Introduction
Hepatocellular carcinoma (HCC) is one of the most
common types of tumor worldwide, with a high mortality rate
(1). Risk factors for HCC
development include infection with hepatitis B or C virus,
cirrhosis, and genetic metabolic diseases (2). As regards treatment, surgical
resection or local ablation is a good choice for non-cirrhotic
patients with small tumors, while liver transplantation is a better
choice for patients with early stages of HCC accompanied by
decompensated cirrhosis (3–4).
However, these treatments and availability of livers for
transplantation are not applicable to the majority of patients.
Therefore, it is necessary to further investigate the potential
molecular mechanisms in HCC development in order to discover novel
intervention targets for HCC.
S100A9 belongs to a family of 25 calcium-binding
proteins and its gene is located on chromosome 1q21, a region that
is unstable and rearrangeable in tumors (5–8).
S100A9 often functions through binding with S100A8 which is another
member of the same family, and has been linked to neoplastic
disorders (9). It has been shown
that S100A9 is upregulated and correlates with poor differentiation
in HCC (10); however, at the same
time, S100A9 upregulation has been found in the HCC cells of humans
and mice and has been shown to protect Hep3B HCC cells from
TNF-γ-induced apoptosis (11). A
recent study demonstrated that extracellular S100A9 protein
secreted from tumor cells plays a dual role, exhibiting anti-tumor
or pro-tumor responses in a dose-dependent manner (12). At a concentration of <20
μg/ml, extracellular S100A9 has been shown to promote tumor
cell proliferation in breast and prostate cancer; however, at a
range of 25–250 μg/ml, it has been shown to exert apoptotic
effects on certain types of tumor cells(13–18).
Nevertheless, the effect of extracellular S100A9 on HCC cells
remains unclear.
Several molecular signaling pathways in HCC,
particularly mitogen-activated protein kinase (MAPK) signaling,
have been reported to play critical roles in carcinogenesis
(19–21). Certain studies have demonstrated
that S100A9 can enhance the activity of MAPK signaling in many
types of cancer, including breast, prostate and colon cancer
(15,16,22).
Whether S100A9 is involved in HCC development via the activation of
the MAPK signaling pathway remains to be further elucidated.
In the present study, we aimed to investigate the
effects of S100A9 on HepG2 HCC cells, as well as the molecular
mechanisms underlying these effects. We found that S100A9 promoted
the proliferation and invasion of HepG2 cells in vitro and
tumor growth in vivo, and that the activation of the MAPK
signaling pathway was involved in the S100A9-induced proliferation
and invasion. These data indicate that S100A9 may play a role in
HCC development.
Materials and methods
Cell culture and reagents
The HepG2 human HCC cell line and the L02 human
normal liver cell line were kindly provided by Professor T.C. He
(The University of Chicago Medical Center, Chicago, IL, USA). The
HepG2 and L02 cells were maintained in Dulbecco's modified Eagle's
medium (DMEM) supplemented with 10% fetal bovine serum (FBS;
HyClone), 100 U/ml of penicillin and 100 μg/ml of
streptomycin. The cells were cultured in a humidified atmosphere of
5% CO2 at 37°C. The primary antibodies used in this
study were as follows: mouse anti-hS100A9 monoclonal antibody (Cat
no. 58706; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA),
rabbit anti-JNK monoclonal antibody (Cat no. 9253; Cell Signaling
Technology, Danvers, MA, USA), rabbit anti-phosphor-JNK monoclonal
antibody (Cat no. 4668; Cell Signaling Technology), rabbit anti-p38
monoclonal antibody (Cat no. 9212; Cell Signaling Technology),
rabbit anti-phosphor-p38 monoclonal antibody (Cat no. 4511; Cell
Signaling Technology), rabbit anti-ERK1/2 monoclonal antibody (Cat
no. 4695; Cell Signaling Technology), rabbit anti-phosphor-ERK1/2
monoclonal antibody (Cat no. 3510; Cell Signaling Technology) and
mouse anti-β-actin monclonal antibody (Cat no. 47778; Santa Cruz
Biotechnology, Inc.). Specific inhibitors of p38 (SB203580) and
ERK1/2 (PD98059) were obtained from Santa Cruz Biotechnology, Inc.
and were used as per the manufacturer's instructions.
Recombinant protein preparation
The pGST-moluc-hS100A9 plasmid used in the current
study has been described previously (23). Briefly, the pGST-moluc-hS100A9 was
transformed into E. coli (BL21) by calcium chloride
transformation. Isopropylthio-β-D-galactoside was used to induce
the expression of GST-hS100A9 protein. The bacteria were then
collected and sonicated on ice, and spun at 4°C. The supernatant
was incubated with glutathione-sepharose 4B beads, and GST-hS100A9
on the beads was eluted by elution buffer with reduced glutathione
on ice. Finally the GST-hS100A9 protein was filtered and stored at
−80°C. The control protein GST was prepared simultaneously
(pGST-moluc plasmid).
Cell viability assay
Cell viability was measured using
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltrazolium bromide (MTT)
assay. A total of 2×103 cells were seeded into each well
of 96-well culture plates, grown for 24 h, then treated with GST or
GST-S100A9 in DMEM containing 1% FBS for 24, 48, 72, and 96 h.
After the indicated hours of incubation, 10 μl of MTT
reagent was added to the cells, followed by another 4 h of
incubation at 37°C. Dimethyl sulfoxide was added to dissolve the
formazan product for 10 min at room temperature. Finally the
absorbance was measured at 492 nm using a micro-plate reader. Each
condition was done in quintuplicate, and the experiment was
repeated 3 times.
Colony forming assay
HepG2 cells during the log growth stage were seeded
in 6-well culture plates (2×102 cells/well) and treated
with GST or GST-S100A9. After incubation for 2 weeks, the cells
were stained with crystal violet and clones were counted. The
colony-forming rate was obtained by the following calculation:
(colony number/seeded cell number) ×100%. The experiment was
repeated 3 times.
Transwell invasion assay
The invasion assay was performed as previously
described (24). The chamber of a
non-type I-collagen-coated 24-well culture insert (Millipore,
Billerica, MA, USA) was used, and the upper side of the insert was
coated with ECM gel (Sigma, St. Louis, MO, USA). Briefly, HepG2
cells were placed in the upper chamber (1×105 cells) and
incubated with GST or GST-S100A9 in serum-free medium, while the
lower chamber contained medium only (600 μl/each insert)
with 20% FBS. After incubation for 24 h, the transmembrane cells
were dried, fixed with methanol, stained with hematoxylin and eosin
(H&E), and counted under a microscope at ×100 magnification.
The experiment was performed 3 times.
Tumorigenicity assays in nude mice
The in vivo experiments were performed in
accordance with the guidelines established by the Animal Care and
Use Committee, University Laboratory Animal Research. The 6-8-week
old female nude mice were randomly divided into 3 groups
(n=5/group). The HepG2 cells treated with GST- or GST-hS100A9
(5×106 cells/cell line) for 3 days were suspended in 200
μl phosphate buffer solution (PBS) and then injected
subcutaneously into the posterior flank position of nude mice. The
nude mice in the blank group were injected with untreated HepG2
cells (5×106/mouse). Subcutaneous tumor growth was
recorded every 5 days with vernier calipers. Tumor volume was
calculated using the formula: π/6 x (Rmax x
Rmin2), where R = tumor diameter. The mice
were sacrificed after 50 days, and the tumor tissues were
collected, fixed in buffered formaldehyde, embedded in paraffin,
and sectioned for further histological and immunohistochemical
analysis.
Western blot analysis
The cells treated with GST or GST-hS100A9 were
harvested and lysed in radio immuno-precipitation assay (RIPA)
buffer. The total cell lysate was centrifuged and the supernatant
was denatured by boiling and loaded onto a 10% gradient SDS-PAGE.
