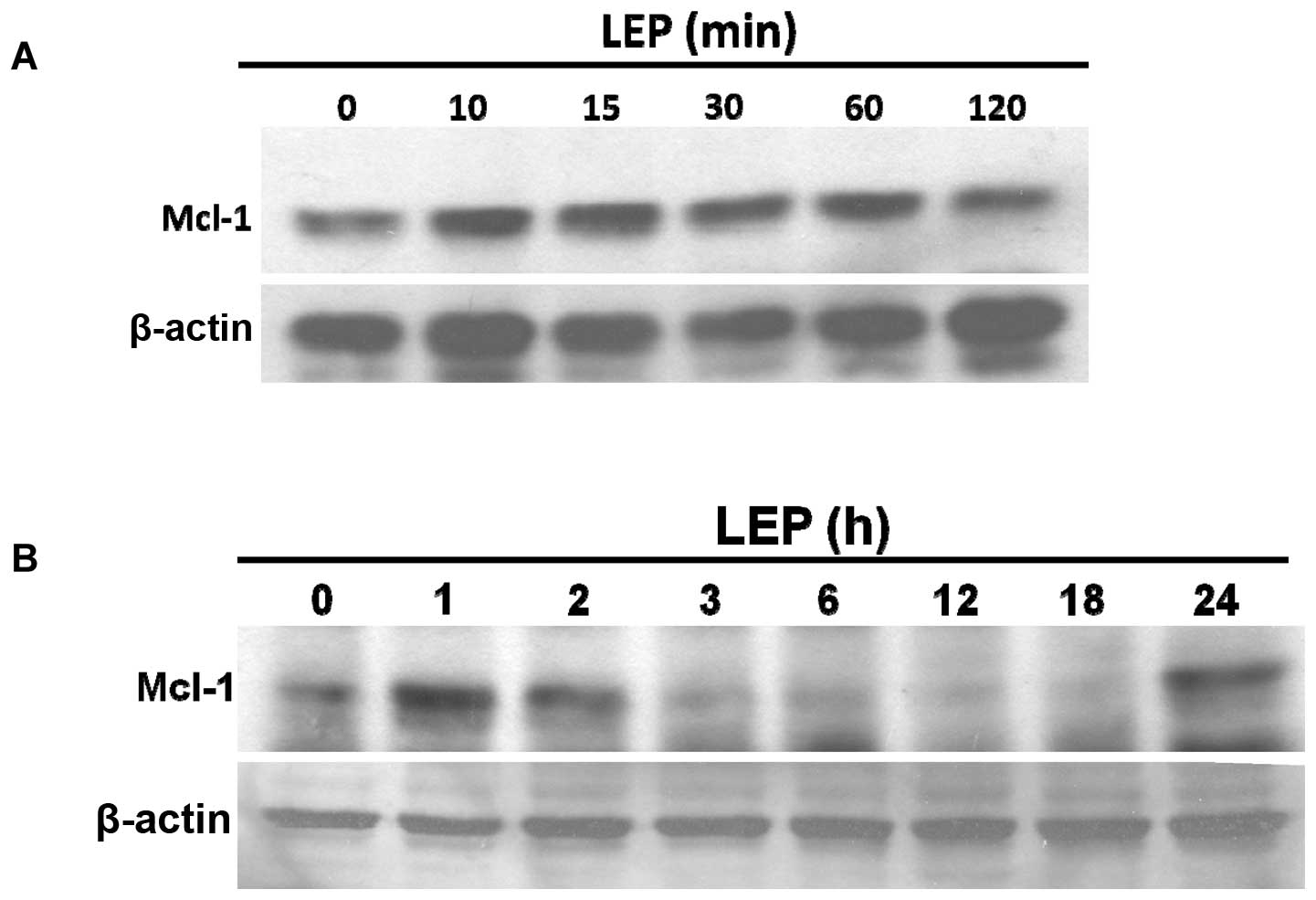
treatment (Fig. 3). As shown in
Fig. 4A, leptin acutely stimulated
Mcl-1 expression within 5–60 min; this effect diminished after 2 h
of treatment. A second peak in leptin-induced Mcl-1 expression was
detected at 24 h (Fig. 4B).
