Introduction
Peyronie’s disease (PD) is an acquired idiopathic
localized fibrosis of the penis involving the tunica albuginea of
the corpus cavernosum. This condition not only gives rise to
palpable plaques in the penis or painful erection but also results
in penile curvature, deformities or sexual dysfunction (1,2).
Although PD once was thought to be a rare disorder, epidemiologic
studies showed a prevalence rate of 3.2–8.9% among adult men
(1,3). Because of our incomplete
understanding of the pathogenesis of PD, however, management of PD
remains a therapeutic dilemma in sexual medicine. Currently,
surgical intervention is the only effective treatment for this
condition, and most available medical treatments have not been
proven to be definitively efficacious. Thus, develop of novel and
effective medical therapies for PD is required.
Fibrosis is a main pathological manifestation of PD,
usually caused by proliferation of fibroblasts and accumulation of
various extracellular matrix (ECM) progresses forming plaques or
even ectopic calcification that appear as scars and impede
expansion of the tunica albuginea during erection (4,5).
Until now, involvement of transforming growth factor (TGF)-β has
been thought to play an important role in PD-related fibrosis
(6) and is also known to be
upregulated in both human PD plaques and animal PD models (7,8). The
binding of active TGF-β to its receptor triggers several signalling
cascades, including the well-characterized smad and
phosphotidylinositol-3-kinase (PI3K)/Akt pathways (9,10).
In particular, the PI3K/Akt pathway has been implicated in the
control of numerous cellular processes including cell
proliferation, survival and inflammation (11,12).
PI3K first activates Akt and subsequently increases the expression
of downstream proteins including mammalian target of rapamycin
(mTOR) and P70S6K. Recently, increasing evidence has shown that the
PI3K/Akt pathway effectively regulates fibrogenic responses such as
ECM remodeling and the proliferation of various types of
fibroblasts (11,13). Indeed, blocking PI3K activity has
been reported to inhibit collagen expression and the proliferation
of fibroblasts such as hepatic stellate cells (14,15),
where the inhibition of Akt has also been found to be associated
with interruption of fibroblast proliferation/differentiation and
collagen type I transcription. Likewise, disturbance of mTOR/P70S6K
suppresses fibroblast proliferation (14,16–18).
Therefore, inhibition of PI3K/Akt signalling pathway, as an
effective way to suppress the activation or proliferation of
fibroblasts, can be a novel therapeutic modality for PD-derived
fibrosis.
Given the emerging importance of PI3K/Akt signalling
in the pathogenesis of fibrosis, we have developed a new
imidazo[1,2-a]pyridine derivative, HS-173, as a novel PI3K
inhibitor. Although the anticancer effect of HS-173 has been
reported in our previous study (19), the therapeutic efficacy of this
compound against PD has not yet been evaluated. In the present
study, we determined whether HS-173 exerts anti-fibrotic effects
and the potential mechanisms underlying these processes in
PD-derived primary fibroblasts. Our results showed that HS-173
alleviated fibrosis by promoting apoptosis and inhibiting the
expression of fibrotic mediators such as collagen, α-SMA and
vimentin by blocking the PI3K/Akt pathway.
Materials and methods
Preparation of HS-173
Ethyl 6-(5-(phenylsulfonamido)
pyridin-3-yl)imidazo[1,2-a]pyridine-3-carboxylate (HS-173) is a new
PI3Kα inhibitor. This imidazopyridine derivative was synthesized as
described in our previous study (20). For all in vitro studies,
HS-173 was dissolved in dimethylsulfoxide (DMSO) before use.
Fibroblast cell culture
Plaque tissue was isolated from 4 PD patients who
underwent surgical correction of their condition. All tissue donors
provided informed consent and the procedures were approved by the
internal review board of our university (21). The tissue was transferred to
sterile vials containing Hank’s balanced salt solution (Gibco,
Carlsbad, CA) and washed three times in phosphate-buffered saline
(PBS). The biopsy tissues were minced into 1-mm2 pieces
and incubated in a shaker with 12.5 ml Dulbecco’s modified Eagle’s
medium (DMEM) supplemented with 0.06% collagenase A (Sigma-Aldrich,
St. Louis, MO) for 1 h. The cells and tissue fragments were
collected by centrifugation (400 × g for 5 min), washed in fresh
culture medium, and placed in 100-mm cell culture dishes (Falcon
Becton-Dickinson Labware, Franklin Lakes, NJ) with DMEM
supplemented with 10% fetal calf serum, penicillin (100 U/ml) and
streptomycin (100 mg/ml). The dishes were incubated in a humidified
incubator at 37°C with 5% CO2.
Measurement of cell proliferation
Cell proliferation was measured with an MTT assay.
Briefly, the fibroblasts were plated at a density of
2×104 cells/well in a 96-well plate and incubated for 24
h. The medium was removed, and cells were treated with either 0.1%
(v/v) DMSO as a control or various concentrations of HS-173. After
the cells were incubated for 72 h, 20 μl of an MTT solution (2
mg/ml) were added to each well and the plate was incubated for
another 4 h at 37°C. The resulting formazan crystals were dissolved
in DMSO (200 μl/well) with constant shaking for 5 min.
Absorbance of the plate was then read with a microplate reader at
540 nm. Three replicate wells were used for each analysis. The
median inhibitory concentration (IC50; defined as the
drug concentration at which cell growth was inhibited by 50%) was
determined based on the dose-response curves.
Terminal deoxynucleotidyl
transferase-mediated nick end labeling (TUNEL) assay
A TUNEL assay was performed using a commercially
available kit (Chemicon, Temecula, CA) following the manufacturer’s
protocol. The cells were plated on an 18-mm cover glass in DMEM at
a density of 1×105 cells/ml and incubated for 24 h. The
cells were then treated with 10 μM HS-173 for 24 h before
being fixed in an ice-cold mixture of acetic acid and ethanol.
After the cells were washed with PBS, they were stained for TUNEL.
The stained cells were examined under a fluorescence microscope for
nuclear fragmentation.
