Introduction
Pancreatic cancer (PC) is currently the leading
cause of cancer-related mortality in western countries and China
(1). Despite improvements in
medical treatment for this cancer, the prognosis for PC patients is
still very poor. Resistance to chemo- and radiotherapy is very
common and directly contributes to the poor outcomes of PC patients
(2). This resistance is thought to
stem from both the intrinsic nature of PC cells (3) and the abundant fibrotic stroma of the
tumors, which favors rapid tumor progression and creates a physical
barrier to prevent drug delivery and immune cell infiltration
(4,5). The mechanism of this
chemoradioresistance, however, remains to be elucidated. Thus, it
is of the utmost clinical importance to determine the molecular
characteristics underlying this resistance and to identify
effective strategies to overcome the resistance.
The main mechanism of radiotherapy involves its
ionization action, which can kill the tumor cell either directly or
indirectly through generation of DNA double-strand breaks (DSB) in
the cells. Tumor cell death via induction of DNA damage is also a
potential mechanism for some chemotherapeutics such as cisplatin
(diamindichloridoplatin, DDP) and camptothecin. Thus, precise
regulation of the DNA damage response is crucial for cellular
survival and can potentially dictate the sensitivity of both
chemotherapy and radiotherapy in different cancers (6).
Methyl-CpG binding domain protein 1 (MBD1), which
binds to methylated CpG islands and couples DNA methylation to
transcriptional repression (7),
has been implicated in gene regulation, chromatin formation and
genome stability (8). Our previous
study showed that MBD1 plays an important role in silencing tumor
suppressor genes in PC cell lines (9). More recently, we found that silencing
MBD1 may restore sensitivity to chemotherapy and therefore enhance
apoptosis in human PC cell lines, however, the molecular mechanism
of MBD1’s involvement in therapy resistance of PC cells is not
clear. Interestingly, Watanabe et al(10) have reported previously that MBD1 is
detached from the methyl-CpG sites under the condition of DNA
damage. Knockdown MBD1 inhibits repair of the damaged DNA and
increases the cell sensitivity to the DNA damage treatment.
Therefore, we hypothesize that MBD1 may affect the sensitivity of
both chemotherapy and radiotherapy through the DNA damage response
in PC.
In this study, we demonstrated that MBD1 was
recruited to sites of DNA damage and was involved in DNA damage
repair in PC cells. Knockdown of MBD1 in PC cells enhanced DNA
damage-induced apoptosis and restored the sensitivity of these
cells to chemoradiotherapy, suggesting that MBD1 may be a potential
therapeutic target to overcome chemoradiotherapy resistance in
PC.
Materials and methods
Cell culture and chemicals
Human PANC-1 and 293T cells were purchased from
Shanghai Institutes for Biological Science (Shanghai, China). Cells
were cultured in Dulbecco’s modified Eagle’s medium (DMEM;
Gibco-BRL) supplemented with 10% fetal bovine serum (FBS;
Gibco-BRL) at 37°C with 5% CO2. DMEM, FBS, horse serum,
L-glutamine (2 mM), penicillin (50 IU/ml) and streptomycin (50
μg/ml) were purchased from Life Technologies Inc.
Lentiviral production and infection of PC
cells
The lentiviral vector pLKO.1 TRC (Addgene Plasmid
10878) was used according to the manufacturer’s instructions
(http://www.addgene.org/tools/protocols/plko/). In
brief, shRNA oligos targeting human MBD1 (sh-MBD1) or MDC1
(sh-MDC1) were designed and cloned into the pLKO.1-TRC cloning
vector digested with EcoRI and Agel. The recombinant construct was
co-transfected together with two packaging vectors psPAX2 and
pMD2.G into 293T cells. A pLKO.1-scramble shRNA (sh-CTL; Addgene
Plasmid 1864) was used as a negative control. Lentiviral particles
were harvested and filtered, and then target PC cells were infected
with these lentiviral particles. For overexpression of MBD1 or
MDC1, FLAG-tagged MBD1 or HA-tagged MDC1 was cloned into the
lentiviral vector pWPI.1. Lentiviral particles were produced by
co-transfection of pWPI.1-MBD1-FLAG, psPAX2, and pMD.G into 293T
cells.
Irradiation (IR) and clonogenic survival
assay
Cell monolayers were grown in vitro and
irradiated using 6 MV X-rays from linear accelerators (Elekta
Synergy, Stockholm, Sweden) with a single dose of 0, 2, 4 or 8 Gy
of IR. A standard colony-forming assay was performed to determine
the surviving fractions. For the clonogenic survival assay, cells
were seeded in 6-well tissue culture dishes. The seeded cell number
was increased with the dose of IR as described (11). After defined time periods, cells
were fixed with 70% ethanol and stained with methylene blue.
Colonies with >50 cells were scored as survivors. Non-irradiated
cultures were used for data normalization.
