Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common cancer worldwide and the third most common cause of death
from cancer (1). Many studies have
shown that hepatitis B virus (HBV) infection is a risk factor for
the development of HCC (2,3). The multifunctional oncoprotein HBx,
encoded by the smallest open reading frame of the HBV and its
interaction with signal transduction pathways are involved in the
initiation of hepatocarcinogenesis (3). In our previous study we reported that
the Notch signaling pathway is activated in HBx-transformed L02
cells and is involved in the malignant transformation of normal
hepatic cells (4). However, the
role of Notch signaling in the development of HCC has not been
fully elucidated and its relationship with HBx still requires
further exploration.
The evolutionarily conserved Notch signaling pathway
mediates cell-to-cell communication and is involved in mediating
binary cell fate decisions. There are four mammalian Notch
receptors (Notch1-4) and two groups of ligands, jagged (JAG1 and
JAG2) and δ-like protein (DLL1, 3 and 4) (5). Upon binding to Notch, the receptor is
cleaved by ADAM10 or ADAM17 (formerly, a disintegrin and
metalloproteinase domain 10 and 17) (6,7).
There is then an intramembranous cleavage by the γ-secretase
protease complex, resulting in the release of the Notch receptor’s
intracellular domain (NICD) (8).
The NICD subsequently translocates to the nucleus, binds to the
transcription factor CSL (i.e., CBF1/RBP-Jκ/suppressor of
hairless/LAG1), which then activates the transcription of a group
of downstream genes (9) including
pro-oncogenes (p21 and c-Myc), cyclin D1, cyclin A and the subunits
of nuclear factor κB (NF-κB) (10–14).
Our previous research indicated that Notch signaling might be one
of the downstream targets through which HBx functions as an
oncoprotein (4).
In addition, HBx interacts with many other signal
pathways in the initiation of hepatocarcinogenesis, including
AKT/PKB, ERK1/2, SAPK, NF-κB signal transduction pathway (15). Many researchers have also shown
that HBx upregulates the activity of the NF-κB transcription factor
(16–18). The NF-κB pathway comprises a family
of transcription factors, namely p50, p52, p65 (RelA), RelB and
c-Rel (19), which homo- or
heterodimerize to form transcriptional regulatory complexes. All
five of these transcription factors contain Rel homology domains
that participate in dimerization and DNA binding. The IκB (i.e.,
nuclear factor of κ light polypeptide gene enhancer in B-cells
inhibitor) family, including IκBα, IκBβ, IκBɛ, Bcl-3, p100 (p52
precursor) and p105 (p50 precursor), are the inhibitors of NF-κB
pathway, each with different roles (20). Activation of NF-κB typically
involves the phosphorylation of IκB by the IκB kinase (IKK)
complex, which results in IκB degradation (20). This releases NF-κB and allows it to
translocate freely to the nucleus (20).
Accumulating evidence indicates that the Notch and
NF-κB pathway are closely related and that they contribute to the
pathogenesis of malignancies. The inhibitor of κB kinase 2 (Ikk2),
a component of the canonical NF-κB signaling pathway, synergizes
with basal Notch signaling to upregulate transcription of primary
Notch target genes, resulting in suppression of anti-inflammatory
protein expression and promotion of pancreatic carcinogenesis in
mice (21). In MDA-MB-231
triple-negative breast cancer cells, genistein inhibited the growth
of cells by inhibiting NF-κB activity via the Notch-1 signaling
pathway in a dose-dependent manner (22). Schwarzer et al(23) reported that Notch is an essential
upstream regulator of alternative NF-κB signaling and confirmed
crosstalk between both pathways in B cell-derived Hodgkin and
Reed-Sternberg cells. However, little is known about the
involvement of the Notch and NF-κB pathways in the pathogenesis of
HBx-associated HCC. This requires further investigation.
In this study we investigated whether HBx directly
binds with components of the Notch and NF-κB pathways and explored
evidence of interactions between these two pathways.
Materials and methods
Cell culture
The human non-tumor hepatic cell line L02/HBx, which
was derived from L02 cells via transfection with an HBx expression
plasmid, was successfully established previously (24). The cells were cultured in
Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Carlsbad, CA, USA)
supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY,
USA) and 250 μg/ml G418 (Invitrogen, Shanghai, China) and
maintained in a humidified incubator at 37°C under a 5%
CO2 atmosphere.
Immunofluorescence assays
Immunofluorescence assays were conducted to
visualize HBx, NICD and cell nuclei in L02/HBx cells. L02/HBx cells
were cultured on glass cover slips for 24 h and fixed with 4%
paraformaldehyde. The fixed cells were incubated with anti-HBx and
anti-NICD (both 1:200; Santa Cruz Biotechnology, Santa Cruz, CA)
for 12 h at 4°C. They were then incubated in CY3-conjugated goat
anti-mouse IgG and fluorescein isothiocyanate (FITC)-conjugated
goat anti-rabbit IgG (both 1:100; Boster, China) for 1 h to stain
the HBx and NICD proteins red and green, respectively. The nuclei
were stained blue with 4′,6-diamidino-2-phenylindole (DAPI;
Boster). The stained cells were observed under a FluoView 1000
laser scanning confocal microscope (Olympus, Japan).
Co-immunoprecipitation (coIP) assays
CoIP was performed to investigate physical
interactions between HBx and components of the Notch signaling
pathway (NICD and JAG1), components of the NF-κB signaling pathway
(p65 and p50), or IκBα (inhibitor of NF-κB pathway) in L02/HBx
cells. L02/HBx cells were lysed with RIPA Lysis buffer and
phenylmethylsulfonyl fluoride (KeyGEN Biotech, China) and the
lysates pretreated with Protein G-Agarose (Santa Cruz) to remove
non-specifically bound proteins. After centrifugation, one third of
the supernatants were immediately boiled for western blot analysis
using antibodies directed serially against NICD, JAG1, p65, or p50,
or IκBα as a positive control. The remaining supernatants were
incubated for 2 h at 4°C with 1 μg of non-immune mouse IgG
(for the negative control) or mouse anti-HBx (experimental group;
Santa Cruz). Then, the mixtures were incubated from 1 h to
overnight at 4°C with 20 μl Protein G-Agarose beads (Santa
Cruz). The immuno-complexes were extensively washed with
phosphate-buffered saline and samples were boiled in
electrophoresis sample buffer, then assayed via western blotting
using antibodies directed against NICD, JAG1, p65, p50, or
IκBα.