Activation of JAK2, PI3K/Akt and
MEK/ERK1/2 pathways mediate leptin-stimulated cell growth of
OVCAR-3 cells
In order to determine the intracellular signaling
pathways responsible for the effect observed with leptin treatment,
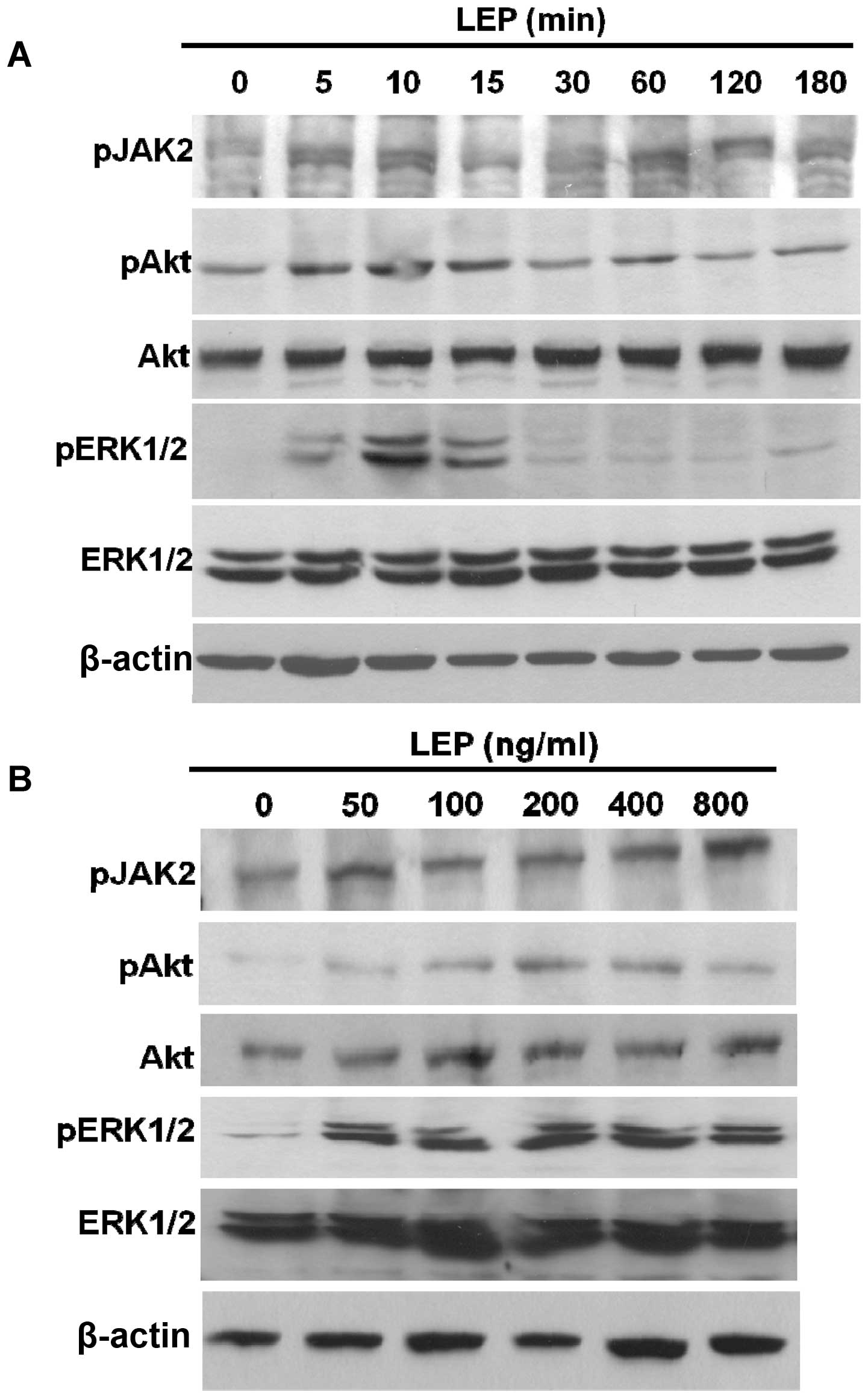
we analyzed the phosphorylation status of JAK2, Akt and ERK1/2. The
phosphorylated forms of JAK2, Akt and ERK1/2 were dose-dependently
increased by leptin in the OVCAR-3 cells (Fig. 5B). Simultaneously, the time-course
experiments showed that the levels of the phosphorylated forms of
JAK2, Akt and ERK1/2 were increased as early as 5 min in the