After SDS-PAGE, the proteins on the gel were blotted onto a PVDF
membrane. The membrane was blocked with 5% bovine serum albumin
(BSA), and incubated with the primary antibodies and then with a
secondary antibody conjugated with horseradish peroxidase. The
proteins of interest were detected using the SuperSignal West Pico
Chemiluminescent Substrate kit. The results were recorded using the
Bio-Rad Electrophoresis Documentation (Gel Doc 1000) and Quantity
One version 4.5.0 software (Bio-Rad, Hercules, CA, USA).
The primary antibodies used this study included
mouse anti-S100A9 monoclonal antibody (1:1,000 dilution), rabbit
anti-phosphor-p38 monoclonal antibody (1:1,000 dilution), rabbit
anti-p38 monoclonal antibody (1:1,000 dilution), rabbit
anti-phosphor-ERK1/2 monoclonal antibody (1:1,000 dilution), rabbit
anti-ERK1/2 monoclonal antibody (1:1,000 dilution), rabbit
anti-phosphor-JNK antibody (1:1,000 dilution), rabbit anti-JNK
monoclonal antibody (1:1,000 dilution) and mouse anti-β-actin
monoclonal antibody (1:1,000 dilution). The secondary antibodies
included goat anti-rabbit IgG serum (1:5,000 dilution; Zhongshan
Golden Bridge, Beijing, China) and goat anti-mouse IgG serum
(1:5,000 dilution; Zhongshan Golden Bridge).
Immunohistochemistry (IHC)
Paraffin-embedded subcutaneous tumor sections were
processed for immunohistochemical analysis. Briefly, the
deparaffinized and dehydrated sections were boiled for 10 min in
0.01 M citrate buffer and incubated with 0.3% hydrogen peroxide
(H2O2) in methanol for 15 min to block
endogenous peroxidase, then with primary and secondary antibodies
tagged with the peroxidase in serial order. The desired brown
reaction product was obtained after incubation with 0.05%
3,3-diaminobenzidine tetrachloride (DAB). Finally, the sections
were counterstained with hematoxylin. The negative control groups
treated as described above, except that the primary antibody was
replaced by PBS.
Primary antibodies used in the assay were the mouse
anti-S100A9 monoclonal antibody (1:300 dilution), rabbit
anti-phosphor-p38 monoclonal antibody (1:300 dilution) and rabbit
anti-phosphor-ERK1/2 monoclonal antibody (1:300 dilution).
Inhibition of p38 and ERK1/2 with
specific inhibitors and its effects on S100A9-induced proliferation
and invasion of HepG2 cells
The cells were treated with 10 μM SB203580
(p38 inhibitor) or 20 μM PD98059 (ERK1/2 inhibitor) for 30
min, followed by treatment with GST or GST-hS100A9 for the
indicated periods of time. After treatment for 30 min, the
phosphorylation of p38 and ERK1/2 was detected by western blot
analysis; after treatment for 24 h, the cell invasion was detected
by transwell invasion assay; after treatment for 72 h, the cell
proliferation was analyzed by MTT assay.
Statistical analysis
All values in the text and figures are presented as
the means ± standard error of mean (SEM). The differences were
analyzed using one-way ANOVA followed by the Student-Newman-Keuls
test, and all statistical analyses were performed using GraphPad
Prism software. Statistical differences are presented at
probability levels of p<0.05, p<0.01 and p<0.001.
Results
Proliferative and invasive activities of
HepG2 human HCC cells and S100A9 expression
In this study, we first used the L02 human normal
liver cell line as a control to characterize the proliferation and
invasive capability of the HepG2 human HCC cell line and to
investigate the change in S100A9 expression in HepG2 cells by MTT
assay, transwell invasion assay, western blot analysis and IHC. It
was found that the absorbance of the formazan product by HepG2
cells at days 3 and 4 was 0.448±0.012 and 0.524±0.017 and the
absorbance by L02 cells was 0.403±0.01 and 0.433±0.012 (p<0.01
and p<0.001, respectively; Fig.
1A). At the same time, the trans-membrane cell numbers were
81±4 cells/field in the HepG2 cells and 24±3 cells/field in the L02
cells (p<0.01, Fig. 1B),
suggesting that the HepG2 cells had strong proliferative and
invasive capabilities. Western blot analysis showed a higher
expression of S100A9 in the HepG2 cells compared with the L02 cells
(p<0.05, Fig. 1C). S100A9
upregulation in HepG2 cells was also confirmed by IHC and S100A9
was mainly localized in the cytoplasm (Fig. 1D).
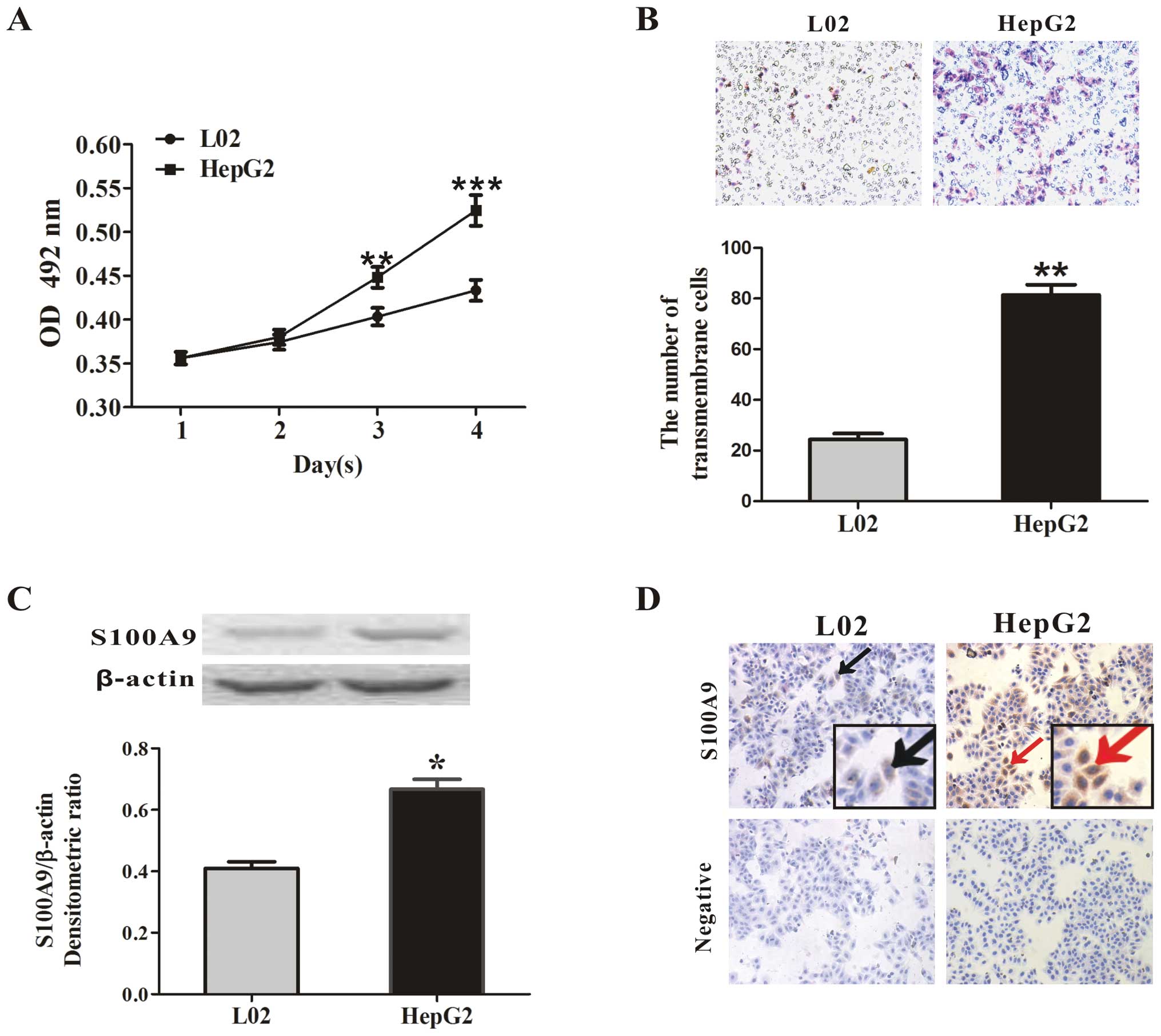 | Figure 1Characterization of the HepG2 human