DAPT treatment
To repress normal activity of the Notch signaling
pathway, cells were treated with the γ-secretase inhibitor DAPT.
DAPT was purchased from Sigma-Aldrich (St. Louis, MO, USA) and
dissolved in 100% dimethyl sulf-oxide (DMSO; Sigma) to make a stock
solution of 10 mM, which was then diluted in culture medium to
obtain the desired final concentration of 20 μM. DMSO was
diluted in culture medium to a final percentage of 0.05% without
DAPT. Untreated cells were incubated in the culture medium without
any additives. Cells with or without DAPT were cultured for 48 h.
Total RNA or protein was then extracted.
Notch1 small interfering RNA (siRNA)
transfection
To block Notch signaling, in another experiment
L02/HBx cells were transfected with Notch1 siRNA. L02/HBx cells
were seeded in 6-well plates. The next day the cells (30–50%
confluence) were treated with Notch1 siRNA (sense
5′-GGUGUCUUCCAGAUCCUGAdTdT-3′; antisense
3′-dTdTCCACAGAAGGUCUAGGACU-5′) or control siRNA (which does not
match any known mammalian GenBank sequences). Notch1 siRNA and
control siRNA were purchased from RiboBio (Guangzhou, China). Cells
were transiently transfected with Notch1 siRNA or control siRNA
using Lipofectamine™ 2000 (Invitrogen). Media were replaced 6 h
after transfection. Cells were allowed to grow for 48 h and
harvested for further analysis.
Quantitative real-time PCR (QRT-PCR)
analysis
To quantify the expression of the genes of interest,
total RNA was isolated from cultured cells using TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) and cDNA was synthesized from 5
μg of total RNA using Moloney murine leukemia virus (MMLV)
reverse transcriptase (Promega, Madison, WI, USA) and
oligo(dT)18 as the primer. Real-time quantitative PCR
was carried out using SYBR Premix Ex Taq (DRR041A, Takara, Japan).
Each real-time PCR (20.0 μl) contained 2.0 μl of
cDNA, 10.0 μl of 2X SYBR Premix Ex Taq, 7.2 μl
nuclease-free water and primers at a final concentration of 0.2
μM. Real-time reverse transcription-polymerase chain
reactions (qRT-PCR) were performed in a Step One Real-Time PCR
system (Applied Biosystems, USA). The expression of RNA was
determined from the threshold cycle (Ct) and the relative
expression levels were calculated by the 2−ΔΔCt method.
All standards and samples were assayed in triplicate. The primer
sequences used to amplify specific target genes (p65, p50, IκBα,
β-actin) are listed in Table
I.
 | Table IPrimer sequences for real-time
polymerase chain reaction analysis. |
Table I
Primer sequences for real-time
polymerase chain reaction analysis.
| Gene | Primer
sequence | PCR product
(bp) | GenBank accession
no. |
|---|
| p65 | f:
5′-GGGGACTACGACCTGAATG-3′ | 118 | NM_021975.3 |
| r:
5′-GGGCACGATTGTCAAAGAT-3′ | | |
| p50 | f:
5′-CGCGGTGACAGGAGACGTGAA-3′ | 162 | NM_003998.2 |
| r:
5′-TGAGAATGAAGGTGGATGATTGCTAATGT-3′ | | |
| IκBα | f: 5′
-TCCACTCCATCCTGAAGGCTACCAA-3′ | 108 | NM_020529.2 |
| r:
5′-GACATCAGCACCCAAGGACACCAAA-3′ | | |
| β-actin | f:
5′-GTTGCGTTACACCCTTTCTTG-3′ | 157 | NM_001101.3 |
| r:
5′-GACTGCTGTCACCTTCACCGT-3′ | | |
Extraction of nuclear and cytoplasmic
proteins
To quantify protein expression changes in the
cytoplasm and nuclei of L02/HBx cells after treatment with the
Notch signal inhibitor DAPT or Notch1 siRNA, L02/HBx cells were
treated with DAPT or DMSO for 48 h, or with Notch1 siRNA or control
siRNA for 48 h. Proteins were subsequently extracted in accordance
with the instructions in a nuclear and cytoplasmic protein
extraction kit (KeyGEN Biotech). The nuclear and cytoplasmic
proteins were measured using the bicinchoninic acid method (Boster,
Wuhan, China). The samples were stored at −80°C and assayed via
western blotting and electrophoretic mobility shift assay
(EMSA).
Western blot analysis
Cells were lysed and the lysates were subjected to
SDS/PAGE. The resolved proteins were transferred to polyvinylidene
fluoride membranes (Millipore, Billerica, MA). The blotted
membranes were blocked and subsequently incubated with rabbit
anti-JAG1 and rabbit anti-NICD (both 1:400; Santa Cruz), rabbit
anti-p65 and rabbit anti-p50 (both 1:1,000; Proteintech Group,
Chicago, IL, USA), rabbit anti-IκBα (1:750; Proteintech) and rabbit
anti-actin (1:1,000; Proteintech) according to the manufacturer’s
instructions. After incubation with horseradish peroxidase-labeled
secondary antibody (1:4,000; Santa Cruz), visualization was
performed with an enhanced chemiluminescence kit (Pierce, Rockford,
IL, USA) and exposure to X-ray film (Kodak, Rochester, NY, USA).
Immunoblotting with anti-actin antibody was used as an internal
control to confirm equivalent protein loading. Western blot
experiments were repeated ≥3 times. The relative intensity of each
protein band was assessed using Quantity One software (Bio-Rad
Laboratories, Hercules, CA, USA).
EMSA for measuring NF-κB activity
To evaluate changes in NF-κB activation after cells
were treated with the Notch signal inhibitor DAPT (as reflected by
DNA-binding activity), cell nuclear extracts from L02/HBx cells,
L02/HBx cells treated with DMSO (0.05%), or L02/HBx cells treated
with DAPT (20 μM) were subjected to EMSA. In addition, a
reaction system sample without cell nuclear extracts was used as a
blank control and with untreated positive cell nuclear extracts
(Pierce) as a positive control. EMSA was performed by incubating 10
μg of nuclear protein extract with biotin-labeled NF-κB
oligonucleotide (Pierce). The mixture included 1 μg of poly
(dI-dC) in a binding buffer. The DNA-protein complex formed was
separated from free oligonucleotides on a 6.0% polyacrylamide gel
using buffer (45 mM Tris, 45 mM boric acid pH 8.3 and 1 mM
ethylenediaminetetraacetic acid) for electrophoretic transfer of
binding reactions to a nylon membrane and then transferred DNA was
UV cross-linked to a membrane at 254 nm and exposed to X-ray film.