OVCAR-3 cells during leptin treatment (Fig. 5A). Leptin treatment resulted in the
activation of JAK2, Akt and ERK1/2 kinases in the ovarian cancer
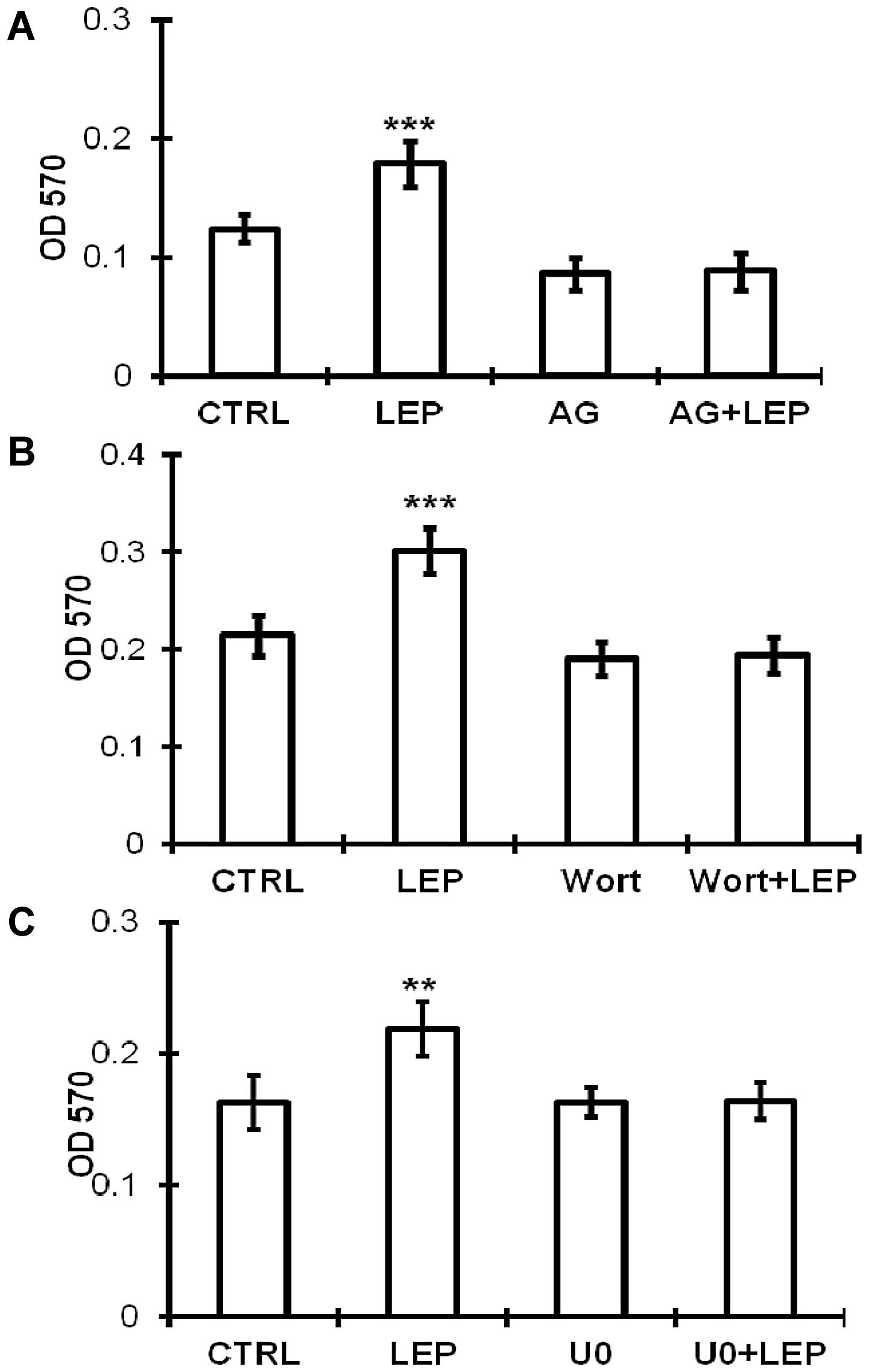
cells. Subsequently, we used pharmacological inhibitors to
determine the involvement of these signaling kinases on the
growth-stimulating effect of leptin. The leptin-stimulated growth
of the OVCAR-3 cells was abolished by the inhibitor specific for
JAK2 (AG490) (Fig. 6A), PI3K/Akt
(wortmannin) (Fig. 6B) and
MEK/ERK1/2 (U0126) (Fig. 6C),
which is consistent with the activation of the JAK2, PI3K/Akt and
MEK/ERK1/2 pathways.