hepatocellular carcinoma cell line. (A) MTT assay. The viability of
the HepG2 human hepatocellular carcinoma cells and the L02 human
normal liver cells was detected by MTT assay at 24, 48, 72 and 96
h, as described in Materials and methods. The absorbance was
measured at 492 nm using a microplate reader. The results represent
the mean absorbance ± SEM of 3 independent experiments. HepG2 cells
had a strong proliferative ability. **p<0.01 or
***p<0.001 vs. L02. (B) Transwell invasion assay.
Equal numbers of HepG2 cells and L02 cells were subjected to cell
invasion using transwell invasion assay. After 24 h, the
transmembrane cells were stained with hematoxylin and eosin
(H&E) and counted under a microscope. Representative images of
transmembrane cells are shown in the upper panel, and the mean
number of transmembrane cells ± SEM per microscopic field of 3
independent experiments are quantified in the lower panel.
Magnification, ×100. **p<0.01 vs. L02. (C) S100A9
expression in HepG2 and L02 cells was measured by western blot
analysis. The densitometric ratios were normalized to those of
β-actin, and the results are expressed as the mean densitometric
ratios ± SEM in the lower panel. *p<0.05 vs. L02. (D)
IHC assay of S100A9 expression in HepG2 and L02 cells.
Representative images are shown. Black arrows, S100A9-expressing
L02 cells (brown); red arrows, S100A9-expressing HepG2 cells
(brown). Magnification, ×100. Inlets represent higher
magnification. |
S100A9-induced proliferation and invasion
of HepG2 cells in vitro
To investigate the effects of extracellular S100A9
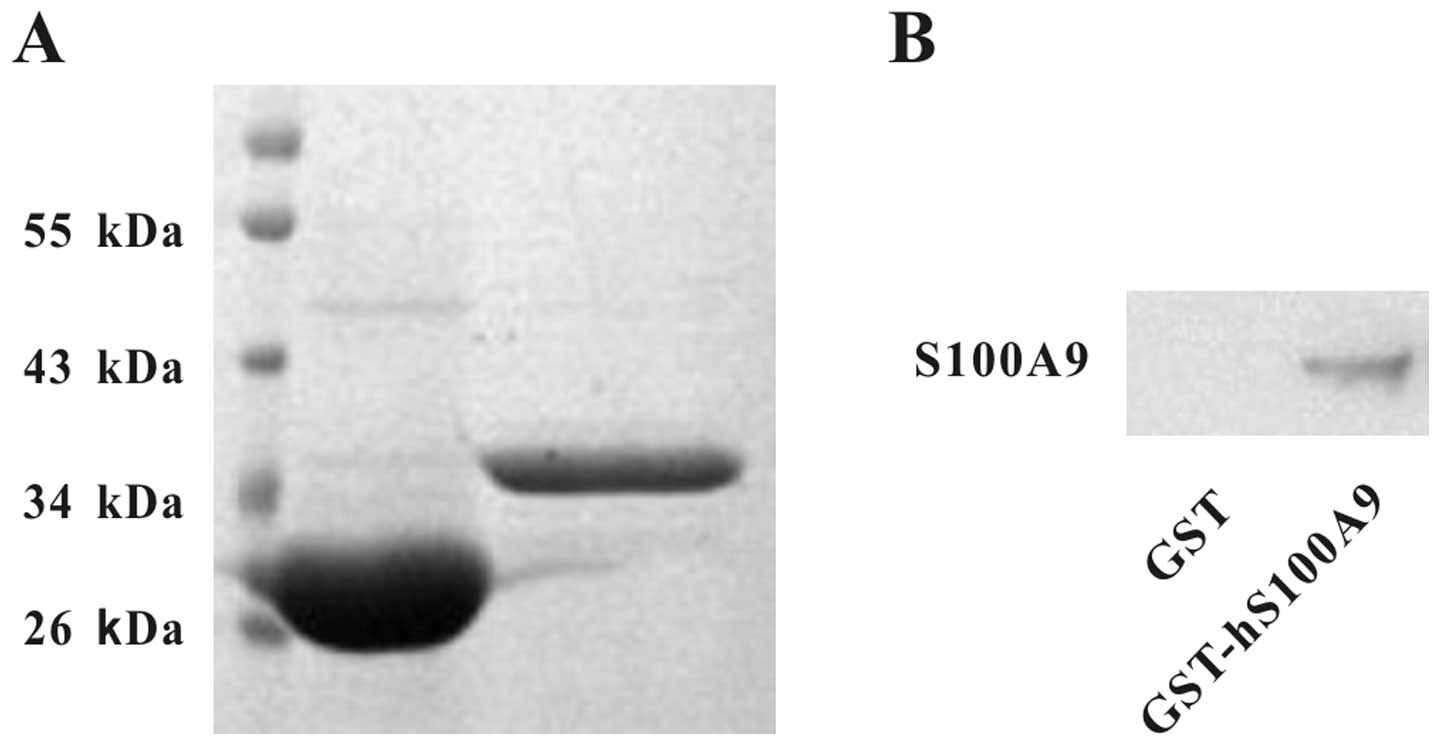
on the proliferation and invasion of HepG2 cells, we first prepared
GST-hS100A9 and GST proteins, which were identified by SDS-PAGE and
western blot analysis (Fig. 2A and
B). Their purities were quantified by Quantity One software
after SDS-PAGE and were found to be >90% (Fig. 2A). The purified proteins were used
to treat cells in our subsequent experiments.
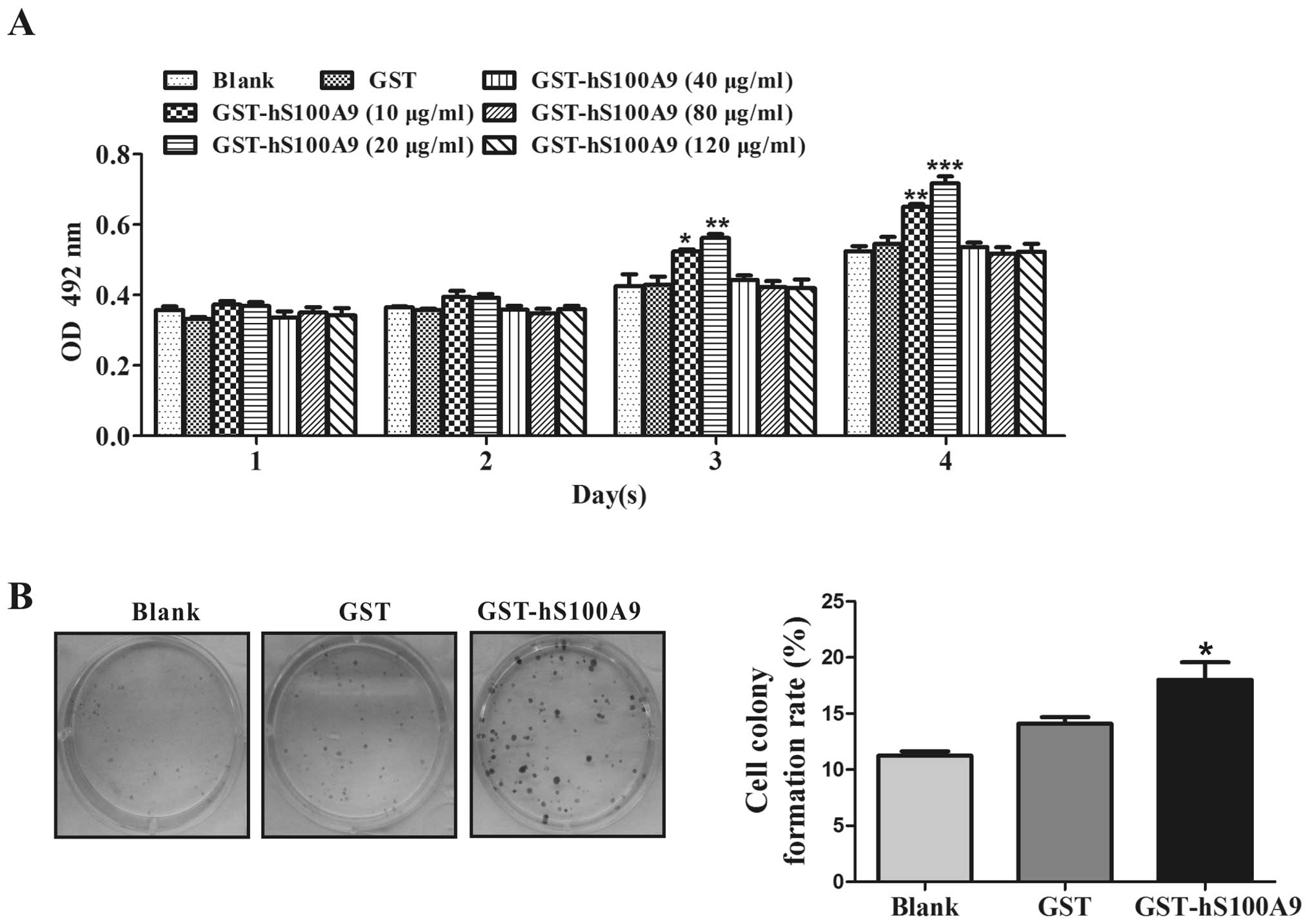
The viability of HepG2 cells was assayed by MTT.