The relative intensity of each protein band was assessed using
Quantity One software (Bio-Rad Laboratories).
Statistical analyses
SPSS version 17.0 software (SPSS for Windows, Inc.
and Chicago, IL, USA) was used for all statistical analyses. All
results are expressed as mean ± standard error of the mean.
Statistical analysis was performed using standard one-way ANOVA or
one-way ANOVA for repeated measures, followed by the least
significant difference post hoc test. Bonferroni’s
correction was used to adjust for multiple comparisons. A 2-tailed
Student’s paired t-test was also used to compare the difference in
values between 2 groups. P<0.05 was considered statistically
significant.
Results
Co-localization and physical interactions
of Notch1 or NF-κB with HBx
To investigate the potential association between
Notch1 or NF-κB and HBx in the development of HCC, we performed
immunofluorescence and coIP assays with HBx-transfected L02 cells
(L02/HBx). In the L02/HBx cells, nuclei, HBx and NICD were stained
blue, red and green, respectively (Fig. 1A, a–c). Yellow areas in
dual-labeling experiments indicated overlapping of red and green
fluorescent labels (Fig. 1A, d),
suggesting the co-localization of NICD with HBx.
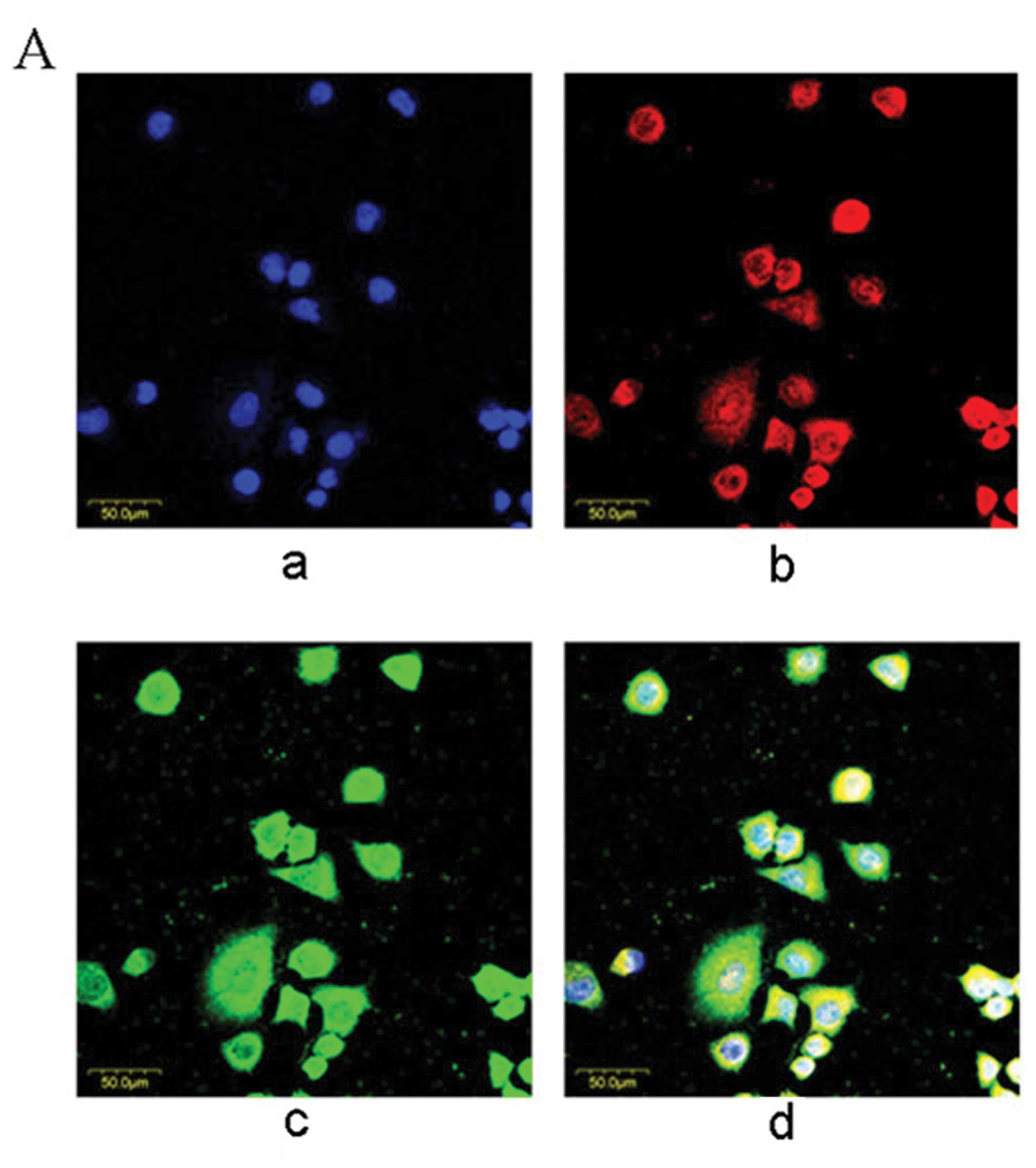 | Figure 1Co-localization and physical
interactions between Notch1 or NF-κB HBx in L02/HBx cells. (A)
Confocal micrograph of co-localization of NICD and HBx. Nuclei were
stained with DAPI (blue) (a), HBx protein was stained with CY3
(red) (b) and the NICD protein was stained with FITC (green) (c).
The yellow indicated overlapping areas of red and green (d)
(magnification ×100). (B) CoIP assay of interactions between NICD,
JAG1, p65, p50, or IκBα and HBx. Data shown are representative of 3
independent experiments. Specific interaction was found between
NICD and HBx and not between JAG1, p65, p50, or IκBα and HBx. PC,
positive control; anti-HBx, coIP proteins treated with anti-HBx;
NC, negative control; coIP proteins treated with non-immune mouse
IgG. |
Then we investigated the possible physical
interaction via the coIP assay. Compounds from L02/HBx cells were
immunoprecipitated with anti-HBx or non-immune mouse IgG, which
were then subjected to western blotting with anti-NICD, anti-JAG1,
anti-p65, anti-p50 and anti-IκBα. NICD was observed to
co-immunoprecipitate with HBx (Fig.