Leptin stimulates the expression of
cyclin D1 and Mcl-1 through the activation of JAK2-associated
signaling pathways
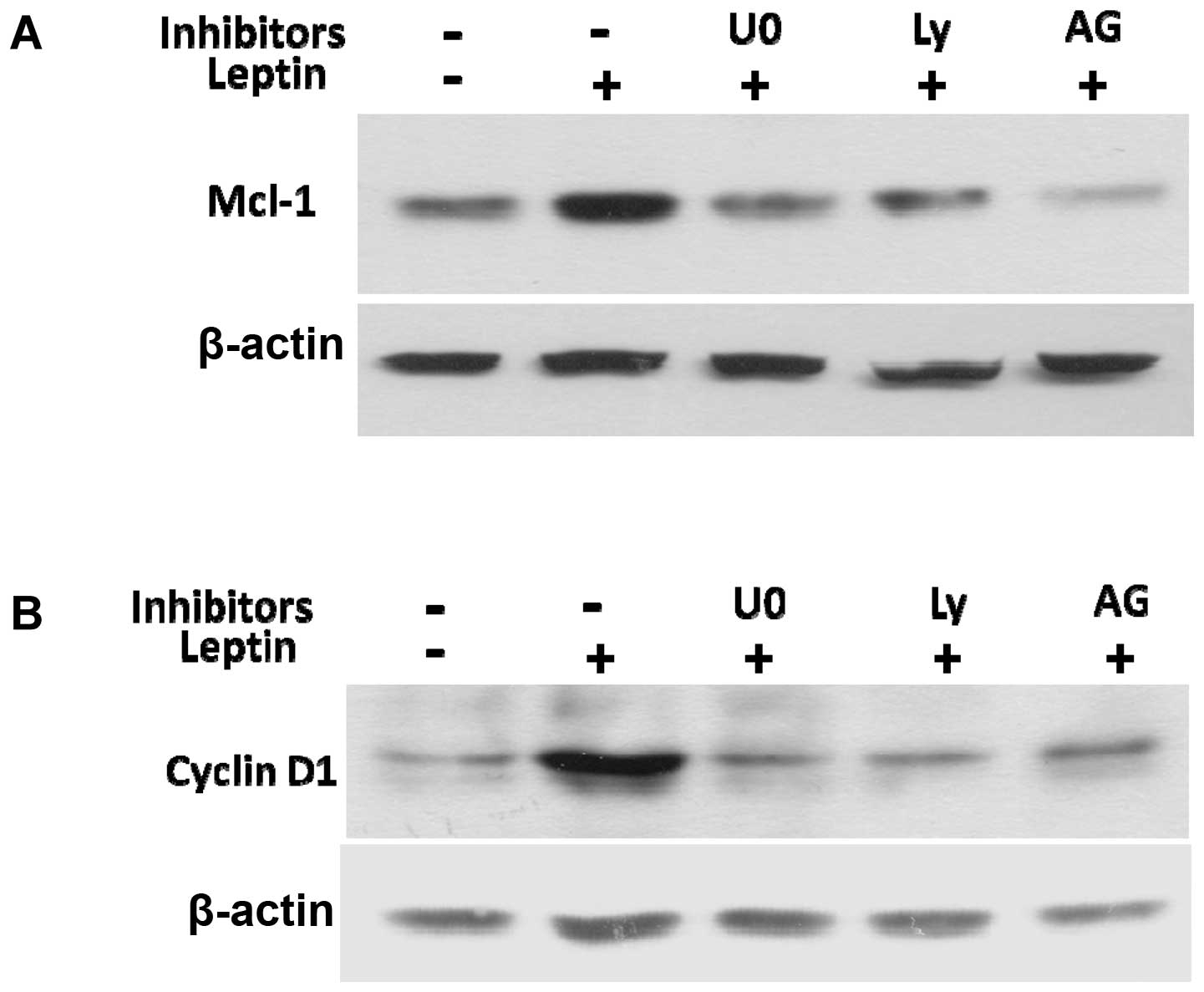
To elucidate the mechanisms by which JAK2, PI3K/Akt
and MEK/ERK1/2 mediate leptin-induced cyclin D1 and Mcl-1
expression, we examined the effects of their inhibitors on cyclin
D1 and Mcl-1 expression. The specific inhibitors for JAK2 (AG490),
PI3K/Akt (Ly294002), or MEK/ERK1/2 (U0126), blocked the leptin
induction of cyclin D1 and Mcl-1 protein expression (Fig. 7A and B). Taken together, these data
reveal that leptin activates the signaling pathways, including
JAK2, PI3K/Akt and MEK/ERK1/2 to enhance the expression of cyclin
D1 and the anti-apoptotic protein, Mcl-1, which subsequently
stimulates OVCAR-3 cell growth and prevents apoptosis.