After the HepG2 cells were treated with GST-hS100A9 at 0, 10, 20,
40, 80 and 120 μg/ml for 4 successive days, we found that
GST-hS100A9 at 10 and 20 μg/ml enhanced the cell
proliferation at day 3 (p<0.05 and p<0.01, respectively) and
day 4 (p<0.01 and p<0.001, respectively), and that
GST-hS100A9 at 20 μg/ml had a more significant effect on
cell proliferation (p<0.001), while the protein at 40, 80 and
120 μg/ml had no effects on cell proliferation, compared
with the GST group (Fig. 3A).
thus, we selected 20 μg/ml as the concentration for all
remaining experiments. Furthermore, after treatment with
GST-hS100A9 for 2 weeks, we found a significant increase in colony
formation in the GST-hS100A9 group and the colony-forming rate
increased by 25.1% compared with that of the GST group (p<0.05)
(Fig. 3B).
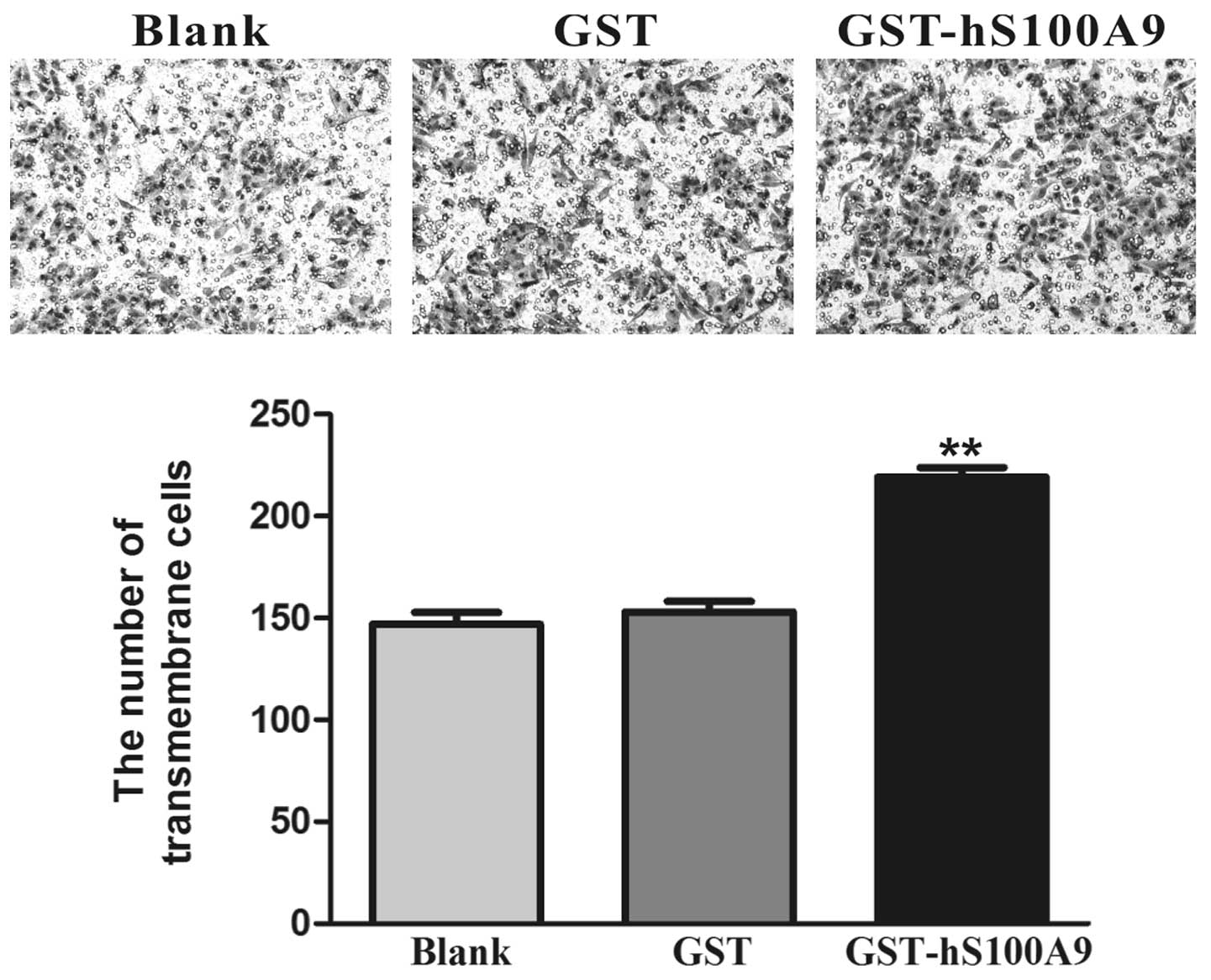
Cell invasiveness plays a crucial role in the
process of tumor metastasis. Transwell invasion assay was used to
detect the change in cellular invasion induced by GST-hS100A9.
After treatment for 24 h, the number of transmembrane cells in the
GST-hS100A9 group increased by 43.1% compared with that of the GST
group (p<0.01, Fig. 4).