1B). No specific interaction was found between JAG1, p65, p50
or IκBα and HBx, or the protein immunoprecipitated with non-immune
IgG, which indicates the specificity of the NICD-HBx
interaction.
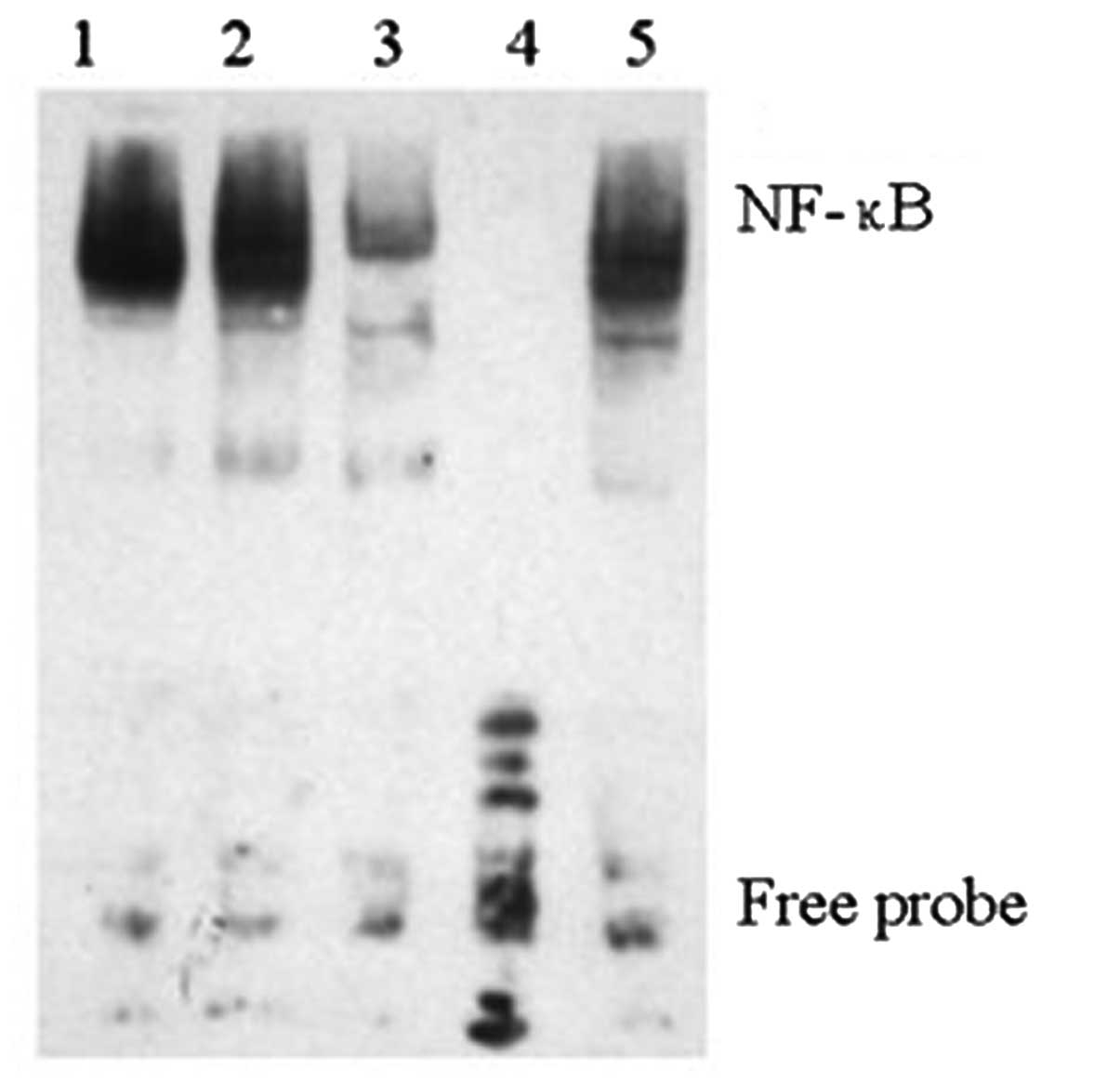
Downregulation of NICD decreases NF-κB
DNA-binding activity
NF-κB (p50/p65) is a ubiquitous, constitutive and
inducible heterodimer and the DNA binding activity of NF-κB
traditionally refers to the p50/p65 (p50/RelA) heterodimer-mediated
binding to DNA (25). In the
present study, we inhibited Notch1 using DAPT in L02/HBx cells
(Fig. 3) to examine the effect of
Notch on NF-κB DNA binding activity. Nuclear proteins from
untreated L02/HBx, DMSO-treated L02/HBx cells and DAPT-treated
L02/HBx cells were subjected to an EMSA for NF-κB DNA-binding
activity (Fig. 2). It was found
that downregulation of NICD via DAPT significantly decreased the
binding of NF-κB p65 to its target gene promoter. These results
revealed that after translocating into the nucleus, NICD could
function as a regulator to regulate the NF-κB DNA-binding activity,
which provided evidence for a mechanistic crosstalk between Notch
and NF-κB in HCC.