Immunofluorescence microscopy
Cells were seeded onto microscope slides and allowed
to adhere overnight. After 24 h, slides were treated with
H2O2 or DDP or irradiated at 8 Gy as
indicated and fixed with 4% paraformaldehyde for 15 min. Cells were
permeabilized with 0.2% Triton X-100/phosphate-buffered saline
(PBS)/1% FBS for 10 min and blocked with 5% bovine serum albumin/1%
FBS in PBS. Slides were incubated with a rabbit anti-phosphohistone
H2AX (γH2AX) antibody (Epitomics) overnight at 4°C. Slides were
incubated with secondary Alexa 488-conjugated mouse antibodies
(Molecular Probes/Invitrogen). Nuclei were counterstained with
4′,6-diamino-2-phenylindole (DAPI) and mounted using Vectashield
(Vector Laboratories, Peterborough, UK). Radiation-induced γH2AX
foci were counted in at least 100 cells per sample using a
fluorescence microscope (Olympus BX 40) and the Leica Application
Suite.
Immunoblot analysis
Cells were exposed to various treatments and
harvested for immunoblot analysis as described (9). Samples were immunoblotted using
antibodies against MDC1, MBD1 (Santa Cruz Biotechnology), total
Chk1, phospho-Chk1 (S345), total Chk2, phospho-Chk2 (S19), NBS1,
phospho-NBS1 (S343), cleaved caspase-3 (Cell Signaling Technology),
total ATM, phospho-ATM (S1981), γH2AX (Epitomics), tubulin, HA and
FLAG (Sigma-Aldrich).
Determination of cell proliferation
Approximately 104 cells/well were seeded
into a 96-well plate and allowed to adhere overnight. After
treatment with DDP for 24–48 h, 10 μl thiazolyl blue
tetrazolium bromide (Sigma-Aldrich) were added, and cells were
incubated at 37°C for 2 h. Colorimetric measurement was performed
at 450 nm in a microplate reader (Spectra Max 190, Molecular
Devices). Experiments were performed in triplicate and repeated at
three different times.
Neutral comet assay
DNA damage repair was measured in PANC-1 cells using
the comet assay system (Trevigen) according to the manufacturer’s
instructions. Comet tail moments were scored using Comet Score
software (TriTek).
Apoptosis assay
At 72 h post-transfection, cells were harvested,