Measurement of mitochondrial membrane
potential
The fibroblasts were plated on 18-mm cover glasses
in DMEM and incubated for 24 h so that approximately 70% confluence
was reached. The cells were then incubated in the presence or
absence of 5 μM HS-173 for 2 h and then incubated with 5
μM JC-1 fluorescence dye (MitoPT, Immunohistochemistry
Technologies, Bloomington, MN) for 20 min in a CO2
incubator. The slides were then washed twice with PBS and covered
with Dabco (Sigma-Aldrich) before being viewed with a confocal
laser scanning microscope (Olympus, Tokyo, Japan).
Western blot analysis
The cells were serum-starved for 24 h and then
treated with various concentrations of HS-173 for 24 h. The cells
were washed three times with ice-cold PBS before being lysed in a
buffer containing 1% Triton X-100, 1% Nonidet P-40, and the
following protease and phosphatase inhibitors: aprotinin (10
mg/ml), leupeptin (10 mg/ml; ICN Biomedicals, Asse-Relegem,
Belgium), phenylmethylsulfonyl fluoride (1.72 mM), NaF (100 mM),
NaVO3 (500 mM) and
Na4P2O7 (500 mg/ml;
Sigma-Aldrich). Equal amounts of protein were separated using 8 or
12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and
transferred onto nitrocellulose membranes. Protein transfer was
confirmed using Ponceau S staining solution (Sigma-Aldrich). The
blots were immunostained using the appropriate primary antibodies
followed by secondary antibodies conjugated to horseradish
peroxidase. Primary antibodies specific for the following factors
were used: β-actin, collagen type I, collagen type IV, plasminogen
activator inhibitor-1 (PAI-1), fibronectin, phosphorylated (p)-Akt
(Abcam, Cambridge, UK), p-mTOR (e-bioscience), p-P70S6K, Akt, mTOR
and P70S6K (Cell Signalling Technology, Beverly, MA). All secondary
antibodies were purchased from Amersham Biosciences. Bands on the
blots were visualized with the enhanced chemiluminescence plus
system (Amersham Biosciences). Band intensities were quantified
with ImageJ software.
Immunofluorescence microscopy
The cells were plated on 18-mm cover glasses in DMEM
at a density of 1×105 cells/well and incubated for 24 h.
Next, the cells were incubated in the presence or the absence of 5
μM HS-173 for 2 h. The cells were washed with PBS and fixed
in an acetic acid:ethanol (2:1) solution for 5 min at −20°C.
Non-specific binding was blocked with 5% goat and horse serum/PBS
for 1 h at room temperature, and the cells were then incubated with
primary antibodies against vimentin, α-SMA (Sigma-Aldrich), p-Akt
(Abcam), p-mTOR, and p-4EBP1 (Cell Signaling Technology) in a
humidified chamber. After washing twice in PBS, the cells were
incubated with fluorescein-labeled secondary antibody (1:50;
Dianova) in antibody dilution solution for 1 h at room temperature
in the dark. The nuclei were stained with
4,6-diamidino-2-phenylindole (DAPI) in the dark for 30 min at room
temperature. The slides were washed twice with PBS, covered with
Dabco (Sigma-Aldrich), and examined with confocal laser scanning
microscopy (Olympus) at 488 and 568 nm.
Measurement of nucleus translocation
The cells were plated on 18-mm cover glasses in DMEM
and grown to approximately 70% confluence for 24 h. Next, the cells
were serum-starved for 24 h and then pretreated with 10 μM
HS-173 for 1 h. The fibroblasts were then treated with 20 ng/ml
TGF-β1 (R&D Systems) for 4 h. The cells were washed with PBS
and fixed in an acetic acid:ethanol (2:1) solution for 5 min at
−20°C. Non-specific binding was blocked in 5% goat and horse
serum/PBS for 1 h at room temperature, and the cells were then
incubated with primary antibodies against smad2/3 (Cell Signaling
Technology) in a humidified chamber. After washing twice in PBS,
the cells were incubated with fluorescein-labeled secondary
antibody (1:200; Dianova) in 1.5% horse serum/PBS at room
temperature in the dark for 1 h at 37°C. The nuclei were stained
with DAPI in the dark for 30 min at room temperature. The slides
were washed twice with PBS, covered with Dabco (Sigma-Aldrich), and
examined with confocal laser scanning microscopy (Olympus) at 488
and at 568 nm.
Statistical analysis
Data are expressed as the mean ± SD, and analyzed
with the ANOVA and unpaired Student’s t-test. A p-value of ≤0.05
was considered statistically significant. Statistical calculations
were performed using SPSS software for the Windows operating system
(version 10.0; SPSS, Chicago, IL).