S100A9 promotes tumorigenicity of HepG2
cells in vivo
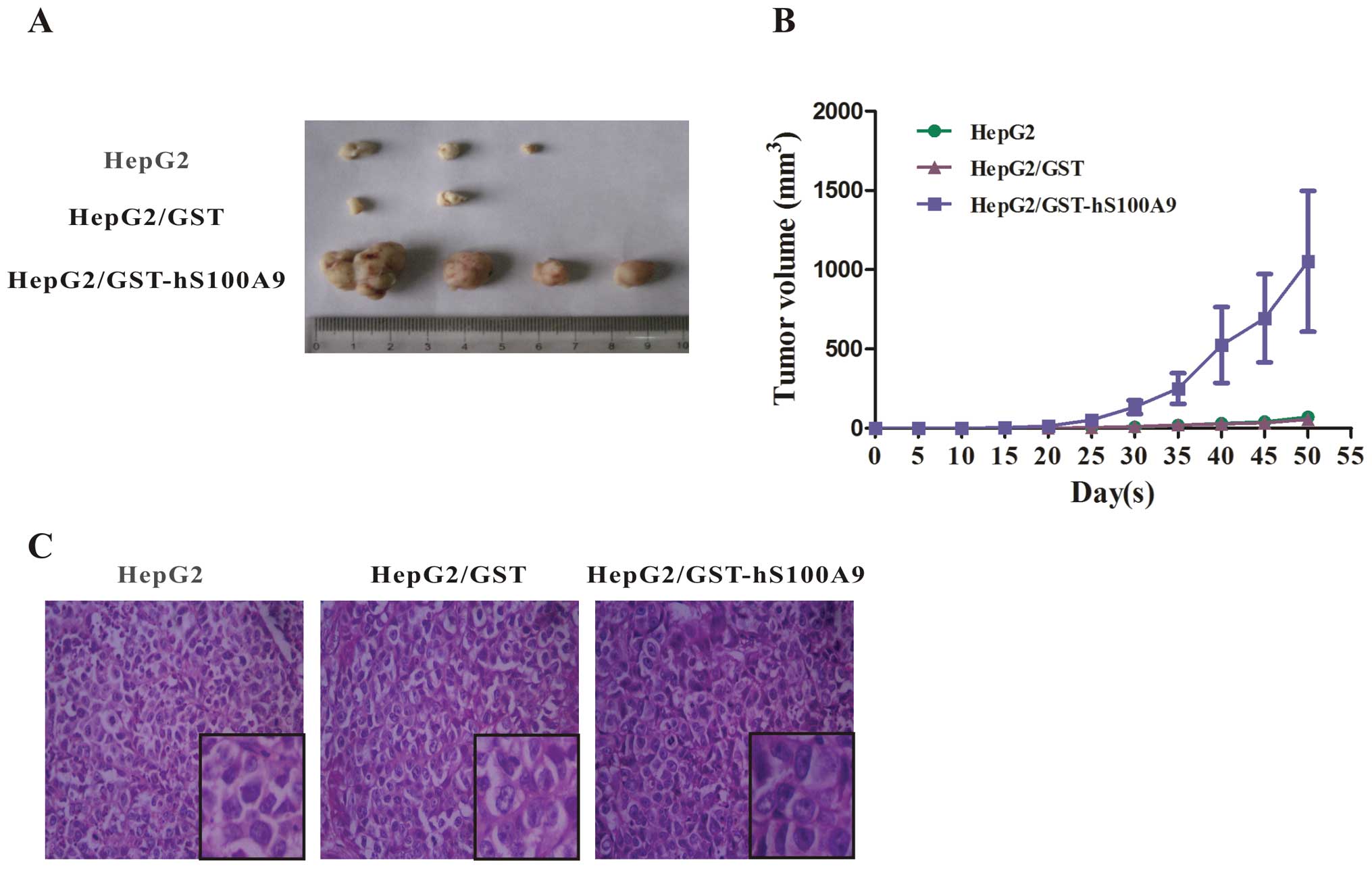
Based on the promotive effect of exogenous S100A9
protein on the proliferation of HepG2 cells in vitro, we
then sought to confirm this effect in vivo. The 3 groups of
HepG2 cells (untreated or treated with GST or GST-hS100A9 for 72 h)
were subcutaneously implanted into nude mice. Tumor volume was
measured every 5 days using a vernier caliper and the tumor tissues
were surgically excised at day 50 after injection (Fig. 5A). At 3 weeks after injection, no
tumors were found in the blank and GST groups, but were found in 4
out of 5 mice in the GST-hS100A9 group. Over the period of 50 days,
the tumor volumes became palpable from day 25 to 50 and grew from
4±1.5 to 71±27.8 mm3 in the blank group, from 5±1.0 to
56±8.0 mm3 in the GST group, and from 60±45.0 to
1,053±444.0 mm3 in the GST-hS100A9 group (Fig. 5B, Table I). The histological examination
(H&E staining) showed that the tumor cells in the 3 groups were
obviously heterogeneous, with a large nucleus, a high
nucleus/cytoplasm ratio, with an irregular nuclear shape and
variable nuclear size (Fig.
5C).
 | Table ITumorigenicity of the 3 groups of
HepG2 cells subcutaneously implanted into nude mice. |
Table I
Tumorigenicity of the 3 groups of
HepG2 cells subcutaneously implanted into nude mice.
| Group | No. (%) of mice
with tumors | Tumor onset
(day) | Average tumor
volume (mm3) |
|---|
| HepG2 | 3 (60) | 28.7±1.7 | 71±27.8 |
| HepG2/GST | 2 (40) | 29.5±2.5 | 56±8.0 |
|
HepG2/GST-S100A9 | 4 (80) | 17.0±0.7 | 1,053±444.0 |
S100A9-induced activation of the MAPK
signaling pathway in HepG2 cells
It has previously been reported that S100A9
activates the MAPK signaling pathway (15–16,22).
To determine whether S100A9 is involved in the activation of the
MAPK signaling pathway in HepG2 cells, we detected and analyzed the
phosphorylation of MAPKs in cell lysates of HepG2 cells treated
with GST-hS100A9 for different periods of time by western blot
analysis. The results showed that GST-hS100A9 had no effect on the
phosphorylation of SAPK/JNK but enhanced the phosphorylation of
ERK1/2 (within 15 min, p<0.01) and p38 (within 30 min,
p<0.01) MAPKs within 60 min (Fig.
6A). Furthermore, the role of S100A9 in the activation of p38
and ERK1/2 was also confirmed in vivo. IHC showed that there
was a higher expression of S100A9, phosphor-p38 and phosphor-ERK1/2
in the subcutaneous tumor tissues in the GST-hS100A9 group,
compared with the other 2 groups (Fig.
6B). These data demonstrate that S100A9 enhances the activity
of the MAPK signaling pathway (p38 and ERK1/2) in HepG2 cells.