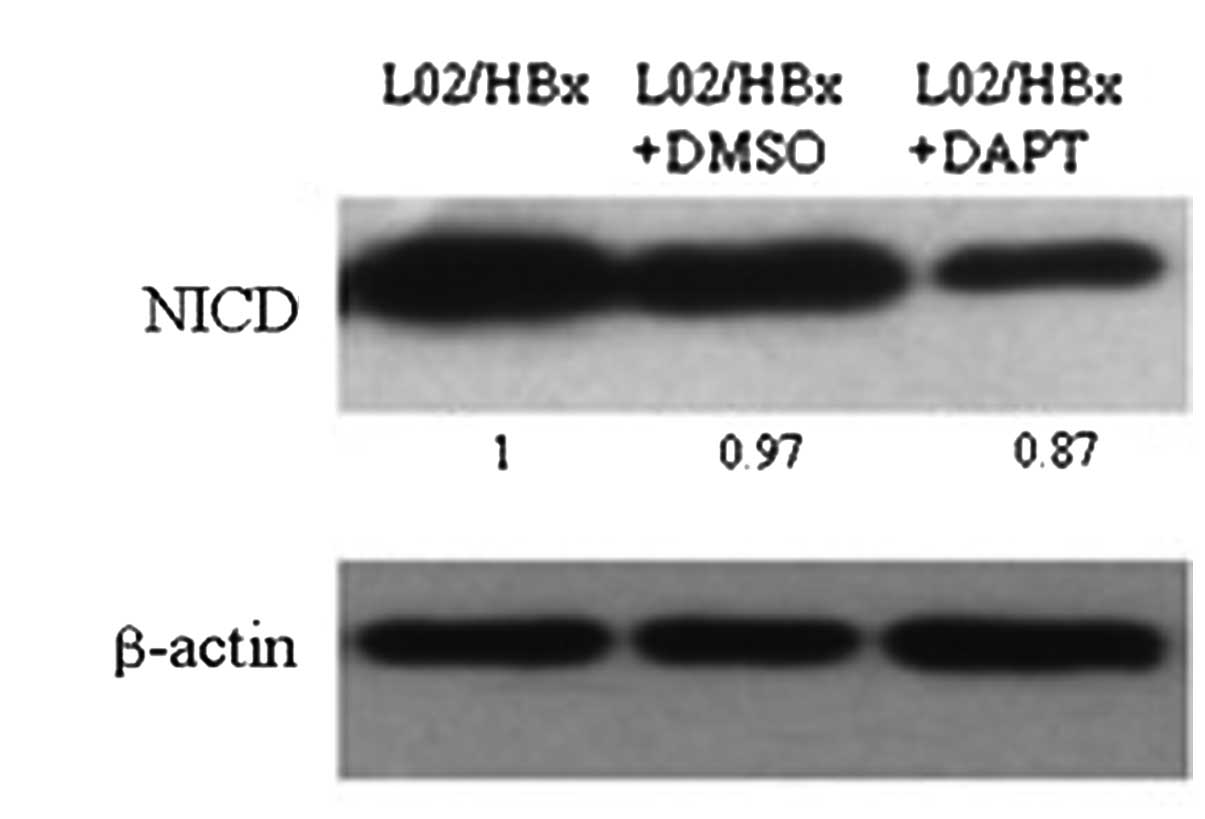
Inhibition of NICD by DAPT decreases the
activation of NF-κB pathway
To investigate whether HBx acted through Notch
signaling to activate the NF-κB pathway, we inhibited the Notch
pathway by using the γ-secretase inhibitor DAPT, which blocks the
processing of transmembrane (TM)-Notch1 to Notch1-IC and has been
widely used for experimental studies of Notch signaling (4). We treated L02/HBx cells with DAPT (20
μM) for 48 h and then used western blotting to determine
NICD protein expression. It turned out that NICD decreased
significantly after DAPT treatment (Fig. 3). Moreover, our data also showed
that inhibition of Notch1 decreased NF-κB DNA-binding activity in
L02/HBx cells.
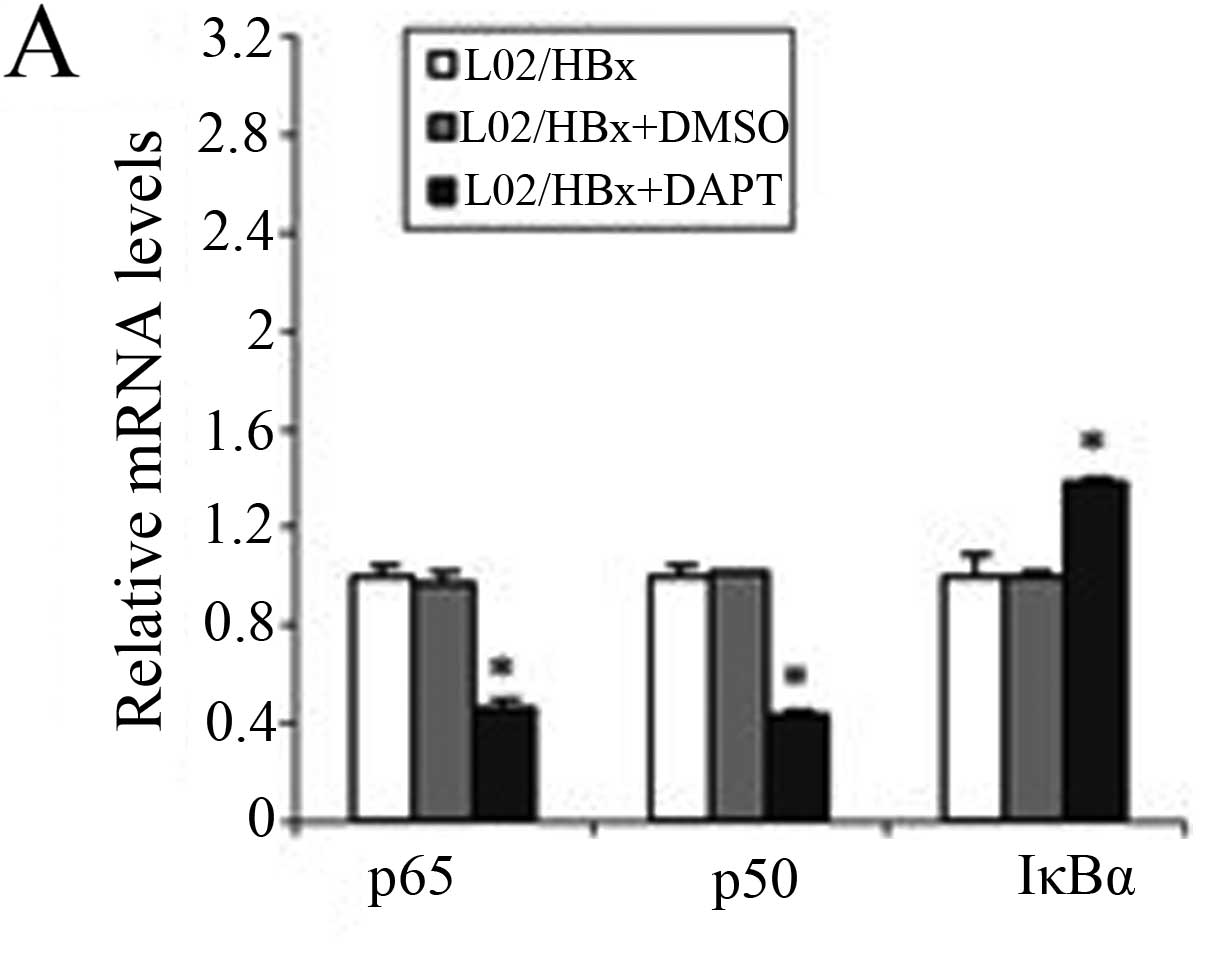
To further study the importance of activated Notch
signaling for the NF-κB pathway, we used qRT-PCR and western blot
analysis to observe changes in the NF-κB pathway after inhibition
of Notch1 (Fig. 4). The mRNA
levels of p65 and p50 were significantly decreased (P<0.001) and
the IκBα mRNA level was increased (P<0.001; Fig. 4A).