Results
HS-173 inhibits the proliferation of
PD-derived primary fibroblasts
We first examined the effect of HS-173 on the
proliferation and viability of fibroblasts derived from human PD
plaques. The cells were exposed to various concentrations (0, 0.1,
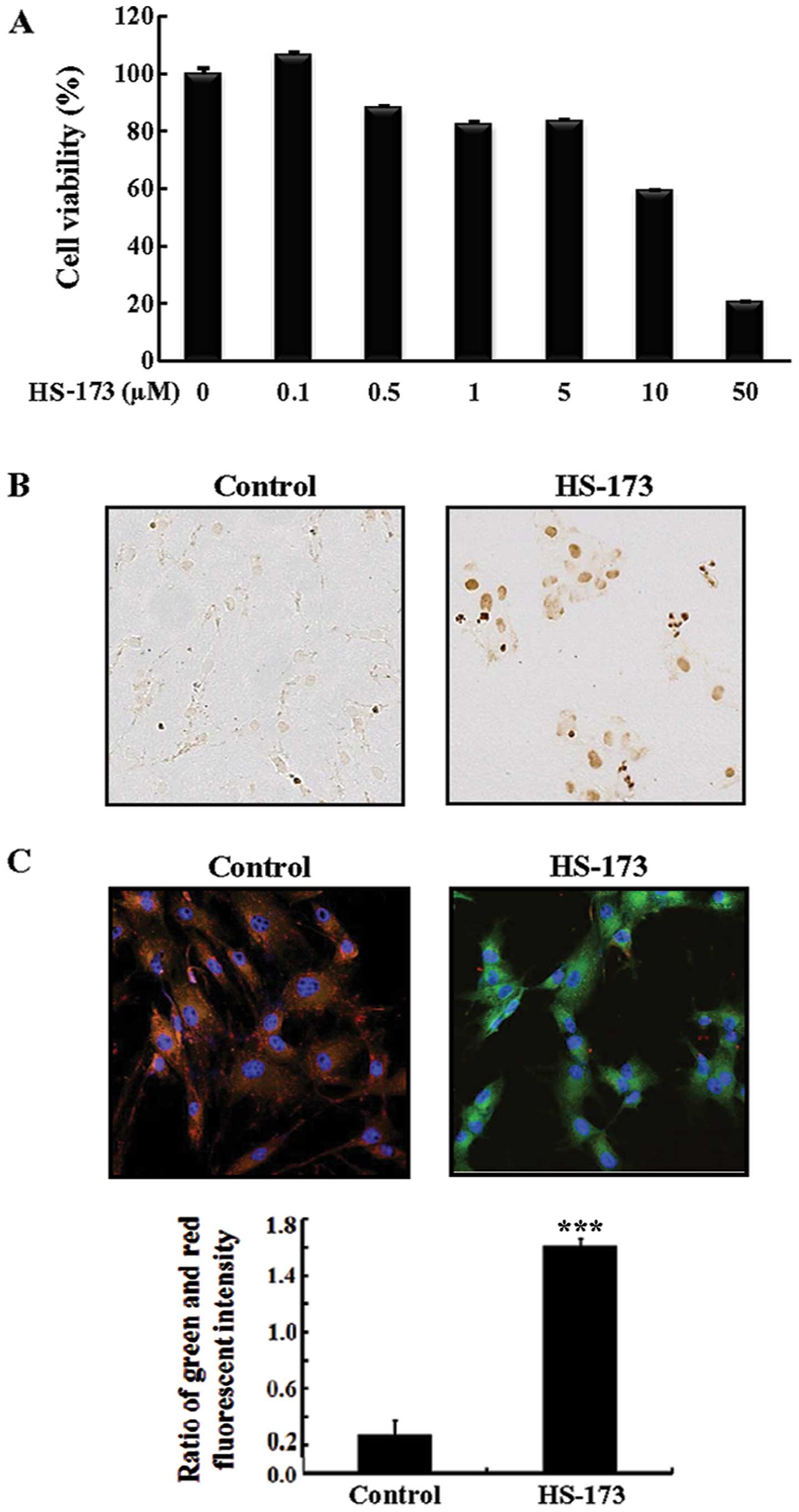
0.5, 1, 5, 10 and 50 μM) of HS-173 for 72 h. As shown in
Fig. 1A, HS-173 inhibited
fibroblast growth in a dose-dependent manner starting at a
concentration of 0.5μM. In particular, 10 and 50 μM
of HS-173 significantly reduced cell growth by 40 and 80%,
respectively.
HS-173 induces apoptotic cell death
To assess the apoptotic effects of HS-173 on
PD-derived primary fibroblasts, we performed a TUNEL assay. When
treated with 10 μM HS-173, the fibroblasts developed
morphological features characteristic of apoptotic cells. As shown
in Fig. 1B, DNA fragmentation was
observed by TUNEL in the cells treated with HS-173. Since the loss
of mitochondrial membrane potential (ψm) is a hallmark
for apoptosis, we also conducted JC-1 staining to identify the
impact of HS-173 on ψm (Fig. 1C). Heterogeneous staining of the
cytoplasm with both red and green fluorescence coexisting in the
same cell was observed in the control cells. Treatment with HS-173
(5 μM) decreased the red fluorescence in the fibroblasts and