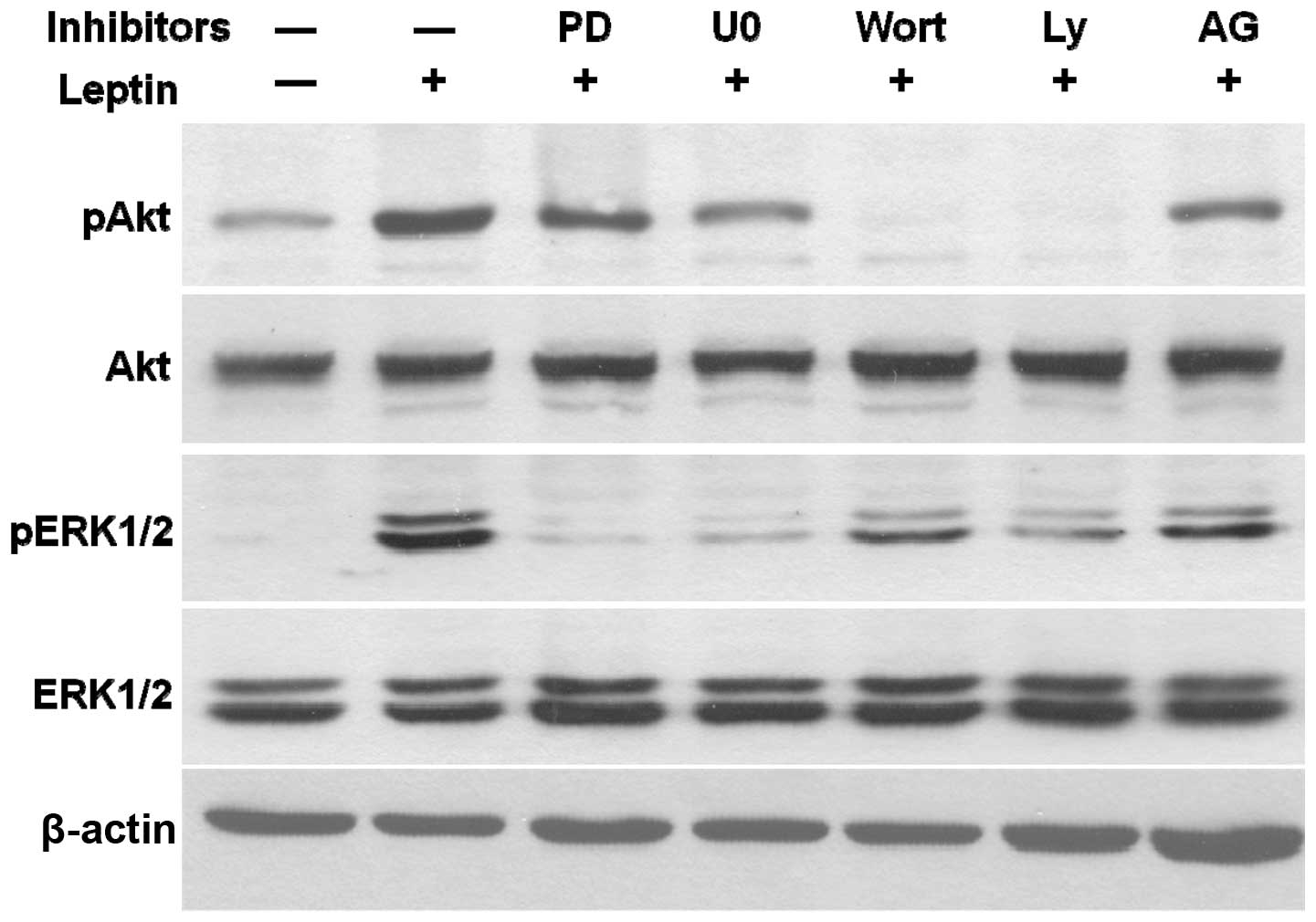
Leptin activates crosstalk among JAK2,
PI3K/Akt and MEK/ERK1/2 signaling pathways
Using AG490, the inhibitor of JAK2, PD98059 and
U0126, the inhibitors of MEK/ERK1/2 and the inhibitors of PI3K/Akt,
wortmannin and Ly294002, we examined the leptin-stimulated
phosphorylation of Akt and ERK1/2 to elucidate the crosstalk among
these signaling pathways in OVCAR-3 cells (Fig. 8). As expected, U0126 and PD98059
specifically blocked the leptin-induced phosphorylation of ERK1/2,
while wortmannin and Ly294002 inhibited the leptin-induced
phosphorylation of Akt. The specific blocker of JAK2, AG490,
decreased leptin-phosphorylated ERK1/2 and Akt. Wortmannin and
Ly294002, not only inhibited leptin-activated Akt but also
completely abrogated the effects of leptin on ERK1/2
phosphorylation. Similar results were observed with U0126 and
PD98059 treatments. The inhibitors of the MEK/ERK1/2 pathway
concomitantly blocked the leptin-activated phosphorylation of
ERK1/2 and partially decreased the level of leptin-induced
phosphorylated Akt. According to these findings, we propose that
leptin triggers a JAK2-initiated signaling cascade, comprised of
the PI3K/Akt and MEK/ERK1/2 pathways in ovarian cancer.