The protein levels of NF-κB were then evaluated
using western blot analysis and found that the total protein level
of IκBα was increased in L02/HBx cells after treating with DAPT
(Fig. 4B). No significant change
was observed in p65 or p50 proteins in the cytoplasm (Fig. 4C) and the nuclear extract levels of
p65 and p50 were decreased (Fig.
4D). These results indicated that inhibition of Notch1
suppressed the activation of the NF-κB pathway in L02/HBx cells by
affecting the transcription of the components of NF-κB and
inhibiting the nuclear transport of NF-κB dimers. This confirmed
that in L02/HBx cells HBx induced Notch signaling, which is
important for stimulating the NF-κB pathway.
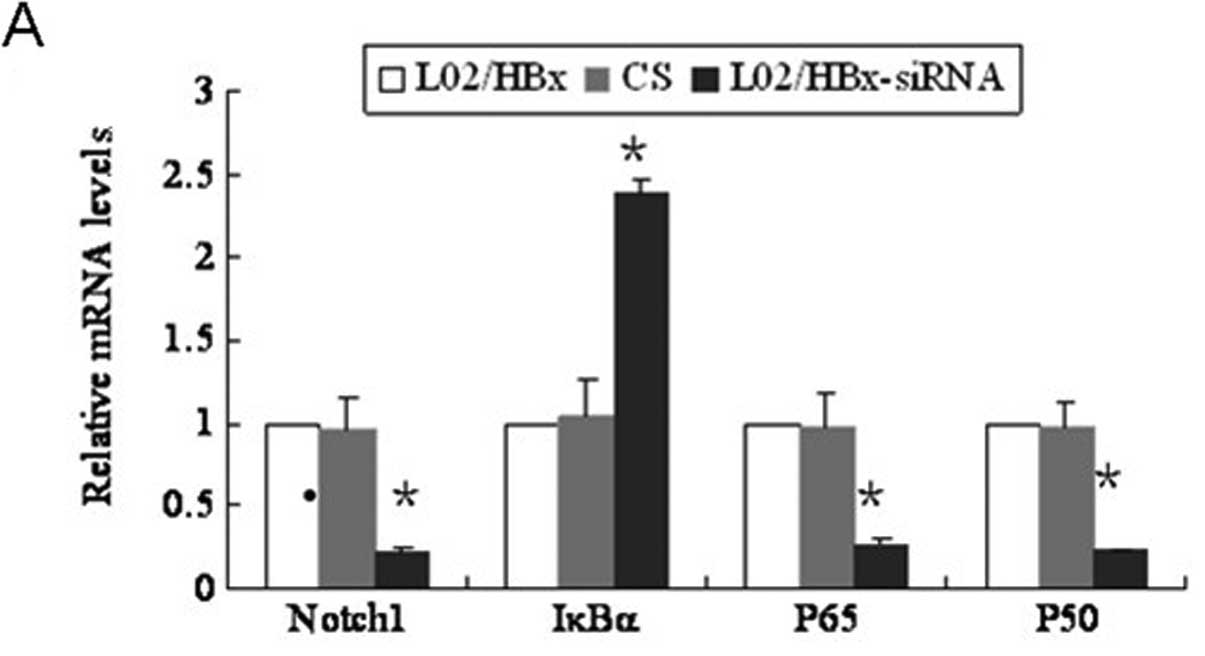
Inhibition of Notch1 by specific siRNA
also decreases the activation of NF-κB pathway
Notch1 siRNA-transfected L02/HBx cells showed
significantly reduced Notch1 mRNA and protein expression
(P<0.001; Fig. 5A and B). We
then detected the expression of NF-κB pathway proteins in the
Notch1 siRNA-transfected L02/HBx cells. In the cytoplasm, p50 and
p65 proteins were not changed compared with either the blank or
control groups (Fig. 5C). However,
these proteins were decreased notably in the nucleus (Fig. 5D).
Consistent with the above results, Q-PCR also showed
that p50 and p65 mRNA levels were significantly decreased
(P<0.001; Fig. 5A). Moreover,
the total protein level of IκBα was increased in the Notch
siRNA-transfected L02/HBx cells (Fig.
5B) and results of the Q-PCR showed that the IκBα mRNA levels
were also increased (Fig. 5A).
Therefore, down-regulation of Notch1 suppressed the activation of
p65 and p50, accompanied by an increase in IκBα. Altogether these
results showed that the NF-κB pathway was regulated by the Notch
signaling pathway.
Discussion
Stably HBx-expressing L02/HBx cells were previously
established and the data indicated that HBx promoted growth and
malignant transformation of the human non-tumor hepatic L02 line
(24). To explore whether
associations among Notch and NF-κB may be involved in the malignant
transformation of hepatic cells induced by HBx, we investigated the
interaction between HBx and Notch, or between HBx and NF-κB, in the
present study. We found that NICD interacted with HBx directly,
while the evidence that HBx acts directly through NF-κB was
lacking. Thus, HBx affected Notch signaling by directly binding to
the NICD upon its release from the cleaved Notch receptor, whereas
HBx did not have an immediate interaction with NF-κB. We then
continued to explore whether the Notch pathway receptor Notch1 and
the NF-κB pathway are interrelated. We found that downregulation of
Notch1 via the γ-secretase inhibitor DAPT decreased the ability of
NF-κB to bind to DNA. RNA-mediated or DAPT inhibition of the Notch1
signaling pathway in HBx-transfected L02 cells reduced NF-κB
expression and enhanced IκBα expression. These results showed that
NF-κB was regulated through the Notch1 signaling pathway.
The development of HCC is a multifactor, multistep,
complex process (26,27). Numerous reports have shown that
hepatocarcinogenesis is associated with the HBx protein. Our
previous research confirmed that HBx induced the malignant
transformation cells of the human non-tumor hepatic cell line L02
(24). Moreover, HBx regulated a
variety of cellular signaling pathways, including the Notch and
NF-κB pathways, thereby contributing to the progression of HCC
(28). In our previous study we
also found that activated Notch signaling is required for HBx to
promote the proliferation and survival of human hepatic cells
(4). However, little is known
about how HBx influences the Notch and NF-κB signaling pathways.