washed, resuspended in the staining buffer and analyzed with the
Vybrant Apoptosis Assay kit (Invitrogen, Carlsbad, CA). Stained
cells were detected with a FACSCalibur system, and data were
analyzed with CellQuest software (both from Becton-Dickinson,
Mountain View, CA). The Annexin V-positive cells were regarded as
apoptotic cells.
Co-immunoprecipitation assays
PANC-1 cells and 293T cells were lysed by brief
sonication in co-immunoprecipitation buffer (20 mM Tris pH 8.0, 150
mM NaCl, 1 mM EDTA, 0.5% NP-40 supplemented with protease
inhibitors). Lysates were centrifuged for 20 min at 10,000 × g, and
the resulting supernatant was pre-cleared by incubation with
immobilized protein A/G gel (Pierce) for 1 h at 4°C. The
pre-cleared supernatant was subjected to overnight
immunoprecipitation using the indicated antibodies or control IgG
antibodies at 4°C. The next day, protein complexes were collected
by incubation with 25 μl immobilized protein A/G gel for 1 h
at 4°C. The collected protein complexes were washed four times with
co-immunoprecipitation buffer and eluted by boiling in protein
sample buffer under reducing conditions. The eluted proteins were
resolved by sodium dodecyl sulfate polyacrylamide gel
electrophoresis and analyzed by western blotting.
Statistical analysis
Statistical analysis was performed using SPSS
software for Windows. All analyses used two-sided hypothesis tests.
Results were expressed as mean ± standard deviation. Differences
between groups were evaluated using the Student’s t-test and
one-way analysis of variance. P-values <0.05 were considered
significant.
Results
MBD1 rapidly accumulates within DNA
damage chromatin
The DNA damage response is characterized by the
accumulation of checkpoint and DNA repair proteins on the damaged
chromatin (12). To investigate
whether MBD1 plays a direct role in the DNA damage response, we
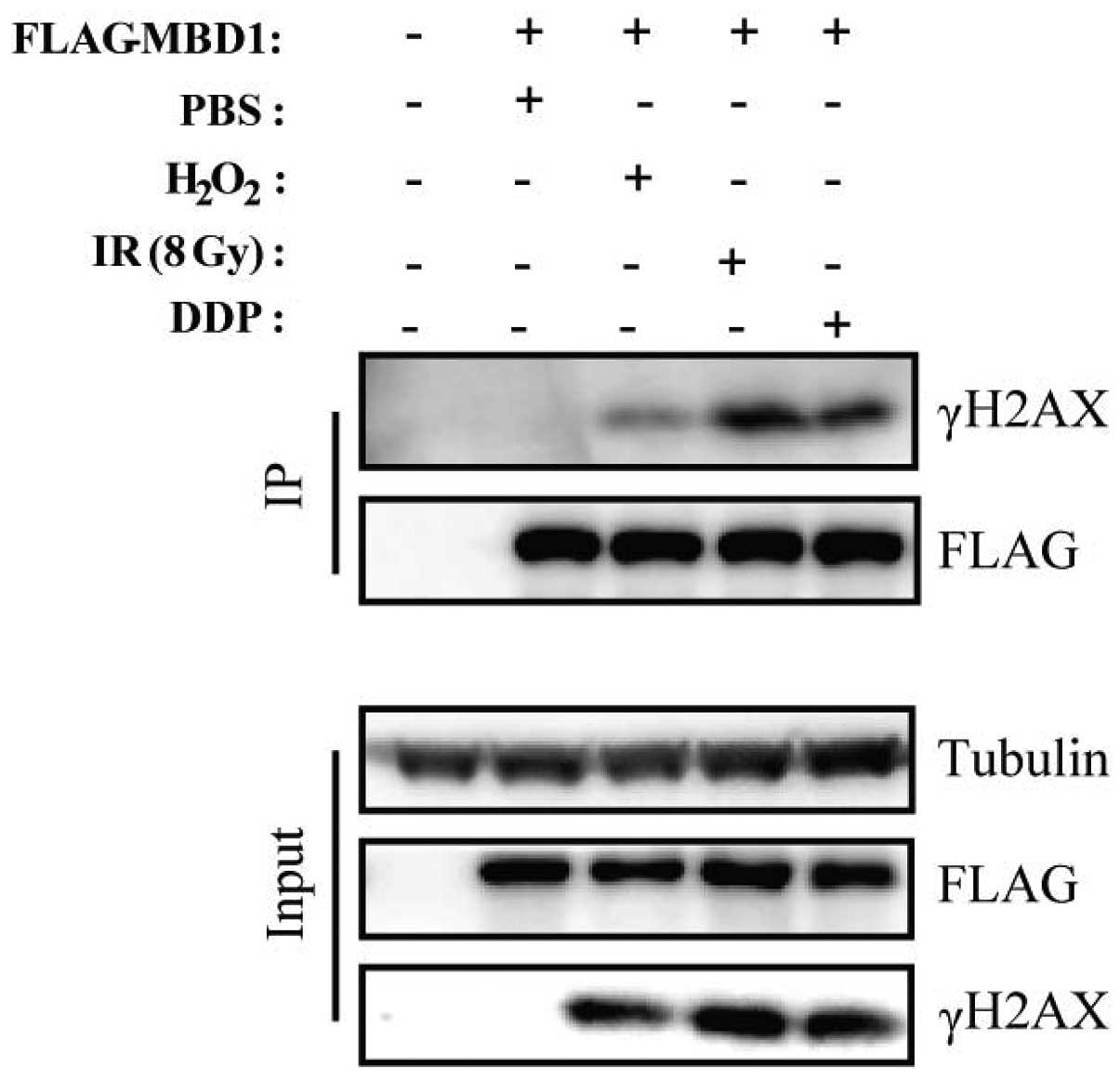
examined recruitment of MBD1 to sites of DNA damage. We first
treated PC cells (PANC-1) with H2O2, DDP or
IR. Each of these treatments induced substantial DNA damage and
increased the number of γH2AX foci, which plays an important role