Discussion
Obesity is a significant health concern in developed
countries with a dramatically increased incidence over the last
decade. In the US, more than one third of the adults (35.7%) are
obese and 68.8% are considered overweight based on their BMI
(33,34). Obesity has been identified as a
risk factor in diabetes, cardiovascular diseases and in many types
of cancer, such as breast, esophageal, prostate, colon and liver
cancer (35). The adipokine,
leptin, may be an important factor in the growth of ovarian cancer
(16). In epidemiological surveys,
obesity has been shown to increase ovarian cancer incidence, and
shorten the time of survival and recurrence (5,9).
However, the molecular mechanisms underlying the clinical
observations remain unclear. In the present study, we investigated
the effects of leptin on ovarian cancer cells. Our results show
that leptin stimulates ovarian cancer cell growth and exerts an
anti-apoptotic effect by increasing the expression of cyclin D1 and
Mcl-1 proteins.
Apart from the effect of leptin on cell growth and
the inhibition of apoptosis, our results showed that leptin induced
cyclin D1 expression, which plays a crucial role in cell
proliferation in a variety of cancer cells, such as breast cancer,
hepatocellular carcinoma and ovarian cancer cells. In addition, to
our knowledge, we are the first to show that leptin directly
stimulates Mcl-1 expression. Mcl-1 is a member of the Bcl-2 family.
The expression of the Bcl-2 family members is frequently
deregulated during carcinogenesis. Mcl-1 is frequently amplified in
human cancers (23). The
downregulation of Mcl-1 is essential to induce cell death in
ovarian cancer cells in response to Bcl-2 inhibitors. siRNA
treatment to inhibit Mcl-1 expression has been shown to induce
apoptosis in ovarian cancer cells (36–38).
In our study, leptin acutely induced Mcl-1 expression within 10–60
min; this effect that rapidly diminished within 3 h (Fig. 4). The half-life of Mcl-1 is around
1 h compared to 10–14 h for Bcl-2 (39). Mcl-1 mRNA expression evolves and
changes rapidly and this feature perhaps explains why Mcl-1
expression may be rapidly increased or downregulated with cytokines
and differentiation factors. Mcl-1 thus differs from other members
of the Bcl-2 family in acting as an immediate response molecule to
protect cells against apoptosis (23). In our experiments, leptin
stimulated Mcl-1 expression in two phases. Apart from acute
stimulation, we found that leptin induced Mcl-1 expression at 24 h
to the same levels observed within 60 min.
The phosphorylation of JAK2 is thought to be the
first event following the binding of leptin to its cellular
receptor. Subsequently, activated JAK2 triggers other signaling
pathways. In our study, AG490, an inhibitor of JAK2, abolished the
leptin-induced activation of the downstream pathways, PI3K/Akt and
MEK/ERK1/2. Previous studies have suggested that the response to
leptin treatment is transmitted via the MAP kinase and PI3K/Akt
pathways in ovarian cancer cells (16,19).
Using specific inhibitors of the MEK/ERK1/2 and PI3K/Akt pathways,
we observed that both pathways interacted with each other. Both
pathways are required to mediate leptin-stimulated cell growth and
inhibition of apoptosis.
In the present study, we demonstrate that leptin
stimulates cyclin D1 and Mcl-1 protein expression. Furthermore,
leptin induces cell proliferation and inhibits apoptosis in ovarian
cancer cells. Our results also identify the signaling pathways
responsible for ovarian cancer cell growth and inhibition of
apoptosis in response to leptin, providing a direct association
between obesity and ovarian carcinogenesis.
Acknowledgements
We thank Dr Ing-Cherng Guo (deceased,
March 2, 2008) for his major contribution in providing us with the
initial experimental design and support. This study was supported
by the Research Institute for Children at the Children’s Hospital,
New Orleans, LA, USA.
References
|
1
|
Beehler GP, Sekhon M, Baker JA, Teter BE,
McCann SE, Rodabaugh KJ and Moysich KB: Risk of ovarian cancer
associated with BMI varies by menopausal status. J Nutr.
136:2881–2886. 2006.PubMed/NCBI
|
|
2
|
Olsen CM, Green AC, Whiteman DC, Sadeghi
S, Kolahdooz F and Webb PM: Obesity and the risk of epithelial
ovarian cancer: a systematic review and meta-analysis. Eur J
Cancer. 43:690–709. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ioka A, Tsukuma H, Ajiki W and Oshima A:
Ovarian cancer incidence and survival by histologic type in Osaka,
Japan. Cancer Sci. 94:292–296. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bray GA: The underlying basis for obesity:
relationship to cancer. J Nutr. 132:3451S–3455S. 2002.PubMed/NCBI
|
|
5
|
Leitzmann MF, Koebnick C, Danforth KN,
Brinton LA, Moore SC, Hollenbeck AR, Schatzkin A and Lacey JV Jr:
Body mass index and risk of ovarian cancer. Cancer. 115:812–822.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schouten LJ, Rivera C, Hunter DJ,
Spiegelman D, Adami HO, Arslan A, Beeson WL, van den Brandt PA,
Buring JE, Folsom AR, Fraser GE, Freudenheim JL, Goldbohm RA,
Hankinson SE, Lacey JV Jr, Leitzmann M, Lukanova A, Marshall JR,
Miller AB, Patel AV, Rodriguez C, Rohan TE, Ross JA, Wolk A, Zhang
SM and Smith-Warner SA: Height, body mass index, and ovarian
cancer: a pooled analysis of 12 cohort studies. Cancer Epidemiol
Biomarkers Prev. 17:902–912. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Calle EE, Rodriguez C, Walker-Thurmond K
and Thun MJ: Overweight, obesity, and mortality from cancer in a
prospectively studied cohort of U.S. adults. N Engl J Med.