frequent clusters of mitochondria were seen. Exposure to HS-173
induced marked changes in ψm as evident from the
disappearance of red fluorescence or increased green fluorescence
in most cells. These results showed that HS-173 induced apoptotic
cell death with the loss of Δψm in PD-derived fibroblast
cells.
HS-173 has anti-fibrotic effects in
PD-derived fibroblasts
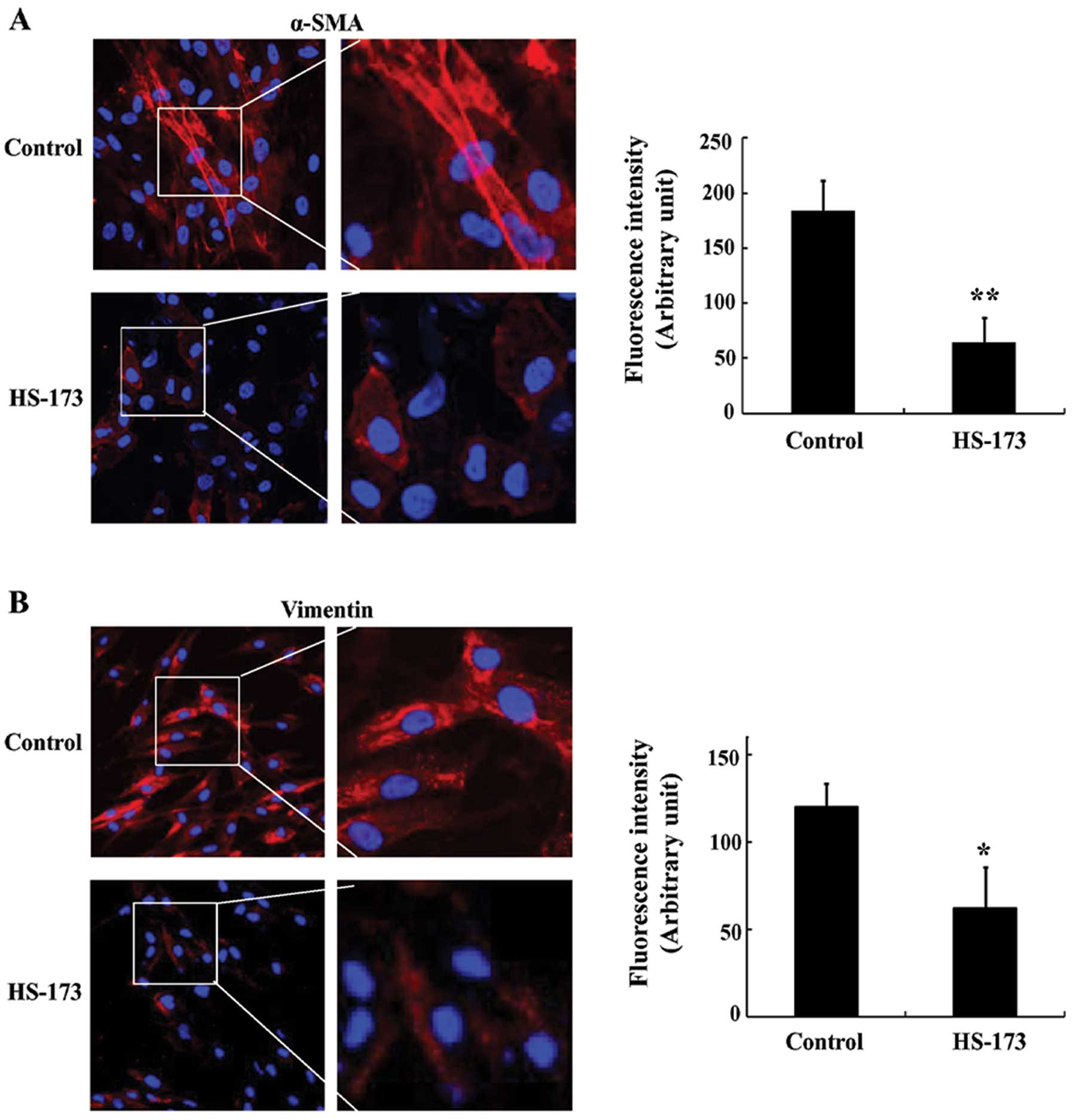
In order to determine whether HS-173 triggers
characteristics of fibroblasts, we stained positive fibroblast
markers in PD-derived fibroblasts. As shown in Fig. 2A and B, PD-derived fibroblasts
highly expressed fibroblast-positive makers such as α-SMA and
vimentin. In contrast, the expression of α-SMA and vimentin in
cells treated with HS-173 was decreased compared to the untreated
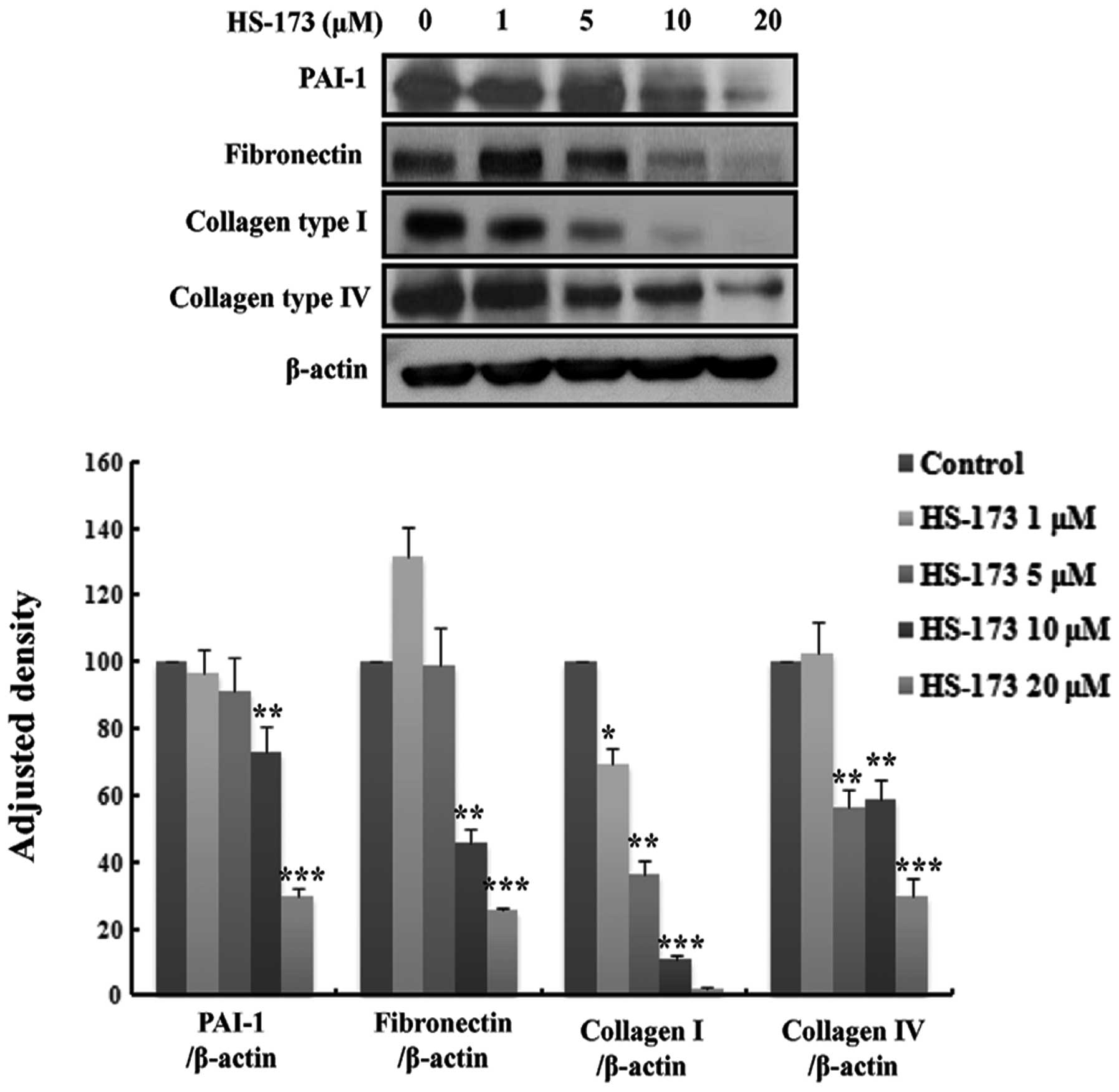
cells. In order to confirm the anti-fibrosis effect of HS-173, we
performed western blot analysis. The fibroblasts were serum-starved
for 24 h and treated with HS-173 (0 to 20 μM) for 24 h. As
shown in Fig. 3A, the expression
of PAI-1, fibronectin, collagen type I and collagen type IV was
decreased by HS-173 in a dose-dependent manner. Our results
revealed that HS-173 had an anti-fibrotic effect by reducing the
expression of fibrosis mediators such as α-SMA, vimentin, PAI-1,
fibronectin and collagen in PD-derived fibroblasts.