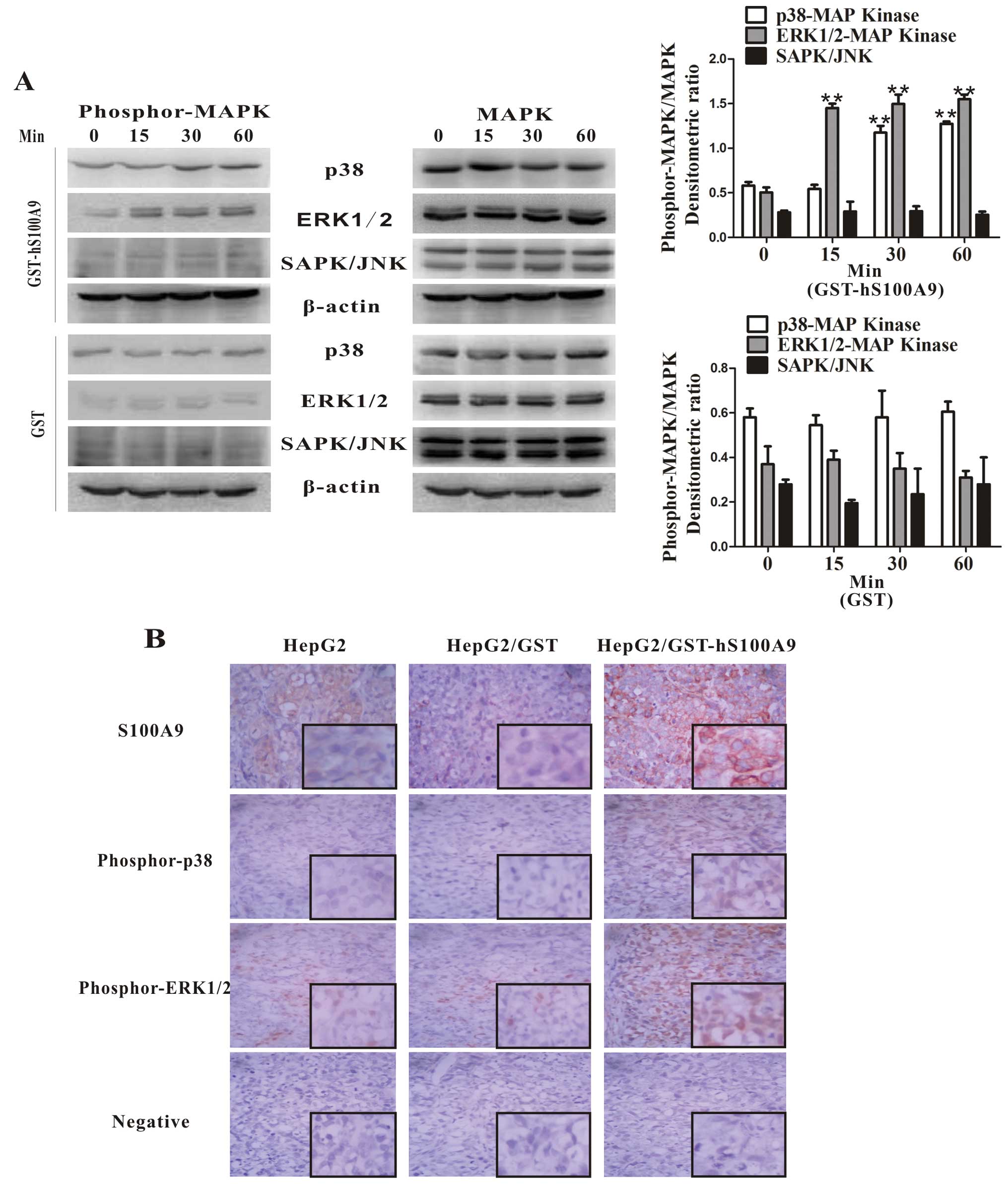 | Figure 6Exogenous S100A9 induces the
activation of the MAPK signaling pathway (p38 and ERK1/2) in HepG2
cells. (A) HepG2 cells were stimulated with GST (20 μg/ml)
and GST-S100A9 (20 μg/ml) for 0, 15, 30 and 60 min, and cell
lysates were analyzed by western blot analysis using respective
antibodies against phosphorylated MAPKs. Total p38, ERK1/2,
SAPK/JNK and β-actin were included as the loading controls. The
densitometric ratios were compared to the controls and then
normalized to the β-actin control, and are shown in the upper right
panel. **p<0.01, GST-hS100A9 (15, 30 or 60 min) vs.
GST-hS100A9 (0 min). (B) Tumor tissues derived from mice in the 3
groups (injected with HepG2 cells, GST-treated HepG2 cells and
GST-hS100A9-treated HepG2 cells) were subjected to
immunohistochemical staining for S100A9, phosphorylated p38 and
ERK1/2. An intense staining for S100A9, phosphorylated p38 and
ERK1/2 was shown in the GST-hS100A9 group compared with the other 2
control groups. Representative images are shown. Magnification,
×400. Inlets represent higher magnification. |
Impact of the inhibition of MAPK
signaling on S100A9-induced proliferation and invasion of HepG2
cells
We further investigated whether the activation of
the MAPK signaling pathway is involved in the S100A9-induced
proliferation and invasion of HepG2 cells. The specific inhibitors
of p38 (SB203580) and ERK1/2 (PD98059) were used to pre-treat the
HepG2 cells and the S100A9-induced phosphorylation of p38 and
ERK1/2 was reversed by SB203580 (p<0.01) and PD98059
(p<0.01), respectively (Fig. 7A and
B). At the same time, we detected changes in cell proliferation
and invasion in the presence and absence of SB203580 or PD98059. We
found that the S100A9-induced proliferation of HepG2 cells was
reversed by PD98059 (p<0.05), but not by SB203580 (Fig. 7C) and that the S100A9-induced cell
invasion of HepG2 cells was reversed by SB203580 (p<0.05), but
not reversed by PD98059 (Fig. 7D).
These data suggest that the promotive role of S100A9 in the
proliferation and invasion of HepG2 cells may be mediated by the
phosphorylation of p38 and ERK1/2, respectively.
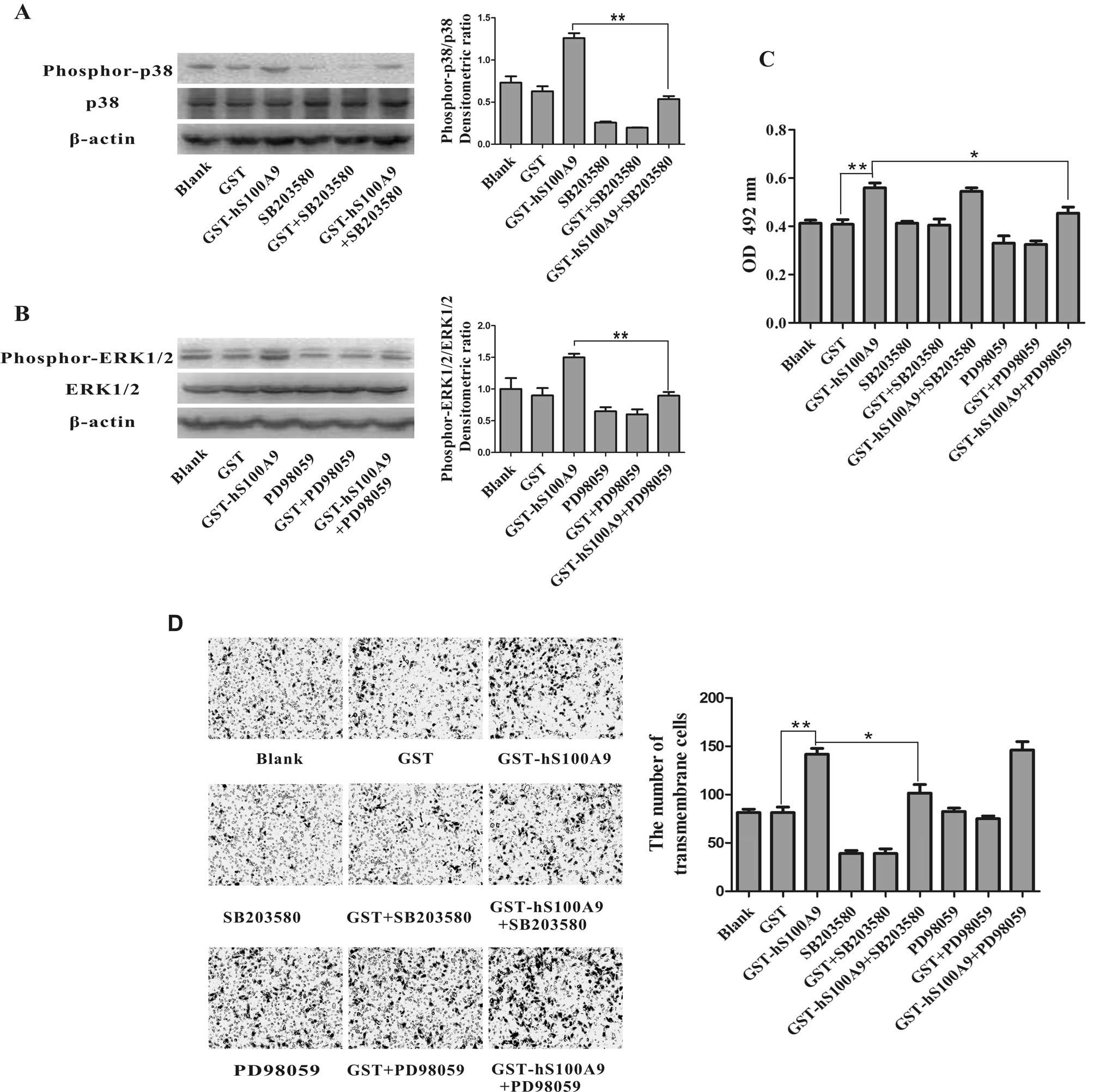 | Figure 7The promotive role of S100A9 in the
proliferation and invasion of HepG2 cells. The proliferation and
invasion was reversed by inhibitors of ERK1/2 (PD98059) and p38
(SB203580), respectively. (A and B) HepG2 cells were pre-treated
with the inhibitors of p38 (SB203580, 10 μM) and ERK1/2
(PD98059, 20 μM) for a period of 30 min prior to treatment
for 30 min with GST (20 μg/ml) or GST-hS100A9 (20
μg/ml). The cell lysates were then analyzed by western blot
analysis using respective antibodies against phosphorylated p38 or
ERK1/2. The densitometric ratios were compared to the control and
then normalized to the β-actin control. The promotive role of
S100A9 in the phosphorylation of (A) p38 and (B) ERK1/2. The
phosphorylation of p38 and ERK1/2 was reversed by SB203580 and
PD98059, respectively. **p<0.01, GST-hS100A9 vs.