in the repair of DNA lesions by recruiting DNA damage signaling and
repair proteins (13). Using a
co-immunoprecipitation assay, we confirmed that FLAG-tagged MBD1
co-precipitated with γH2AX following treatment with different DNA
damaging agents as described above (Fig. 1). Thus, we conclude that MBD1 is
one of the factors that assemble at sites of DNA damage after
treated with H2O2, DDP or IR.
Silencing MBD1 impairs DNA damage repair
in PANC-1 cells
Given that MBD1 accumulates in the damaged
chromatin, we next examined whether MBD1 is involved in DNA damage
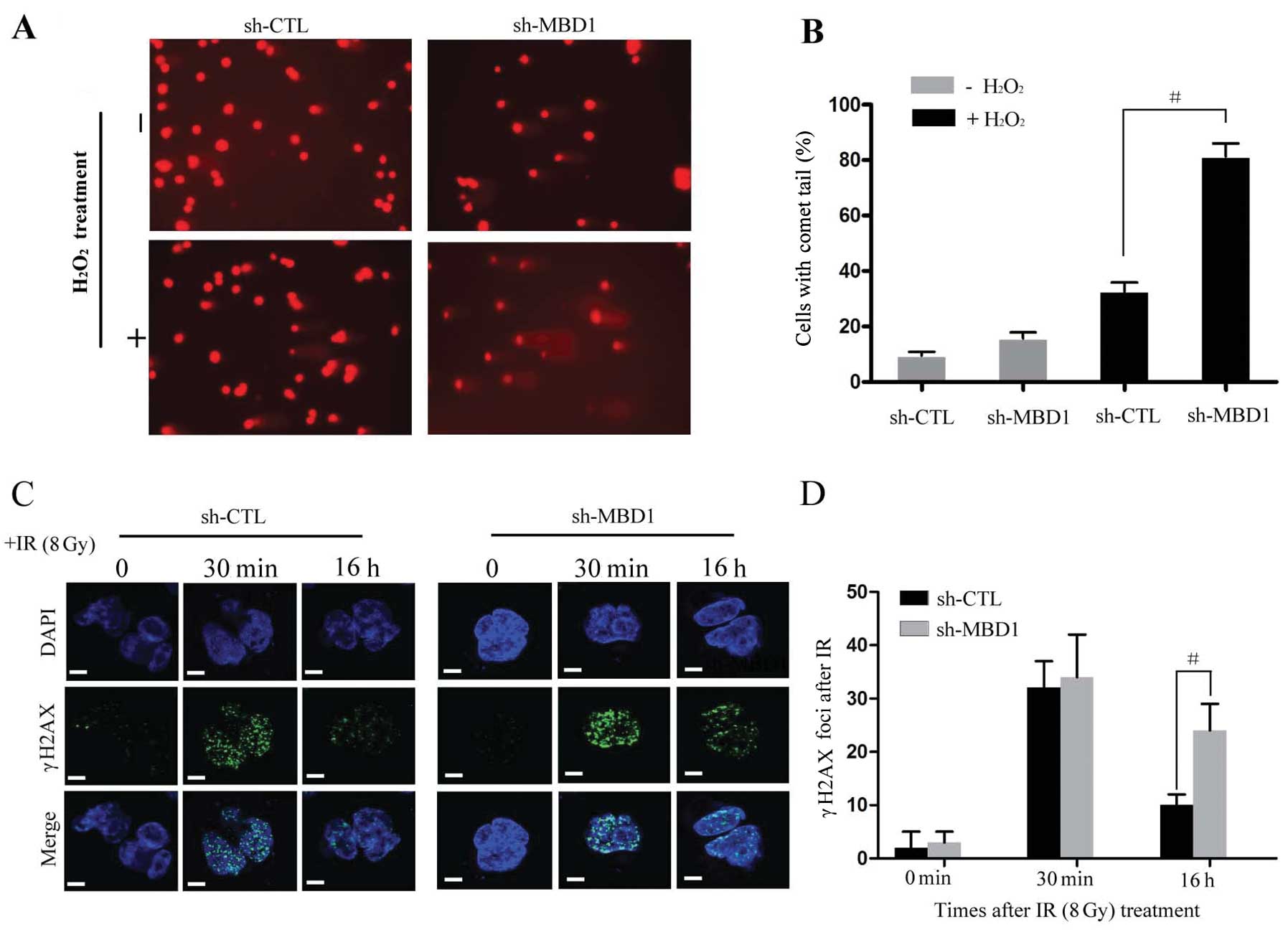
repair in PC cells. We used a comet assay (14) to directly assess the efficiency of
DNA damage repair in MBD1-depleted cells. PANC-1 cells treated with
sh-MBD1 or control (CTL) RNAs were exposed to
H2O2 and subjected to a neutral comet
analysis 6 h later. As shown in Fig.
2A and B, we detected a slight increase in the level of damaged
DNA in untreated MBD-depleted cells. This implies that spontaneous
DNA damage accumulates in the absence of MBD1. Importantly, the
level of H2O2-induced DNA damage remained
higher at 6 h in MBD1 knockdown cells, suggesting that MBD1
promotes proper DNA damage repair (Fig. 2A and 2B). This result was further
confirmed by assessing the resolution of phosphorylated histone
H2AX (γH2AX) nuclear foci following IR (15). In PANC-1 cells, few γH2AX foci were
observed in unirradiated cells (both sh-CTL-and sh-MBD1-treated
cells), and comparable levels of foci were induced in sh-CTL and
sh-MBD1-treated cells 30 min after IR at 8 Gy (Fig. 2C). Importantly, 16 h after IR, few
foci remained in the non-silenced control cells, whereas the number
of γH2AX foci in MBD1-depleted PANC-1 cells persisted at
considerably higher levels (Fig.
2C). This difference was statistically significant (P<0.01;
Fig. 2D).
MBD1 depletion restores
chemoradiosensitivity of PANC-1 cells
Since the DNA damage response is tightly connected
to the resistance of both chemotherapy and radiotherapy and the
fact that MBD1 is an important regulator in PC DNA damage repair,