HS-173 suppresses the expression of
smad2/3 in TGF-β1-treated PD-derived fibroblasts
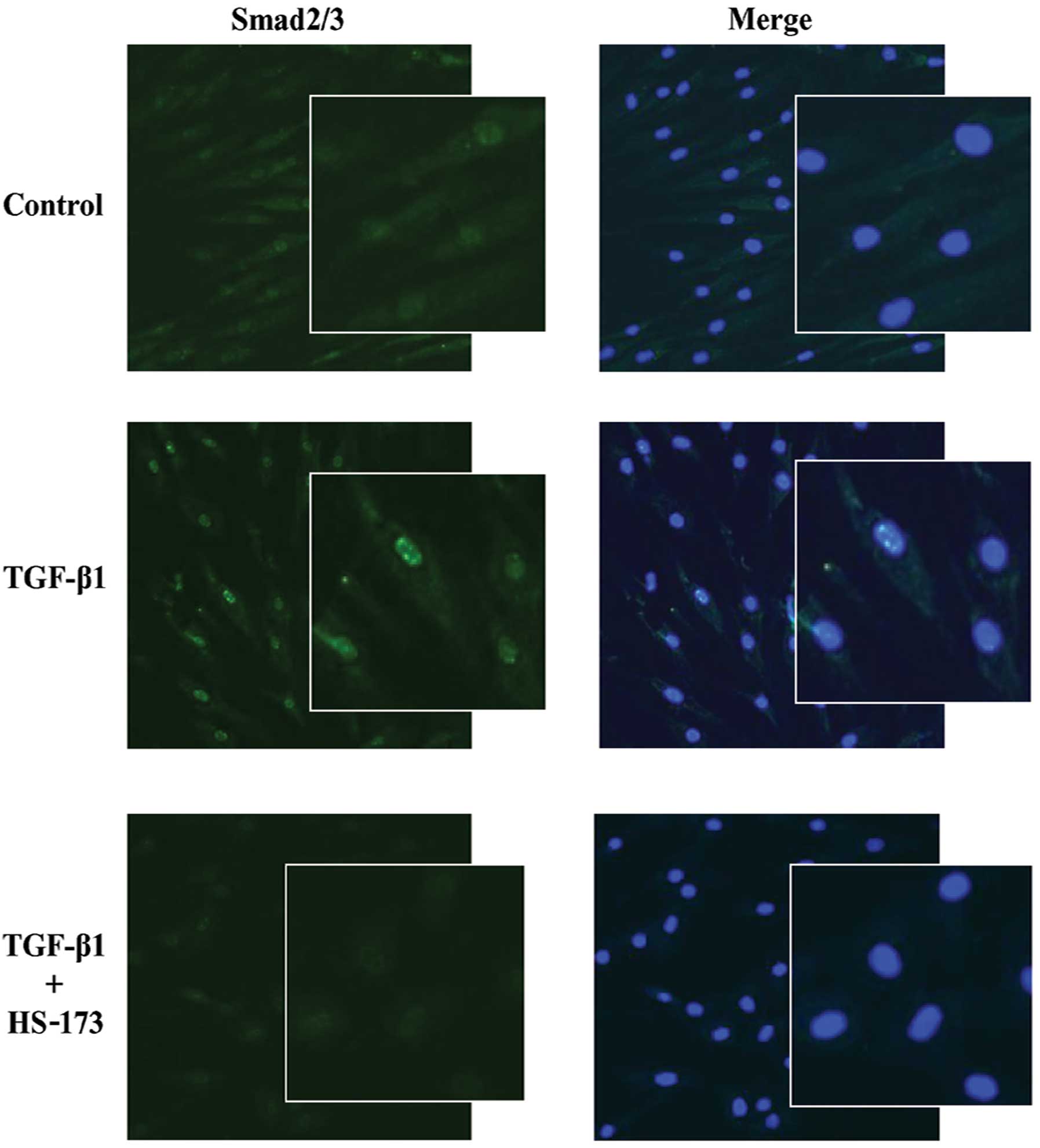
In the TGF-β signalling pathway, TGF-β binds to its
receptor on the plasma membrane and smad2/3 moves from the
cytoplasm to nucleus resulting in the upregulation of
transcription. We investigated whether HS-173 suppressed
TGF-β-induced nuclear translocation of smad2/3 in PD-derived
fibroblasts. Fibroblasts treated with TGF-β1 showed high expression
of smad2/3 in the nucleus compared to the control cells (Fig. 4). Interestingly, HS-173
significantly decreased the nuclear translocation of smad2/3 in
TGF-β-treated fibroblasts. Moreover, the cells treated with HS-173
showed low expression of smad2/3 not only in the nucleus but also
in the cytoplasm. Our results demonstrated that HS-173 inhibited
the translocation of smad2/3 in fibroblasts exposed to TGF-β.