Therefore, in the present study we continued to investigate the
mechanisms by which HBx directly regulates Notch1 and NF-κB and how
Notch1 affects the NF-κB signaling pathway.
The possibility of crosstalk between the HBx and
Notch signaling pathways has become an important focus of research.
One recent report showed that the protein and mRNA expressions of
Notch1 and JAG1 were upregulated in HBx-stably transfected L02
cells (4), which was also
consistent with the report of Gao et al(29). But the underlying molecular
mechanisms are still unknown. The present research revealed a
direct association between HBx and the Notch signaling pathway and
showed that NICD interacts with HBx in HBx-transformed L02 cells,
while the ligand JAG1 did not co-precipitate with HBx. Here we
could deduce that the malignant function of HBx is directly
associated with the activity ofNotch1 signaling pathway, since HBx
could immediately regulate the Notch1 signaling pathway and the key
factor is NICD instead of other components of the Notch1 signaling
pathway. Our study thus provides some novel clues to the mechanism
by which HBx affects the Notch signaling pathway.
NF-κB is involved in the biological processes of
cancers as an important modulator of genes that promote cell
survival, proliferation, migration, inflammation, angiogenesis and
metastasis (30), including the
malignant transformation of hepatocytes (31). NF-κB (p50/p65) is a ubiquitous,
constitutive and inducible heterodimer and the DNA binding activity
of NF-κB traditionally refers to the p50/p65 (p50/RelA)
heterodimer-mediated binding to DNA (25). IκBα interacts with and sequesters
NF-κB in the cytosol (32). It can
also export NF-κB from the nucleus to the cytoplasm (19) and directly inhibits the DNA-binding
activity of NF-κB. Thus p50, p65 and IκBα play crucial roles in the
NF-κB pathway. Recent studies have reported that HBx stimulates the
phosphorylation of IκBα via the IκB kinase complex (28), thereby upregulating the activity of
the NF-κB signaling pathway (29).
In the present study our objective was to discover the mechanisms
of HBx-induced activation of the NF-κB pathway. We found that HBx
could not co-precipitate with the subunits p65 or p50 and the
suppressor IκBα of NF-κB, suggesting that HBx does not activate
NF-κB signaling by directly binding to p65, p50, or IκBα. Reports
have described the regulation of NF-κB by Notch, the mechanisms of
which are dependent on cell type. Notch1 upregulates the expression
of the NF-κB subunits p50, p65, RelB and c-Rel in murine bone
marrow hematopoietic precursors and therefore it can be concluded
that Notch1 upregulates NF-κB activity (13). In satellite liver cells, Notch1
reversed the repression of IκBα expression and lowered NF-κB
activity (33). Since numerous
studies have reported crosstalk between Notch and the NF-κB
pathway, we hypothesize that HBx may promote NF-κB signaling by
activating the Notch pathway. Our studies attempted to reveal the
molecular mechanism of the interaction between Notch1 and NF-κB in
HBx-transformed L02 cells.
Our EMSA results showed that the NF-κB DNA-binding
activity was decreased after Notch signaling was inhibited by DAPT.
To learn more about how Notch1 influence NF-κB in HBx-related HCC,
we used the γ-secretase inhibitor DAPT and Notch1 siRNA to block
Notch signaling. We found that when Notch signaling was inhibited
in HBx-transformed L02 cells, p50 and p65 mRNA levels were
significantly decreased, while IκBα mRNA level was notably
increased. This suggests that Notch activates the NF-κB pathway at
the transcription level. At the same time, in the nucleus the
proteins p50 and p65 were decreased, although their levels had not
changed in the cytoplasm. The results imply that NICD stimulated
the expression of the functional subunits and the nuclear transport
of NF-κB dimers and inhibited the suppressor of NF-κB to active the
NF-κB pathway. To our knowledge, several mechanisms potentially may
explain the effect of Notch1 on NF-κB activity. These include
transcriptional effects, physical binding between NF-κB and Notch1
and indirect effects on IκBα/β phosphorylation (13). There have been studies reported
that Notch1 signaling promoted NF-κB translocation to the nucleus
and DNA binding by increasing both phosphorylation of the IκBα/β
complex and the expression of some NF-κB family members (34). However, the details on the
molecular mechanism still required further confirmation, including
in vivo experiments and in different kinds of HBx-expressing
hepatic cells.
In conclusion, the results of this study showed that
HBx could promote Notch signaling by binding to NICD, which then
activates the NF-κB pathway. Therefore, crosstalk between the NF-κB
and Notch1 pathways is partially responsible for the progression of
HBx-induced HCC. However, further investigations are needed to
delineate the exact mechanisms by which Notch1 effects IκBα/β
phosphorylation and promotes NF-κB translocation into the
nucleus.
Abbreviations:
|
ACTB
|
β-actin
|
|
ADAM
|
a disintegrin and metalloproteinase
domain
|
|
BCL
|
B-cell CLL/lymphoma
|
|
coIP
|
co-immunoprecipitation
|
|
CSL
|
CBF1/RBP-Jκ/suppressor of
hairless/LAG1
|
|
DAPI
|
4′,6-diamidino-2-phenylindole
|
|
DAPT
|
N-[N-(3,5-difluorophenacetyl)-1-alanyl]-S-phenylglycine t-butyl
ester
|
|
DLL
|
δ-like protein
|
|
DMSO
|
dimethyl sulfoxide
|
|
EMSA
|
electrophoretic mobility shift
assay
|
|
FITC
|
fluorescein isothiocyanate
|
|
HBV
|
hepatitis B virus
|
|
HBx
|
hepatitis B virus X protein
|
|
HCC
|
hepatocellular carcinoma
|
|
IκB
|
nuclear factor of κ light polypeptide
gene enhancer in B-cells inhibitor
|
|
JAG
|
jagged
|
|
MMLV
|
Moloney murine leukemia virus
|
|
NF-κB
|
nuclear factor κB
|
|
NICD
|
Notch receptor’s intracellular
domain
|
|
qRT-PCR
|
quantitative real-time reverse
transcription PCR
|
|
RHD
|
Rel homology domain
|
Acknowledgements
This study was supported by the
National Science Foundation of China, nos. 81172063 and
30971352.