GST-hS100A9+SB203580 or GST-hS100A9+PD98059. (C) HepG2 cells were
treated with GST-S100A9 (20 μg/ml) in the presence of
SB203580 or PD98059 for 72 h. Cell viability was measured by MTT
assay. The promotive role of S100A9 in cell proliferation was
reversed by PD98059. The results represent the mean absorbance ±
SEM of 3 independent experiments. **p<0.01,
GST-hS100A9 vs. GST control; *p<0.01, GST-hS100A9 vs.
GST-hS100A9 + PD98059. (D) HepG2 cells were treated with GST-S100A9
(20 μg/ml) in the presence of SB203580 or PD98059 for 24 h.
Cell invasion was measured by transwell invasion assay. The
transmembrane cells were stained with H&E and were counted
under a microscope. The results weere obtained from 5 randomly
selected fields for each well, and representative images of
transmembrane cells are shown in the left panel; the mean number of
transmembrane cells ± SEM per microscopic field of 3 independent
experiments are quantified in the right panel. Magnification, ×100.
**p<0.01, GST-hS100A9 vs. GST control;
*p<0.05, GST-hS100A9 vs. GST-hS100A9+SB203580. |
Discussion
As mentioned in the Introduction, it is necessary to
elucidate the potential molecular mechanisms involved in HCC
development in order to discover novel therapeutic strategies
against HCC. S100A9 has been shown to play a role in various types
of tumor (14–16,22).
A previous study demonstrated that S100A9 expression was associated
with tumor differentiation and vascular invasion in HCC (10). Nonetheless, the exact role of
S100A9 in the progression of HCC has not been extensively studied.
Therefore, in this study, we investigated the effects of S100A9
protein on the cell proliferation and invasion of HepG2 HCC cells
and the possible underlying mechanisms.
Our results showed that S100A9 was upregulated in
the HepG2 HCC cells compared with the L02 normal liver cells. These
results are consistent with those from previous studies
demonstrating that S100A9 is upregulated in other HCC cell lines,
including Hep3B and HuH-7 cells (11). A number of studies have
demonstrated that extracellular S100A9 secreted from the tumor
cells functions as a danger signal by activating signaling cascades
and promoting tumor cell proliferation in breast, colon and
pancreatic cancer (14–16). However, little is known about the
effect of extracellular S100A9 on HCC cells. Our data showed that
exogenous S100A9 protein promoted the proliferation and growth of
HepG2 HCC cells in vitro and in vivo, indicating that
S100A9 is involved in HCC development. We also found that treatment
with S100A9 resulted in an increase in cell invasion, suggesting
that S100A9 may be involved in the metastasis of HCC cells. These
results are consistent with those from previous reports,
demonstrating that S100A9 plays a role in the metastasis of
prostate, colorectal and breast cancer (16,25–26).
Of note, our results showed that higher concentrations of S100A9
(40–120 μg/ml) had no apoptotic effects on HepG2 cells,
which vary from the results of previous reports demonstrating that
S100A9 at the range of 25–250 μg/ml exerted apoptotic
effects on certain types of tumor cells (13–18).
This discrepancy suggests the apoptotic effects exerted by S100A9
are dependent on the tumor cell type.
Activated MAPK signaling plays a central role in HCC
development (27–31), providing us with a novel target for
intervention in HCC treatment. A number of previous studies have
shown that extracellular S100A9 activates the MAPK signaling
pathway in a variety of cell lines, mostly through the receptor for
advanced glycation end-products (RAGE) (14–16,32–34).
Therefore, we investigated the effect of S100A9 on MAPK signaling
in HCC and found that S100A9 enhanced the phosphorylation of p38
and ERK1/2 MAPKs in HepG2 cells in vitro. This
phosphorylation of p38 and ERK1/2 was inhibited by the specific
inhibitors, SB203580 and PD98059, respectively; this effect was
also demonstrated in vivo. These data suggest that S100A9 is
involved in HCC development by the activation of the MAPK signaling
pathway. A previous study demonstrated that the expression of RAGE
mRNA was higher in HCC compared to normal liver tissues (35), and that the binding of RAGE with
its ligands activated the MAPK signaling pathway (36). However, further studies are
required to elucidate whether the S100A9-enhanced phosphorylation
of p38 and ERK1/2 MAPKs in HepG2 cells is mediated by RAGE.
The results from the present study demonstrated that
the S100A9-induced cell proliferation was reversed by PD98059 (an
ERK1/2 inhibitor), suggesting that S100A9 promotes the
proliferation of HepG2 cells via the ERK1/2 signal transduction
pathway. These results are consistent with those from previous
studies, demonstrating that the ERK1/2 signal transduction pathway
plays a crucial role in liver tumor cell proliferation and
tumorigenesis (29,37). At the same time, we found that the
S100A9-induced cell invasion was reversed by SB203580 (a p38
inhibitor), which suggests that S100A9 promotes the invasion of
HepG2 cells via the p38 signal transduction pathway. These results
are supported by previous studies showing that the p38 signal
transduction pathway plays an important role in the upregulation of
the expression of matrix metalloproteinases (MMPs), correlating
with an increased invasive phenotype of tumor cells (38–41).
In conclusion, the current observations indicate
that S100A9 is upregulated in the HepG2 HCC cell line, and that
exogenous S100A9 promotes the proliferation and invasion of HepG2
HCC cells by activating ERK1/2 and p38 MAPKs, respectively.
Therefore, S100A9 may be a novel intervention target for HCC
treatment.
Acknowledgements
The authors would like to thank
Professor T.C. He (The University of Chicago, Medical center) for
his kind provision of the HepG2 and L02 cell lines. The present
study was supported by the National Natural Science Foundation of
China (grant no. 30772548) and the Foundation for Excellent Master
Dissertation of College of Laboratory Medicine at Chongqing Medical
University.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Cornella H, Alsinet C and Villanueva A:
Molecular pathogenesis of hepatocellular carcinoma. Alcohol Clin
Exp Res. 35:821–825. 2011. View Article : Google Scholar
|
|
3
|
Bruix J and Sherman M: Management of
hepatocellular carcinoma. Hepatology. 42:1208–1236. 2005.
View Article : Google Scholar
|
|
4
|
Rossi L, Zoratto F, Papa A, et al: Current
approach in the treatment of hepatocellular carcinoma. World J
Gastrointest Oncol. 2:348–359. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Donato R: S100: a multigenic family of
calcium-modulated proteins of the EF-hand type with intracellular
and extracellular functional roles. Int J Biochem Cell Biol.
33:637–668. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Heizmann CW, Fritz G and Schafer BW: S100
proteins: structure, functions and pathology. Front Biosci.