we next examined whether knockdown of MBD1 restores the sensitivity
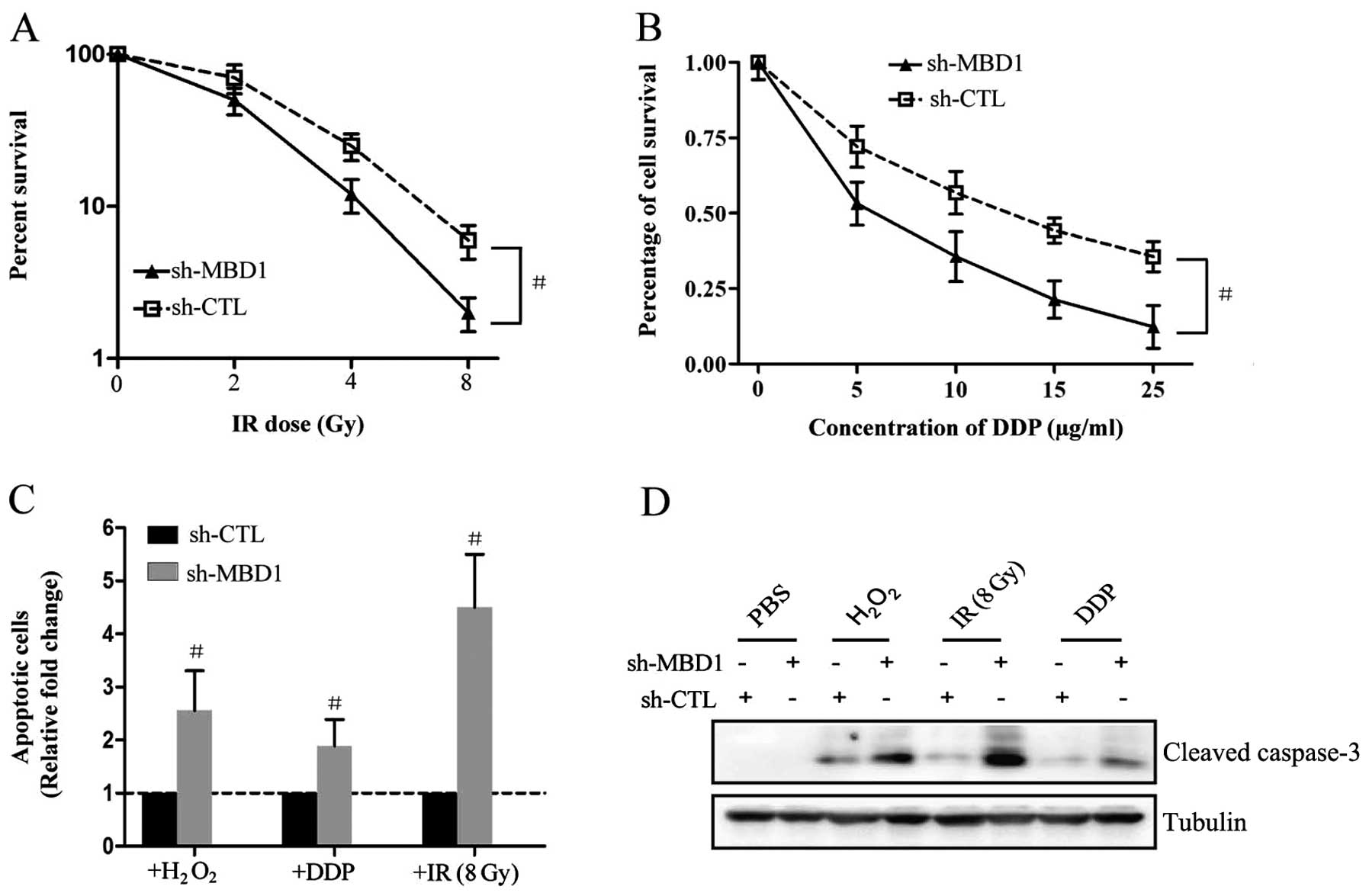
of PC cells to both chemotherapy and radiotherapy. A colony
survival assay revealed that PANC-1sh-MBD1 cells were more
sensitive to IR (Fig. 3A) or DDP
(Fig. 3B) than control cells
(PANC-1sh-CTL). Thus, downregulation of MBD1 resulted in increased
sensitivity of PC cells to both chemotherapy and radiotherapy. In
addition, flow cytometry analysis also confirmed that this
increased sensitivity was partially dependent on inducing apoptosis
of PC cells (Fig. 3C), as the
expression of the pro-apoptotic protein cleaved caspase-3 was
increased in PANC-1sh-MBD1 cells after treatment with
H2O2, IR or DDP compared with PANC-1sh-CTL
cells (Fig. 3D).
MBD1 activates the DNA damage checkpoint
through interaction with MDC1
Although IR and DDP damage tumor cells by way of
several mechanisms, these agents kill cancer cells primarily via
DNA damage (6,16). Thus, DNA damage checkpoint
responses play essential roles in cellular chemoradiosensitivity
(17). To determine the role of
MBD1 in the DNA damage checkpoint response in PC
chemoradioresistance, we examined the DNA damage checkpoint
responses in both PANC-1sh-MBD1 and PANC-1sh-CTL cells after
treatment with either IR or DDP. Activating phosphorylation of
canonical DNA damage response factors, such as checkpoint protein 1
(pChk1), pChk2, and the checkpoint protein pNBS1 were significantly
greater in PANC-1sh-CTL cells than in PANC-1sh-MBD1 cells following
exposure to IR or DDP (Fig. 4A and
4B). As MDC1 is known to play an important role in DNA damage
response through binding and phosphorylation of NBS1 (18), we hypothesized that the promotion
of DNA repair by MBD1 contributes to its interaction with MDC1. The
interaction between MBD1 and MDC1 occurred with both overexpressed
(Fig. 4C) and endogenous (Fig. 4D) protein, and this interaction was
induced by radiation and strengthened with increased exposure to IR
treatment (Fig. 4E). Our findings
support the notion that inhibition of DNA damage response by
silencing MBD1 may be driven in collaboration with effects of
MDC1.