References
|
1.
|
Jemal A, Bray F, Melissa M, et al: Global
Cancer Statistics. CA Cancer J Clin. 55:74–108. 2005. View Article : Google Scholar
|
|
2.
|
Hashem BE and Rudolph KL: Hepatocellular
carcinoma epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar
|
|
3.
|
Feitelson MA and Duan LX: Hepatitis B
virus X antigen in the pathogenesis of chronic infections and the
development of hepatocellular carcinoma. Am J Pathol.
150:1141–1157. 1997.PubMed/NCBI
|
|
4.
|
Wang F, Zhou H, Xia X, et al: Activated
Notch signaling is required for hepatitis B virus X protein to
promote proliferation and survival of human hepatic cells. Cancer
Lett. 298:64–73. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Lai EC: Notch signaling: control of cell
communication and cell fate. Development. 131:965–973. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Miele L: Notch signaling. Clin Cancer Res.
12:1074–1079. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Miele L, Golde T and Osborne B: Notch
signaling in cancer. Curr Mol Med. 6:905–918. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Kopan R and Ilagan MX: Gamma-secretase:
proteasome of the membrane? Nat Rev Mol Cell Biol. 5:499–504. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Berman JN and Look AT: Targeting
transcription factors in acute leukemia in children. Curr Drug
Targets. 8:727–737. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Rangarajan A, Talora C, Okuyama R, et al:
Notch signaling is a direct determinant of keratinocyte growth
arrest and entry into differentiation. EMBO J. 20:3427–3436. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Ronchini C and Capobianco AJ: Induction of
cyclin D1 transcription and CDK2 activity by Notch(ic): implication
for cell cycle disruption in transformation by Notch(ic). Mol Cell
Biol. 1:5925–5934. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Baonza A and Freeman M: Control of cell
proliferation in the Drosophila eye by Notch signaling. Dev Cell.
8:529–539. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Cheng P, Zlobin A, Volgina V, et al:
Notch-1 regulates NF-kappaB activity in hemopoietic progenitor
cells. J Immunol. 167:4458–4467. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Klinakis A, Szabolcs M, Politi K, et al:
Myc is a Notch1 transcriptional target and a requisite for
Notch1-induced mammary tumorigenesis in mice. Proc Natl Acad Sci
USA. 103:9262–9267. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Diao J, Garces R and Richardson CD: X
protein of hepatitis B virus modulates cytokine and growth factor
related signal transduction pathways during the course of viral
infections and hepatocarcinogenesis. Cytokine Growth Factor Rev.
12:189–205. 2001. View Article : Google Scholar
|
|
16.
|
Seto E, Mitchell PJ and Yen TS:
Transactivation by the hepatitis B virus X protein depends on AP-2
and other transcription factors. Nature. 344:72–74. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Lucito R and Schneider RJ: Hepatitis B
virus X protein activates transcription factor NF-kappa B without a
requirement for protein kinase C. J Virol. 66:983–991.
1992.PubMed/NCBI
|
|
18.
|
Meyer M, Caselmann WH, Schluter V, et al:
Hepatitis B virus transactivator MHBst: activation of NF-kappa B,
selective inhibition by antioxidants and integral membrane
localization. EMBO J. 11:2991–3001. 1992.PubMed/NCBI
|
|
19.
|
Campbell KJ and Perkins ND: Regulation of
NF-kappaB function. Biochem Soc Symp. 73:165–180. 2006.
|
|
20.
|
Perkins ND: Integrating cell-signalling
pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol.
8:49–62. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Maniati E, Bossard M, Cook N, et al:
Crosstalk between the canonical NF-κB and Notch signaling pathways
inhibits PPARγ expression and promotes pancreatic cancer
progression in mice. J Clin Invest. 121:4685–4699. 2011.
|
|
22.
|
Pan H, Zhou W, He W, et al: Genistein
inhibits MDA-MB-231 triple-negative breast cancer cell growth by
inhibiting NF-κB activity via the Notch-1 pathway. Int J Mol Med.
30:337–343. 2012.PubMed/NCBI
|
|
23.
|
Schwarzer R, Dörken B and Jundt F: Notch
is an essential upstream regulator of NF-κB and is relevant for
survival of Hodgkin and Reed-Sternberg cells. Leukemia. 26:806–813.
2012.PubMed/NCBI
|
|
24.
|
Cheng B, Zheng Y, Guo XR, et al: Hepatitis
B viral X protein alters the biological features and expressions of
DNA repair enzymes in LO2 cells. Liver Int. 30:319–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Wang Z, Banerjee S, Ahmad A, et al:
Activated K-ras and INK4a/Arf deficiency cooperate during the
development of pancreatic cancer by activation of Notch and NF-κB
signaling pathways. PLoS One. 6:e205372011.PubMed/NCBI
|
|
26.
|
Berasain C, Castillo J, Perugorria MJ, et
al: Infammation and liver cancer: new molecular links. Ann NY Acad
Sci. 1155:206–221. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Lupberger J and Hildt E: Hepatitis B
virus-induced concogenesis. World J Gastroenterol. 13:74–81. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Qiao L, Zhang H, Yu J, et al: Constitutive
activation of NF-kappaB in human hepatocellular carcinoma: evidence
of a cytoprotective role. Hum Gene Ther. 17:280–290. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Gao J, Chen C, Hong L, et al: Expression
of Jagged1 and its association with hepatitis B virus X protein in
hepatocellular carcinoma. Biochem Biophys Res Commun. 356:341–347.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Karin M: NF-kappaB and cancer: mechanisms
and targets. Mol Carcinog. 45:355–361. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Luedde T and Schwabe RF: NF-κB in the
liver - linking injury, fibrosis and hepatocellular carcinoma. Nat
Rev Gastroenterol Hepatol. 8:108–118. 2011.
|
|
32.
|
Beg AA and Baldwin JR: The I kappa B
proteins: multifunctional regulators of Rel/NF-kappa B
transcription factors. Genes Dev. 7:2064–2070. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Oakley F, Mann J, Ruddell RG, et al: Basal
expression of IkappaBalpha is controlled by the mammalian
transcriptional repressor RBP-J (CBF1) and its activator Notch1. J
Biol Chem. 278:24359–24370. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Monsalve E, Ruiz-García A, Baladrón V, et
al: Notch1 upregulates LPS-induced macrophage activation by
increasing NF-kappaB activity. Eur J Immunol. 39:2556–2570. 2009.
View Article : Google Scholar : PubMed/NCBI
|