7:d1356–d1368. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Donato R: Intracellular and extracellular
roles of S100 proteins. Microsc Res Tech. 60:540–551. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marenholz I, Heizmann CW and Fritz G: S100
proteins in mouse and man: from evolution to function and pathology
(including an update of the nomenclature). Biochem Biophys Res
Commun. 322:1111–1122. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Benedyk M, Sopalla C, Nacken W, et al:
HaCaT keratinocytes overexpressing the S100 proteins S100A8 and
S100A9 show increased NADPH oxidase and NF-kappaB activities. J
Invest Dermatol. 127:2001–2011. 2007. View Article : Google Scholar
|
|
10
|
Arai K, Yamada T and Nozawa R:
Immunohistochemical investigation of migration inhibitory
factor-related protein (MRP)-14 expression in hepatocellular
carcinoma. Med Oncol. 17:183–188. 2000. View Article : Google Scholar
|
|
11
|
Nemeth J, Stein I, Haag D, et al: S100A8
and S100A9 are novel nuclear factor kappa B target genes during
malignant progression of murine and human liver carcinogenesis.
Hepatology. 50:1251–1262. 2009. View Article : Google Scholar
|
|
12
|
Srikrishna G: S100A8 and S100A9: new
insights into their roles in malignancy. J Innate Immun. 4:31–40.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ghavami S, Chitayat S, Hashemi M, et al:
S100A8/A9: a Janus-faced molecule in cancer therapy and
tumorgenesis. Eur J Pharmacol. 625:73–83. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Turovskaya O, Foell D, Sinha P, et al:
RAGE, carboxylated glycans and S100A8/A9 play essential roles in
colitis-associated carcinogenesis. Carcinogenesis. 29:2035–2043.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ghavami S, Rashedi I, Dattilo BM, et al:
S100A8/A9 at low concentration promotes tumor cell growth via RAGE
ligation and MAP kinase-dependent pathway. J Leukoc Biol.
83:1484–1492. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hermani A, De Servi B, Medunjanin S,
Tessier PA and Mayer D: S100A8 and S100A9 activate MAP kinase and
NF-kappaB signaling pathways and trigger translocation of RAGE in
human prostate cancer cells. Exp Cell Res. 312:184–197. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ghavami S, Kerkhoff C, Chazin WJ, et al:
S100A8/9 induces cell death via a novel, RAGE-independent pathway
that involves selective release of Smac/DIABLO and Omi/HtrA2.
Biochim Biophys Acta. 1783:297–311. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ghavami S, Eshragi M, Ande SR, et al:
S100A8/A9 induces autophagy and apoptosis via ROS-mediated
cross-talk between mitochondria and lysosomes that involves BNIP3.
Cell Res. 20:314–331. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schmidt CM, McKillop IH, Cahill PA and
Sitzmann JV: The role of cAMP-MAPK signalling in the regulation of
human hepatocellular carcinoma growth in vitro. Eur J Gastroenterol
Hepatol. 11:1393–1399. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huynh H, Nguyen TT, Chow KH, Tan PH, Soo
KC and Tran E: Over-expression of the mitogen-activated protein
kinase (MAPK) kinase (MEK)-MAPK in hepatocellular carcinoma: its
role in tumor progression and apoptosis. BMC Gastroenterol.
3:192003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nakagawa H and Maeda S: Molecular
mechanisms of liver injury and hepatocarcinogenesis: focusing on
the role of stress-activated MAPK. Patholog Res Int.
2012:1728942012.PubMed/NCBI
|
|
22
|
Ichikawa M, Williams R, Wang L, Vogl T and
Srikrishna G: S100A8/A9 activate key genes and pathways in colon
tumor progression. Mol Cancer Res. 9:133–148. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
You L, Xu LL, Guo YY, et al: Prokaryotic
expression, purification and identification of GST-human S100A9
fusion protein. Chin J Biochem Pharm. 32:253–256. 2011.
|
|
24
|
Punathil T, Tollefsbol TO and Katiyar SK:
EGCG inhibits mammary cancer cell migration through inhibition of
nitric oxide synthase and guanylate cyclase. Biochem Biophys Res
Commun. 375:162–167. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ang CW, Nedjadi T, Sheikh AA, et al: Smad4
loss is associated with fewer S100A8-positive monocytes in
colorectal tumors and attenuated response to S100A8 in colorectal
and pancreatic cancer cells. Carcinogenesis. 31:1541–1551. 2010.
View Article : Google Scholar
|
|
26
|
Arai K, Takano S, Teratani T, Ito Y,
Yamada T and Nozawa R: S100A8 and S100A9 overexpression is
associated with poor pathological parameters in invasive ductal
carcinoma of the breast. Curr Cancer Drug Targets. 8:243–252. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sakurai T, He G, Matsuzawa A, et al:
Hepatocyte necrosis induced by oxidative stress and IL-1 alpha
release mediate carcinogen-induced compensatory proliferation and
liver tumorigenesis. Cancer Cell. 14:156–165. 2008. View Article : Google Scholar
|
|
28
|
Spaziani A, Alisi A, Sanna D and Balsano
C: Role of p38 MAPK and RNA-dependent protein kinase (PKR) in
hepatitis C virus core-dependent nuclear delocalization of cyclin
B1. J Biol Chem. 281:10983–10989. 2006. View Article : Google Scholar
|
|
29
|
Gailhouste L, Ezan F, Bessard A, et al:
RNAi-mediated MEK1 knock-down prevents ERK1/2 activation and
abolishes human hepatocarcinoma growth in vitro and in vivo. Int J
Cancer. 126:1367–1377. 2010.PubMed/NCBI
|
|
30
|
Guo L, Guo Y, Xiao S and Shi X: Protein
kinase p-JNK is correlated with the activation of AP-1 and its
associated Jun family proteins in hepatocellular carcinoma. Life
Sci. 77:1869–1878. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Min L, He B and Hui L: Mitogen-activated
protein kinases in hepatocellular carcinoma development. Semin
Cancer Biol. 21:10–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Simard JC, Girard D and Tessier PA:
Induction of neutrophil degranulation by S100A9 via a
MAPK-dependent mechanism. J Leukoc Biol. 87:905–914. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sunahori K, Yamamura M, Yamana J, et al:
The S100A8/A9 heterodimer amplifies proinflammatory cytokine
production by macrophages via activation of nuclear factor kappa B
and p38 mitogen-activated protein kinase in rheumatoid arthritis.
Arthritis Res Ther. 8:R692006. View
Article : Google Scholar
|
|
34
|
Ehlermann P, Eggers K, Bierhaus A, et al:
Increased proinflammatory endothelial response to S100A8/A9 after
preactivation through advanced glycation end products. Cardiovasc
Diabetol. 5:62006. View Article : Google Scholar
|
|
35
|
Hiwatashi K, Ueno S, Abeyama K, et al: A
novel function of the receptor for advanced glycation end-products
(RAGE) in association with tumorigenesis and tumor differentiation
of HCC. Ann Surg Oncol. 15:923–933. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hoefen RJ and Berk BC: The role of MAP
kinases in endothelial activation. Vascul Pharmacol. 38:271–273.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee HC, Tian B, Sedivy JM, Wands JR and
Kim M: Loss of Raf kinase inhibitor protein promotes cell
proliferation and migration of human hepatoma cells.
Gastroenterology. 131:1208–1217. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Montesano R, Soriano JV, Hosseini G,
Pepper MS and Schramek H: Constitutively active mitogen-activated
protein kinase kinase MEK1 disrupts morphogenesis and induces an
invasive phenotype in Madin-Darby canine kidney epithelial cells.
Cell Growth Differ. 10:317–332. 1999.
|
|
39
|
Behren A, Binder K, Vucelic G, et al: The
p38 SAPK pathway is required for Ha-ras induced in vitro invasion
of NIH3T3 cells. Exp Cell Res. 303:321–330. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang X, Chen S, Xu L, et al: Genistein
inhibits p38 map kinase activation, matrix metalloproteinase type
2, and cell invasion in human prostate epithelial cells. Cancer
Res. 65:3470–3478. 2005.
|
|
41
|
Ringshausen I, Dechow T, Schneller F, et
al: Constitutive activation of the MAPkinase p38 is critical for
MMP-9 production and survival of B-CLL cells on bone marrow stromal
cells. Leukemia. 18:1964–1970. 2004. View Article : Google Scholar : PubMed/NCBI
|