MBD1 regulation of chemoradiosensitivity
depends on DNA damage repair
Because the DNA damage response was highly
correlated with the sensitivity of both chemotherapy and
radiotherapy, we asked whether restoration of chemoradiotherapy
sensitivity in PC was at least partly dependent on the inhibition
of DNA damage response by the interaction between MBD1 and MDC1. We
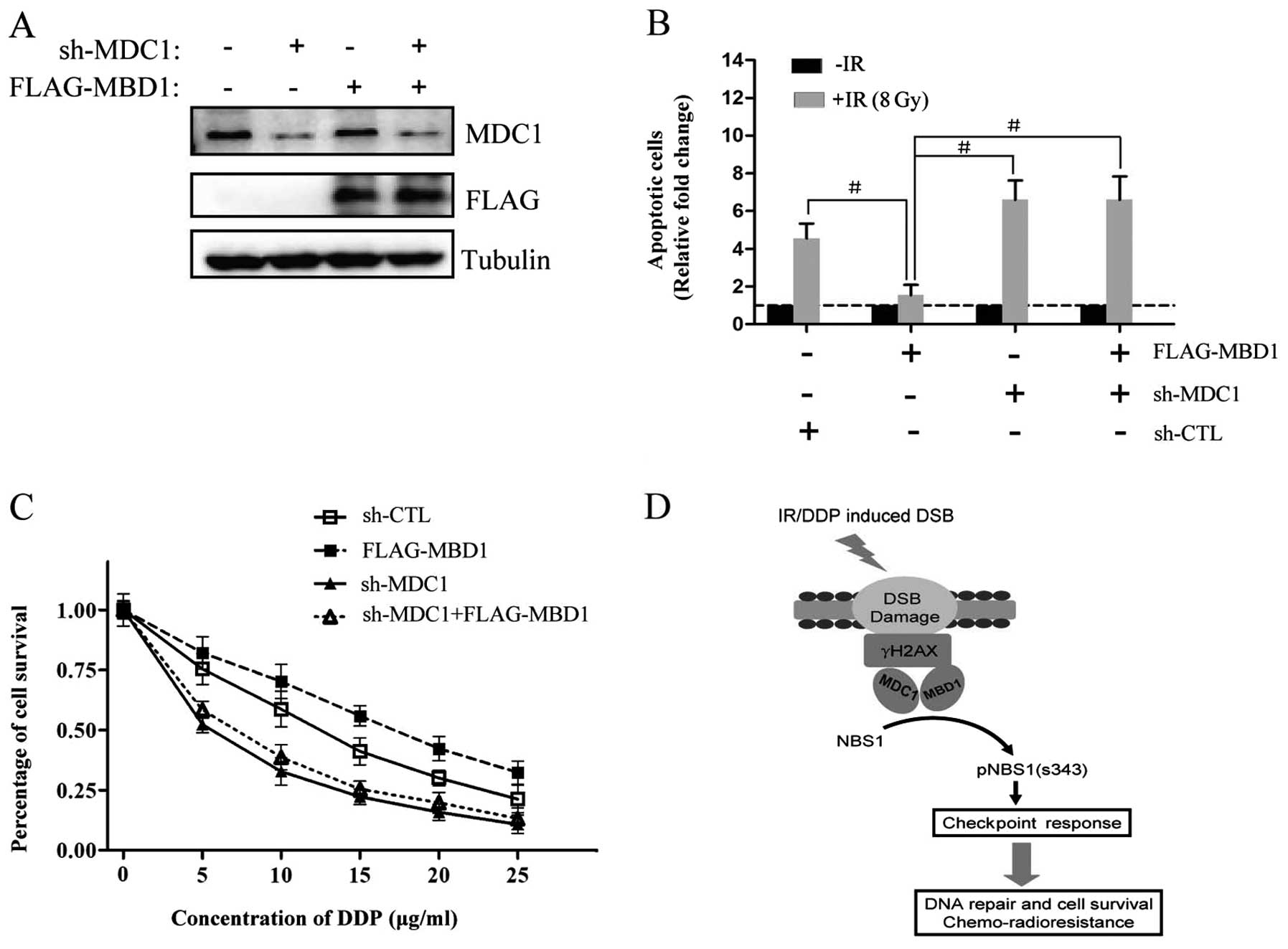
introduced MBD1 into cells that had stable silencing of MDC1 via
shRNA (PANC-1sh-MDC1) along with an MBD1-expressing plasmid
(Fig. 5A). These cells were used
in the subsequent experiments to evaluate the degree of inhibition
after treatment with chemo- or radiotherapeutic agents (DDP or IR).
Cells with MBD1 overexpression were resistant to both radiotherapy
(Fig. 5B) and chemotherapy
(Fig. 5C), but these cells became
sensitive to either radiotherapy or chemotherapy following MDC1
knockdown (Fig. 5B and 5C).
Furthermore, MDC1-silenced cells did not display a remarkable
increase of chemo- or radio-sensitivity immediately upon
reintroduction of MBD1, suggesting that MBD1 may regulate cell fate
following DNA damage through an interaction with MDC1 and
subsequent acceleration of a downstream check point response. These
effects may, therefore, increase DNA repair activity and promote
chemoradioresistance (Fig.
5D).
Discussion
Genomic integrity is critical to organismal survival
and is controlled by the DNA damage response network, an elaborate
signal transduction system that senses DNA damage and recruits
appropriate repair factors (19).
This global signaling network senses the different types of DNA
lesions and coordinates a response that includes activation of
transcription, cell cycle control, apoptosis, senescence, and/or
DNA repair processes (20). It has
been hypothesized that one of the most important determinants of
chemoradiotherapy resistance in cancer cells may stem from an
overall resistance to DNA damage-induced apoptosis (21). PC remains one of the deadliest of
all cancers despite aggressive surgical treatment combined with
chemo- and radiotherapy. Chemoradioresistance is the principal
cause of treatment failure in PC patients and leads to the poor
prognosis for patients with the disease (22,23).
Strategies to find candidate targets linking DNA damage repair to
chemoradioresistance in order to sensitize PC cells to
chemoradiotherapy are well underway.
In this study, we report a novel role for MBD1 in
the DNA damage repair network in PC cells. We find that MBD1 is
recruited to DNA damage sites under DNA damage conditions.