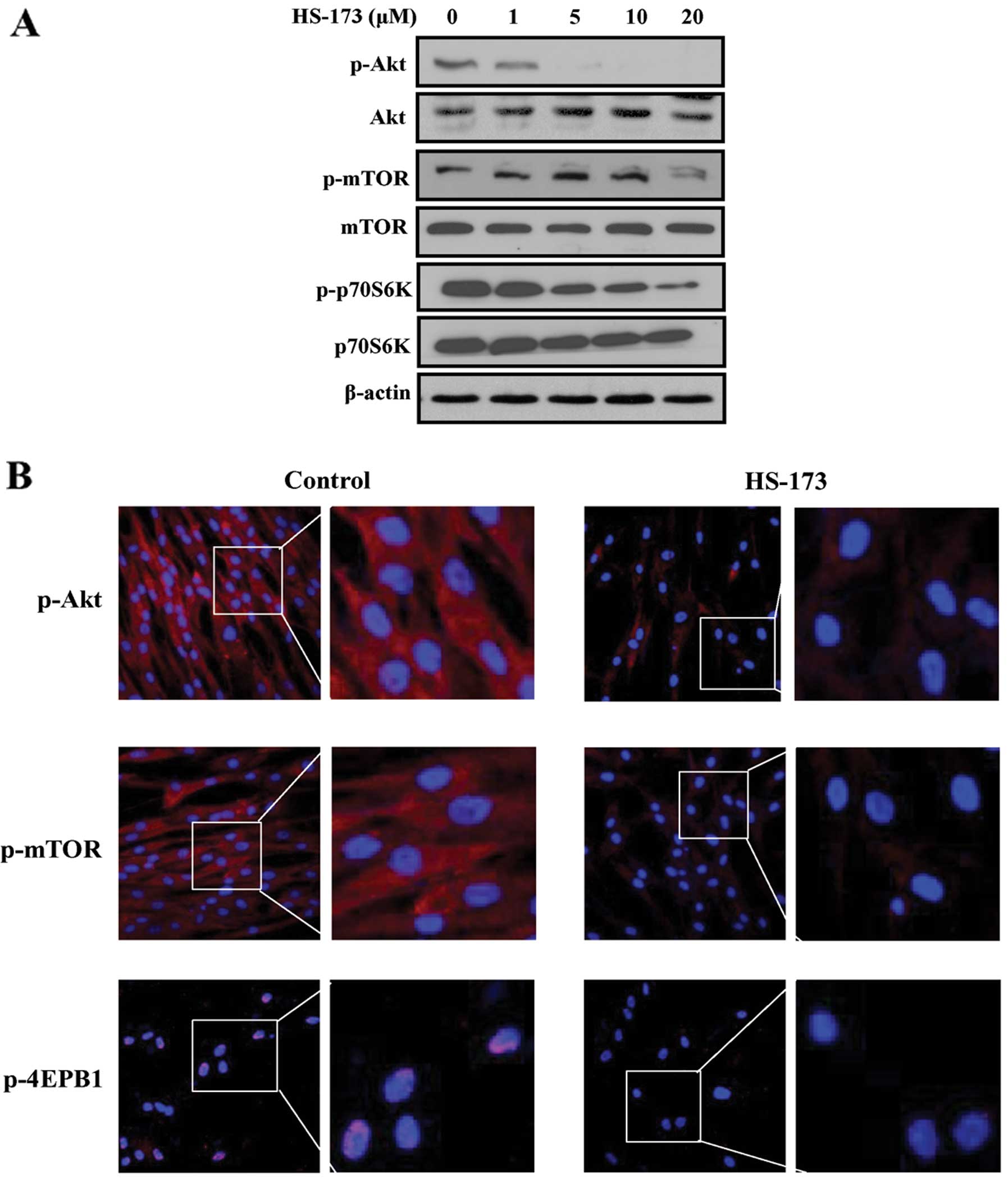
HS-173 inhibits the PI3K/Akt/mTOR
signalling pathway in PD-derived fibroblasts
Since HS-173 is a novel PI3K inhibitor, we
determined whether this compound blocked the PI3K/Akt/mTOR pathway
in PD-derived fibroblasts. Fibroblasts were treated with various
concentrations of HS-173 and the protein expression of
PI3K/Akt/mTOR signalling factors was observed by western blot
analysis. We found that the expression of p-Akt, p-mTOR and
p-P70S6K was obviously inhibited by HS-173 in PD-derived
fibroblasts (Fig. 5A). To confirm
these results, we performed immunofluorescence analyses. Consistent
with the western blot analysis data, we found that HS-173
effectively blocked the PI3K/Akt/mTOR pathway by decreasing the
levels of p-mTOR, p-Akt and p-4EBP1 in PD-derived fibroblasts
(Fig. 5B).
Discussion
Surgical correction remains the gold standard for
treating PD in cases of severe deformity. However, surgical
correction is available in limited cases because it is highly
invasive procedure and may give rise to shortening of the penis or
recurrence of penile curvature as a complication after surgery
(22,23). Currently, a variety of oral,
injectable and topical agents are available for the treatment PD
(24,25). Unfortunately, most of medical
treatment options did not demonstrate conclusive effects in PD
patients, although these therapies have been proven to be effective
at preclinical levels.
Based on recent studies showing that PI3K/Akt
signalling is involved in the development and progression of
fibrogenesis during cell proliferation and collagen production
(26,27), we synthesized HS-173, a novel PI3K
inhibitor and evaluated its anti-fibrosis effects on PD-derived
fibroblasts along with the underlying mechanisms. Our study showed
that HS-173 exerted anti-fibrotic effects by decreasing the
expression of collagen, α-SMA and vimentin in PD-derived primary
fibroblasts. Additionally, this compound inhibited the growth and
proliferation of the cells. Taken together, our findings suggest
that the anti-fibrotic effects of HS-173 were mediated by
inhibition of the PI3K/Akt signalling pathway.
PD is characterized by the proliferation of tunical
fibroblasts, subsequent differentiation of these cells into
contractile myofibroblasts and excessive deposition of collagen
(28). Aberrant proliferation of
PD fibroblasts has also been detected in PD patients (28). To inhibit PD fibroblast
proliferation, many agents with anti-fibrotic potential have been
employed to downregulate or neutralize proliferative, fibrogenic
and contractile responses of myofibroblasts (29). Thus, we first investigated the
anti-proliferation effect of HS-173 on PD-derived fibroblasts. In
our study, HS-173 inhibited the growth of PD-derived fibroblasts by
20–80% starting at a concentration of 0.5μM. HS-173 did not
only inhibit cell growth and proliferation, but also induced
apoptosis in the PD-derived fibroblasts. This is of particular
interest because there is an emerging evidence showing that the
process of apoptosis is defective in PD plaques. It has been
demonstrated that the rate of proliferation of fibroblasts derived
from plaque tissue is faster than that of apoptosis (30). Additionally, PD plaque-derived
fibroblasts have higher DNA production levels than control cells
(31). Since the biological
features of apoptosis include DNA fragmentation and increase
mitochondrial membrane permeability (32), we identified the apoptotic effects
of HS-173 using the TUNEL assay and JC-1 staining. Our result
showed that HS-173 induced apoptosis by increasing DNA
fragmentation. In addition, a loss of Δψm following
HS-173 treatment led to the induction of apoptosis in PD-derived
fibroblasts.