348:1625–1638. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rodriguez C, Jacobs EJ, Patel AV, Calle
EE, Feigelson HS, Fakhrabadi-Shokoohi D and Thun MJ: Jewish
ethnicity and prostate cancer mortality in two large US cohorts.
Cancer Causes Control. 13:271–277. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pavelka JC, Brown RS, Karlan BY, Cass I,
Leuchter RS, Lagasse LD and Li AJ: Effect of obesity on survival in
epithelial ovarian cancer. Cancer. 107:1520–1524. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Key T, Appleby P, Barnes I and Reeves G;
Endogenous Hormones and Breast Cancer Collaborative Group:
Endogenous sex hormones and breast cancer in postmenopausal women:
reanalysis of nine prospective studies. J Natl Cancer Inst.
94:606–616. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Risch HA: Hormonal etiology of epithelial
ovarian cancer, with a hypothesis concerning the role of androgens
and progesterone. J Natl Cancer Inst. 90:1774–1786. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hu X, Juneja SC, Maihle NJ and Cleary MP:
Leptin - a growth factor in normal and malignant breast cells and
for normal mammary gland development. J Natl Cancer Inst.
94:1704–1711. 2002. View Article : Google Scholar
|
|
13
|
Chen C, Chang YC, Liu CL, Chang KJ and Guo
IC: Leptin-induced growth of human ZR-75-1 breast cancer cells is
associated with up-regulation of cyclin D1 and c-Myc and
down-regulation of tumor suppressor p53 and p21WAF1/CIP1. Breast
Cancer Res Treat. 98:121–132. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sharma D, Saxena NK, Vertino PM and Anania
FA: Leptin promotes the proliferative response and invasiveness in
human endometrial cancer cells by activating multiple
signal-transduction pathways. Endocr Relat Cancer. 13:629–640.
2006. View Article : Google Scholar
|
|
15
|
Somasundar P, Frankenberry KA, Skinner H,
Vedula G, McFadden DW, Riggs D, Jackson B, Vangilder R, Hileman SM
and Vona-Davis LC: Prostate cancer cell proliferation is influenced
by leptin. J Surg Res. 118:71–82. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Uddin S, Bu R, Ahmed M, Abubaker J,
Al-Dayel F, Bavi P and Al-Kuraya KS: Overexpression of leptin
receptor predicts an unfavorable outcome in Middle Eastern ovarian
cancer. Mol Cancer. 8:742009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Garofalo C and Surmacz E: Leptin and
cancer. J Cell Physiol. 207:12–22. 2006. View Article : Google Scholar
|
|
18
|
Chen C, Chang YC, Liu CL, Liu TP, Chang KJ
and Guo IC: Leptin induces proliferation and anti-apoptosis in
human hepatocarcinoma cells by up-regulating cyclin D1 and
down-regulating Bax via a Janus kinase 2-linked pathway. Endocr
Relat Cancer. 14:513–529. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Choi JH, Park SH, Leung PC and Choi KC:
Expression of leptin receptors and potential effects of leptin on
the cell growth and activation of mitogen-activated protein kinases
in ovarian cancer cells. J Clin Endocrinol Metab. 90:207–210. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Buettner R, Mora LB and Jove R: Activated
STAT signaling in human tumors provides novel molecular targets for
therapeutic intervention. Clin Cancer Res. 8:945–954.