Silencing MBD1 significantly impairs activation of the DNA damage
checkpoint response and inhibits DNA repair capacity in PC. Our
data support the earlier report that MBD1 is detached away from the
damaged methyl-CpG sites and may serve as a sensor for damaged
bases during the DNA damage response to promote chromatin
remodeling and repair of the damaged DNA (10), however, the mechanism of MBD1
involvement in DNA damage repair requires further elaboration.
Moreover, previous study from our group also demonstrated that the
expression levels of pro-apoptosis proteins, such as cleaved
caspase-3, -9 and bax, were significantly elevated in MBD1-silenced
PANC-1 cells after treatment with gemcitabine, which has been
reported as an inhibitor of DNA damage repair (24). Taken together, our recent findings
together with the previous reports suggest that MBD1 is involved in
DNA damage repair of PC cells and may be a chemosensitizing target
for PC treatment.
We also explored the potential mechanism and
involvement of MBD1 in regulation of DNA damage repair in PC cells.
Knockdown of MBD1 significantly impaired activation of the DNA
damage checkpoint response. Following DNA damage, DNA repair and
cell cycle checkpoints are the main means of maintaining genomic
stability and cell survival (25).
Several checkpoints are activated at different stages of the cell
cycle. Chk1 is a serine/threonine kinase that is primarily
responsible for initiating cell cycle arrest in order to allow
adequate time for DNA repair (26), while the serine/threonine kinase
Chk2 is activated when phosphorylated by ataxia telangiectasia
mutated (ATM) following generation of DNA double-strand breaks. The
effects of activated Chk2 on the effector protein Cdc25A
phosphatase are similar to those mediated by Chk1 (27,28).
Indeed, changes in MBD1 expression led to alterations in the
expression of these cell cycle checkpoint factors in PC cells and
these findings further supported our hypothesis that MBD1 affects
the DNA damage response through effects on the checkpoint
response.
Critical to the recruitment of DNA repair proteins
to sites of DNA damage (nuclear foci) is phosphorylation of histone
H2AX (γH2AX) on Ser139 by the protein kinases ATM and ataxia
telangiectasia and Rad3-related protein (ATR), which are activated
by DNA damage at the core of the DNA damage signaling apparatus
(29), leading to the accumulation
of repair proteins at the sites of damaged DNA (30,31).
Several proteins involved in the DNA damage response contain
specific H2AX recognition domains and MDC1 is one such protein. A
body of evidence indicates that the interaction between MDC1 and
H2AX is the first step in preparing the DNA damage signaling and
repair (26,32). We noted in our study that MBD1 was
physically binding with both γH2AX and MDC1 when exposed to DNA
damage agents. Importantly, MDC1 has been reported to act upstream
of NBS1 and regulate the intra-S-phase checkpoint in response to
DNA damage through targeting and activating NBS1 to damage DNA
sites directly (33). Considering
that knockdown of MBD1 also significantly abrogate NBS1 activation
and down-stream checkpoint response following IR or DDP. Thus our
results indicated that MBD1 may contribute to the observed
chemoradioresistance in PC cells via interference with the DNA
damage response in association with MDC1 although further
investigation is needed.
In conclusion, our study provides the first evidence
that the methyl-CpG binding domain protein MBD1 is closely involved
in mediating the resistance of PC cell lines to chemoradiotherapy.
MBD1, therefore, represents a promising molecular target to
sensitize a priori-resistant PC to IR. Although these findings have
begun to uncover the underlying cellular mechanisms of this
chemoradioresistance (Fig. 5D),
future studies will ultimately be required to elucidate the
mechanism of MBD1 regulation on the DNA damage response.
Furthermore, these studies indicated that MBD1 inhibition may be an
effective strategy to increase the fraction of patients that
respond to multimodal treatment and thus improve overall
survival.
Acknowledgements
This study was partially supported by
the National Natural Science Foundation of China (NSFC-30901435,
NSFC-30972905, NSFC-81172276 and NSFC-81001058) and Joint Project
for Emerging Frontier Technology of Shanghai Hospitals of Municipal
Level (SHDC12010120).
References
|
1
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: the impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Erkan M, Hausmann S, Michalski CW, et al:
The role of stroma in pancreatic cancer: diagnostic and therapeutic
implications. Nat Rev Gastroenterol Hepatol. 9:454–467. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wong HH and Lemoine NR: Pancreatic cancer:
molecular pathogenesis and new therapeutic targets. Nat Rev
Gastroenterol Hepatol. 6:412–422. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Olive KP, Jacobetz MA, Davidson CJ, et al:
Inhibition of Hedgehog signaling enhances delivery of chemotherapy
in a mouse model of pancreatic cancer. Science. 324:1457–1461.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Garrido-Laguna I, Uson M, Rajeshkumar NV,
et al: Tumor engraftment in nude mice and enrichment in
stroma-related gene pathways predict poor survival and resistance
to gemcitabine in patients with pancreatic cancer. Clin Cancer Res.
17:5793–5800. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Terry SY and Vallis KA: Relationship
between chromatin structure and sensitivity to molecularly targeted
auger electron radiation therapy. Int J Radiat Oncol Biol Phys.