Considering the above results, the effect of HS-173
on proliferation and apoptosis was expected to influence
anti-fibrotic responses in PD-derived fibroblasts. Furthermore,
previous studies have reported that the fibroblasts secrete
ECM-related proteins such as collagen and proteoglycans, and in
turn the ECM itself regulates fibroblast proliferation, apoptosis
and migration (27,28). We therefore determined whether
HS-173 inhibited the expression of α-SMA, vimentin, PAI-1,
fibronectin and collagen type I/IV. As expected, HS-173 reduced the
expression of fibrosis-related mediators in PD-derived fibroblasts.
These results were consistent with those of Mikulec et al
showing that tamoxifen used to treat PD inhibits the proliferation
of fibroblasts and decreases collagen synthesis (33). Our findings suggest that HS-173
exerts anti-fibrotic effects by regulating the proliferation and
apoptosis of PD-derived fibroblasts.
PI3K is well known to control many cellular
functions such as proliferation, survival and migration (14). Recently, the PI3K/Akt pathway has
been reported to represent a mechanism critical for the
proliferation of fibroblasts in various organs including the lung,
liver and heart (26,34,35).
Furthermore, inhibition of PI3K/Akt activation decreases both
fibroblast proliferation and differentiation into myofibroblasts in
human lung (36). Thus, PI3K
signalling is activated in fibroblasts and correlates with collagen
production (34,37). If PI3K is targeted, the expression
or activation of various fibrogenic mediators might be reduced. In
this regard, the PI3K/Akt signalling pathway could represent a
therapeutic target for PD-derived fibrosis. However, it has been
not reported whether this signalling pathway contributes to the
regulation of PD.
We therefore evaluated the activation of the
PI3K/Akt signalling pathway in PD-derived fibroblasts and
suppression of this pathway by HS-173. We observed that the
expression of Akt, mTOR and P70S6K was notably increased in
PD-derived fibroblasts, indicating that the PI3K signalling pathway
was activated in PD-derived fibroblasts. On the contrary, HS-173
significantly inhibited PI3K signalling. These results led us to
hypothesize that the anti-fibrotic effects of HS-173 might be
mediated by inhibition of the PI3K/Akt pathway. It was previously
reported that LY294002 (a pan-inhibitor of PI3K) abrogates cell
proliferation as well as α-SMA expression and collagen synthesis in
embryonic fibroblasts (38). This
is consistent with data from our present study showing that PI3K
inhibition by HS-173 decreased the proliferation of PD-derived
fibroblasts and collagen expression.
In conclusion, the present study shows that HS-173,
targeting the PI3K/Akt pathway, has an anti-fibrotic action. To our
knowledge, this is the first report showing that inhibition of
PI3K/Akt signalling pathway may be potentially useful for the
treatment of PD although the effect remains to be evaluated in
vivo.
Acknowledgements
This study was supported by a grant
from the Korea Health Technology R&D Project, Ministry of
Health and Welfare, Republic of Korea (J.-K.S. A110076).
References
|
1
|
Schwarzer U, Sommer F, Klotz T, Braun M,
Reifenrath B and Engelmann U: The prevalence of Peyronie’s disease:
results of a large survey. BJU Int. 88:727–730. 2001.
|
|
2
|
Ralph D, Gonzalez-Cadavid N, Mirone V,
Perovic S, Sohn M, Usta M and Levine L: The management of
Peyronie’s disease: evidence-based 2010 guidelines. J Sex Med.
7:2359–2374. 2010.
|
|
3
|
Mulhall JP, Creech SD, Boorjian SA, Ghaly
S, Kim ED, Moty A, Davis R and Hellstrom W: Subjective and
objective analysis of the prevalence of Peyronie’s disease in a
population of men presenting for prostate cancer screening. J Urol.
171:2350–2353. 2004.PubMed/NCBI
|
|
4
|
Dean RC and Lue TF: Physiology of penile
erection and pathophysiology of erectile dysfunction. Urol Clin
North Am. 32:379–395. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gonzalez-Cadavid NF and Rajfer J:
Mechanisms of disease: new insights into the cellular and molecular
pathology of Peyronie’s disease. Nat Clin Pract Urol. 2:291–297.
2005.PubMed/NCBI
|
|
6
|
Haag SM, Hauck EW, Szardening-Kirchner C,
Diemer T, Cha ES, Weidner W and Eickelberg O: Alterations in the
transforming growth factor (TGF)-beta pathway as a potential factor
in the pathogenesis of Peyronie’s disease. Eur Urol. 51:255–261.
2007.PubMed/NCBI
|
|
7
|
Domes T, De Young L, O’Gorman DB, Gan BS,
Bella AJ and Brock G: Is there a role for proteomics in Peyronie’s
disease? J Sex Med. 4:867–877. 2007.PubMed/NCBI
|
|
8
|
El-Sakka AI, Hassoba HM, Chui RM,
Bhatnagar RS, Dahiya R and Lue TF: An animal model of
Peyronie’s-like condition associated with an increase of
transforming growth factor beta mRNA and protein expression. J
Urol. 158:2284–2290. 1997.
|
|
9
|
Assinder SJ, Dong Q, Kovacevic Z and
Richardson DR: The TGF-beta, PI3K/Akt and PTEN pathways:
established and proposed biochemical integration in prostate
cancer. Biochem J. 417:411–421. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Danielpour D: Functions and regulation of
transforming growth factor-beta (TGF-beta) in the prostate. Eur J
Cancer. 41:846–857. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cantley LC: The phosphoinositide 3-kinase
pathway. Science. 296:1655–1657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Carracedo A and Pandolfi PP: The PTEN-PI3K
pathway: of feedbacks and cross-talks. Oncogene. 27:5527–5541.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Andre E, Gazzieri D, Bardella E, Ferreira
J, Mori MA, Saul VV, Bader M, Calixto JB, De Giorgio R, Corinaldesi
R, Geppetti P and Trevisani M: Expression and functional
pharmacology of the bradykinin B1 receptor in the normal and
inflamed human gallbladder. Gut. 57:628–633. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Reif S, Lang A, Lindquist JN, Yata Y,
Gabele E, Scanga A, Brenner DA and Rippe RA: The role of focal
adhesion kinase-phosphatidylinositol 3-kinase-akt signaling in
hepatic stellate cell proliferation and type I collagen expression.