2002.PubMed/NCBI
|
|
21
|
Heiser D, Labi V, Erlacher M and Villunger
A: The Bcl-2 protein family and its role in the development of
neoplastic disease. Exp Gerontol. 39:1125–1135. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Reynolds JE, Yang T, Qian L, Jenkinson JD,
Zhou P, Eastman A and Craig RW: Mcl-1, a member of the Bcl-2
family, delays apoptosis induced by c-Myc overexpression in Chinese
hamster ovary cells. Cancer Res. 54:6348–6352. 1994.PubMed/NCBI
|
|
23
|
Michels J, Johnson PW and Packham G:
Mcl-1. Int J Biochem Cell Biol. 37:267–271. 2005. View Article : Google Scholar
|
|
24
|
Shigemasa K, Katoh O, Shiroyama Y, Mihara
S, Mukai K, Nagai N and Ohama K: Increased MCL-1 expression is
associated with poor prognosis in ovarian carcinomas. Jpn J Cancer
Res. 93:542–550. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kuo ML, Chuang SE, Lin MT and Yang SY: The
involvement of PI 3-K/Akt-dependent up-regulation of Mcl-1 in the
prevention of apoptosis of Hep3B cells by interleukin-6. Oncogene.
20:677–685. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Leu CM, Chang C and Hu C: Epidermal growth
factor (EGF) suppresses staurosporine-induced apoptosis by inducing
mcl-1 via the mitogen-activated protein kinase pathway. Oncogene.
19:1665–1675. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wei LH, Kuo ML, Chen CA, Chou CH, Cheng
WF, Chang MC, Su JL and Hsieh CY: The anti-apoptotic role of
interleukin-6 in human cervical cancer is mediated by up-regulation
of Mcl-1 through a PI 3-K/Akt pathway. Oncogene. 20:5799–5809.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rouet-Benzineb P, Aparicio T, Guilmeau S,
Pouzet C, Descatoire V, Buyse M and Bado A: Leptin counteracts
sodium butyrate-induced apoptosis in human colon cancer HT-29 cells
via NF-κB signaling. J Biol Chem. 279:16495–16502. 2004.PubMed/NCBI
|
|
29
|
Nicholson DW, Ali A, Thornberry NA,
Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle
M, Lazebnik YA, et al: Identification and inhibition of the
ICE/CED-3 protease necessary for mammalian apoptosis. Nature.
376:37–43. 1995. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Germain M, Affar EB, D’Amours D, Dixit VM,
Salvesen GS and Poirier GG: Cleavage of automodified
poly(ADP-ribose) polymerase during apoptosis. Evidence for
involvement of caspase-7. J Biol Chem. 274:28379–28384. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Knudsen KE, Diehl JA, Haiman CA and
Knudsen ES: Cyclin D1: polymorphism, aberrant splicing and cancer
risk. Oncogene. 25:1620–1628. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang JM, Chao JR, Chen W, Kuo ML, Yen JJ
and Yang-Yen HF: The antiapoptotic gene mcl-1 is up-regulated by
the phosphatidylinositol 3-kinase/Akt signaling pathway through a
transcription factor complex containing CREB. Mol Cell Biol.
19:6195–6206. 1999.PubMed/NCBI
|
|
33
|
Ogden CL, Carroll MD, Kit BK and Flegal
KM: Prevalence of obesity in the United States, 2009–2010. NCHS
Data Brief. 1–8. 2012.
|
|
34
|
Flegal KM, Carroll MD, Kit BK and Ogden
CL: Prevalence of obesity and trends in the distribution of body
mass index among US adults, 1999–2010. JAMA. 307:491–497. 2012.
|
|
35
|
Bray GA: Drug treatment of obesity. Rev
Endocr Metab Disord. 2:403–418. 2001. View Article : Google Scholar
|
|
36
|
Simonin K, Brotin E, Dufort S, Dutoit S,
Goux D, N’Diaye M, Denoyelle C, Gauduchon P and Poulain L: Mcl-1 is
an important determinant of the apoptotic response to the
BH3-mimetic molecule HA14-1 in cisplatin-resistant ovarian
carcinoma cells. Mol Cancer Ther. 8:3162–3170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Varin E, Denoyelle C, Brotin E,
Meryet-Figuière M, Giffard F, Abeilard E, Goux D, Gauduchon P,
Icard P and Poulain L: Downregulation of Bcl-xL and Mcl-1 is
sufficient to induce cell death in mesothelioma cells highly
refractory to conventional chemotherapy. Carcinogenesis.
31:984–993. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Brotin E, Meryet-Figuière M, Simonin K,
Duval RE, Villedieu M, Leroy-Dudal J, Saison-Behmoaras E, Gauduchon
P, Denoyelle C and Poulain L: Bcl-XL and MCL-1 constitute pertinent
targets in ovarian carcinoma and their concomitant inhibition is
sufficient to induce apoptosis. Int J Cancer. 126:885–895.
2010.PubMed/NCBI
|
|
39
|
Weng C, Li Y, Xu D, Shi Y and Tang H:
Specific cleavage of Mcl-1 by caspase-3 in tumor necrosis
factor-related apoptosis-inducing ligand (TRAIL)-induced apoptosis
in Jurkat leukemia T cells. J Biol Chem. 280:10491–10500. 2005.
View Article : Google Scholar : PubMed/NCBI
|