83:1298–1305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ng HH, Jeppesen P and Bird A: Active
repression of methylated genes by the chromosomal protein MBD1. Mol
Cell Biol. 20:1394–1406. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lopez-Serra L, Ballestar E, Fraga MF,
Alaminos M, Setien F and Esteller M: A profile of methyl-CpG
binding domain protein occupancy of hypermethylated promoter CpG
islands of tumor suppressor genes in human cancer. Cancer Res.
66:8342–8346. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu C, Chen Y, Yu X, et al: Proteomic
analysis of differential proteins in pancreatic carcinomas: effects
of MBD1 knockdown by stable RNA interference. BMC Cancer.
8:1212008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Watanabe S, Ichimura T, Fujita N, et al:
Methylated DNA-binding domain 1 and methylpurine-DNA glycosylase
link transcriptional repression and DNA repair in chromatin. Proc
Natl Acad Sci USA. 100:12859–12864. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang Y, Chen LH, Wang L, Wang HM, Zhang
YW and Shi YS: Radiation-inducible PTEN expression radiosensitises
hepatocellular carcinoma cells. Int J Radiat Biol. 86:964–974.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K
and Linn S: Molecular mechanisms of mammalian DNA repair and the
DNA damage checkpoints. Annu Rev Biochem. 73:39–85. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bekker-Jensen S, Lukas C, Kitagawa R, et
al: Spatial organization of the mammalian genome surveillance
machinery in response to DNA strand breaks. J Cell Biol.
173:195–206. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jirawatnotai S, Hu Y, Michowski W, et al:
A function for cyclin D1 in DNA repair uncovered by protein
interactome analyses in human cancers. Nature. 474:230–234. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Löbrich M, Shibata A, Beucher A, et al:
gammaH2AX foci analysis for monitoring DNA double-strand break
repair: strengths, limitations and optimization. Cell Cycle.
9:662–669. 2010.PubMed/NCBI
|
|
16
|
Bartkova J, Horejsí Z, Koed K, et al: DNA
damage response as a candidate anti-cancer barrier in early human
tumorigenesis. Nature. 434:864–870. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kastan MB and Bartek J: Cell-cycle
checkpoints and cancer. Nature. 432:316–323. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chapman JR and Jackson SP:
Phospho-dependent interactions between NBS1 and MDC1 mediate
chromatin retention of the MRN complex at sites of DNA damage. EMBO
Rep. 9:795–801. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harper JW and Elledge SJ: The DNA damage
response: ten years after. Mol Cell. 28:739–745. 2007.PubMed/NCBI
|
|
20
|
Zhou BB and Elledge SJ: The DNA damage
response: putting checkpoints in perspective. Nature. 408:433–439.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yao W, Yu X, Fang Z, et al: Profilin1
facilitates staurosporine-triggered apoptosis by stabilizing the
integrin betal-actin complex in breast cancer cells. J Cell Mol
Med. 16:824–835. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Shibamoto Y, Kubota T, Kishii K and
Tsujitani M: Radiosensitivity of human pancreatic cancer cells in
vitro and in vivo, and the effect of a new hypoxic cell sensitizer,
doranidazole. Radiother Oncol. 56:265–270. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim MP and Gallick GE: Gemcitabine
resistance in pancreatic cancer: picking the key players. Clin
Cancer Res. 14:1284–1285. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Parsels LA, Morgan MA, Tanska DM, et al:
Gemcitabine sensitization by checkpoint kinase 1 inhibition
correlates with inhibition of a Rad51 DNA damage response in
pancreatic cancer cells. Mol Cancer Ther. 8:45–54. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Krempler A, Deckbar D, Jeggo PA and
Lobrich M: An imperfect G2M checkpoint contributes to chromosome
instability following irradiation of S and G2 phase cells. Cell
Cycle. 6:1682–1686. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bartek J and Lukas J: Mammalian G1- and
S-phase checkpoints in response to DNA damage. Curr Opin Cell Biol.
13:738–747. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Taverna SD, Li H, Ruthenburg AJ, Allis CD
and Patel DJ: How chromatin-binding modules interpret histone
modifications: lessons from professional pocket pickers. Nat Struct
Mol Biol. 14:1025–1040. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mailand N, Falck J, Lukas C, et al: Rapid
destruction of human Cdc25A in response to DNA damage. Science.
288:1425–1429. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Helt CE, Cliby WA, Keng PC, Bambara RA and
O’Reilly MA: Ataxia telangiectasia mutated (ATM) and ATM and
Rad3-related protein exhibit selective target specificities in
response to different forms of DNA damage. J Biol Chem.
280:1186–1192. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Falck J, Mailand N, Syljuasen RG, Bartek J
and Lukas J: The ATM-Chk2-Cdc25A checkpoint pathway guards against
radioresistant DNA synthesis. Nature. 410:842–847. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kouzarides T: Snapshot: histone-modifying
enzymes. Cell. 131:8222007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stucki M, Clapperton JA, Mohammad D, Yaffe
MB, Smerdon SJ and Jackson SP: MDC1 directly binds phosphorylated
histone H2AX to regulate cellular responses to DNA double-strand
breaks. Cell. 123:1213–1226. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu L, Luo K, Lou Z and Chen J: MDC1
regulates intra-S-phase checkpoint by targeting NBS1 to DNA
double-strand breaks. Proc Natl Acad Sci USA. 105:11200–11205.
2008. View Article : Google Scholar : PubMed/NCBI
|