J Biol Chem. 278:8083–8090. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gentilini A, Marra F, Gentilini P and
Pinzani M: Phosphatidylinositol-3 kinase and extracellular
signal-regulated kinase mediate the chemotactic and mitogenic
effects of insulin-like growth factor-I in human hepatic stellate
cells. J Hepatol. 32:227–234. 2000. View Article : Google Scholar
|
|
16
|
Kulik G, Klippel A and Weber MJ:
Antiapoptotic signalling by the insulin-like growth factor I
receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol.
17:1595–1606. 1997.PubMed/NCBI
|
|
17
|
Kim AH, Khursigara G, Sun X, Franke TF and
Chao MV: Akt phosphorylates and negatively regulates apoptosis
signal-regulating kinase 1. Mol Cell Biol. 21:893–901. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Coffer PJ, Jin J and Woodgett JR: Protein
kinase B (c-Akt): a multifunctional mediator of
phosphatidylinositol 3-kinase activation. Biochem J. 335(Pt 1):
1–13. 1998.PubMed/NCBI
|
|
19
|
Lee H, Jung KH, Jeong Y, Hong S and Hong
SS: HS-173, a novel phosphatidylinositol 3-kinase (PI3K) inhibitor,
has anti-tumor activity through promoting apoptosis and inhibiting
angiogenesis. Cancer Lett. 328:152–159. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hong S, Lee S, Kim B, Lee H and Hong SS:
Discovery of new azaindole-based PI3Kalpha inhibitors: apoptotic
and antiangiogenic effect on cancer cells. Bioorg Med Chem Lett.
20:7212–7215. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Piao S, Choi MJ, Tumurbaatar M, Kim WJ,
Jin HR, Shin SH, Tuvshintur B, Yin GN, Song JS, Kwon MH, Lee SJ,
Han JY, Kim SJ, Ryu JK and Suh JK: Transforming growth factor
(TGF)-beta type I receptor kinase (ALK5) inhibitor alleviates
profibrotic TGF-beta1 responses in fibroblasts derived from
Peyronie’s plaque. J Sex Med. 7:3385–3395. 2010.PubMed/NCBI
|
|
22
|
LaRochelle JC and Levine LA: A survey of
primary-care physicians and urologists regarding Peyronie’s
disease. J Sex Med. 4:1167–1173. 2007.PubMed/NCBI
|
|
23
|
Sasso F, Gulino G, Falabella R, D’Addessi
A, Sacco E, D’Onofrio A and Bassi PF: Peyronie’s disease: lights
and shadows. Urol Int. 78:1–9. 2007.
|
|
24
|
Shindel AW and Lue TF: Peyronie’s disease:
past, present, future? Curr Urol Rep. 9:425–427. 2008.
|
|
25
|
Hauck EW, Diemer T, Schmelz HU and Weidner
W: A critical analysis of nonsurgical treatment of Peyronie’s
disease. Eur Urol. 49:987–997. 2006.PubMed/NCBI
|
|
26
|
Son G, Hines IN, Lindquist J, Schrum LW
and Rippe RA: Inhibition of phosphatidylinositol 3-kinase signaling
in hepatic stellate cells blocks the progression of hepatic
fibrosis. Hepatology. 50:1512–1523. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Conte E, Fruciano M, Fagone E, Gili E,
Caraci F, Iemmolo M, Crimi N and Vancheri C: Inhibition of PI3K
prevents the proliferation and differentiation of human lung
fibroblasts into myofibroblasts: the role of class I P110 isoforms.
PLoS One. 6:e246632011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Abdel-Hamid IA and Anis T: Peyronie’s
disease: perspectives on therapeutic targets. Expert Opin Ther
Targets. 15:913–929. 2011.
|
|
29
|
Bobustuc GC, Smith JS, Maddipatla S, Jeudy
S, Limaye A, Isley B, Caparas ML, Constantino SM, Shah N, Baker CH,
Srivenugopal KS, Baidas S and Konduri SD: MGMT inhibition restores
ERalpha functional sensitivity to antiestrogen therapy. Mol Med.
18:913–929. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Anderson MS, Shankey TV, Lubrano T and
Mulhall JP: Inhibition of Peyronie’s plaque fibroblast
proliferation by biologic agents. Int J Impot Res. 12(Suppl 3):
S25–S31. 2000.PubMed/NCBI
|
|
31
|
Mulhall JP, Nicholson B, Pierpaoli S,
Lubrano T and Shankey TV: Chromosomal instability is demonstrated
by fibroblasts derived from the tunica of men with Peyronie’s
disease. Int J Impot Res. 16:288–293. 2004.PubMed/NCBI
|
|
32
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mikulec AA, Hanasono MM, Lum J, Kadleck
JM, Kita M and Koch RJ: Effect of tamoxifen on transforming growth
factor beta1 production by keloid and fetal fibroblasts. Arch
Facial Plast Surg. 3:111–114. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
He Z, Gao Y, Deng Y, Li W, Chen Y, Xing S,
Zhao X, Ding J and Wang X: Lipopolysaccharide induces lung
fibroblast proliferation through Toll-like receptor 4 signaling and
the phosphoinositide3-kinase-Akt pathway. PLoS One. 7:e359262012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Voloshenyuk TG, Landesman ES, Khoutorova
E, Hart AD and Gardner JD: Induction of cardiac fibroblast lysyl
oxidase by TGF-beta1 requires PI3K/Akt, Smad3, and MAPK signaling.
Cytokine. 55:90–97. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xia H, Khalil W, Kahm J, Jessurun J,
Kleidon J and Henke CA: Pathologic caveolin-1 regulation of PTEN in
idiopathic pulmonary fibrosis. Am J Pathol. 176:2626–2637. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Marra F, Gentilini A, Pinzani M, Choudhury
GG, Parola M, Herbst H, Dianzani MU, Laffi G, Abboud HE and
Gentilini P: Phosphatidylinositol 3-kinase is required for
platelet-derived growth factor’s actions on hepatic stellate cells.
Gastroenterology. 112:1297–1306. 1997.
|
|
38
|
Foukas LC, Berenjeno IM, Gray A, Khwaja A
and Vanhaesebroeck B: Activity of any class IA PI3K isoform can
sustain cell proliferation and survival. Proc Natl Acad Sci USA.
107:11381–11386. 2010. View Article : Google Scholar : PubMed/NCBI
|