Introduction
Double-stranded RNA-dependent protein kinase (PKR)
is an abundantly expressed serine/threonine protein kinase which is
activated by double-stranded RNA (dsRNA), interferons, cytokines,
stress signals, and viral infection (1,2). PKR
is also involved in several signal transduction pathways, such as
mitogen-activated protein kinase (MAPK), nuclear factor of κB
(NF-κB), inhibitor of NF-κB (IκB) and Smad (3–5). PKR
is activated through autophosphorylation and once activated the
enzyme phosphorylates certain substrates including the α-subunit of
eukaryotic initiation factor 2 (eIF-2α) (6,7). The
PKR-eIF-2α cascade has been implicated as a general transducer of
apoptosis in response to a variety of stimuli (8–12).
It was reported that PKR was dephosphorylated by serine/threonine
protein phosphatases type 1 (PP1) (13,14).
PP1 binds directly to PKR and reduces dsRNA-mediated
auto-activation of PKR (15). PP1
may regulate the activities of both PKR and eIF-2α by
dephosphorylating them and thus might block the protein synthesis
and apoptosis.
Apoptosis is one of the essential steps in the
maintenance of normal cell populations of adult mammals and occurs
continually in various cell populations. Apoptosis is a
morphologically and biochemically distinct mode of cell death that
plays major roles during embryogenesis, carcinogenesis, cancer
treatment, or immune and toxic cell killing (16–20).
The cytological apparent stages of apoptosis are rapid condensation
of chromatin and fragmentation of the cells with membrane-enclosed
apoptotic bodies that are phagocytosed and digested by nearby
resident cells (21). A
biochemical characteristic feature of the process is double-strand
cleavage of nuclear DNA at the linker regions between nucleosomes,
leading to the production of oligonucleosomal fragments with
180–200 bp, which results in a characteristic laddering pattern on
agarose gel electrophoresis (22,23).
Okadaic acid (OA) is a toxic polyether fatty acid
produced by several dinoflagellates and is a potent inhibitor of
PP1 and PP2A. The use of this agent has led to the understanding
that the phosphorylation and dephosphorylation status is related to
cellular regulation, including the biological end-point, apoptosis
(24–26). We previously reported that OA
induced apoptosis in human osteoblastic cells (27–29).
Protein kinases and phosphatases were reported to be involved in
transcriptional stimulation through activation of the NF-κB pathway
(15,30). We reported that the PKR/eIF-2α
pathway was activated and that NF-κB translocation occurred during
the OA-induced apoptosis (7,31).
However, details of the mechanisms of OA-mediated expression and
phosphorylation of IκB and NF-κB are still obscure. The
relationship between IκB or NF-κB and PKR in apoptosis is also to
be determined.
Materials and methods
Reagents
G418 Geneticin, cycloheximide (CHX), and
anti-β-actin antibody were obtained from Sigma-Aldrich (St. Louis,
MO, USA). α-modification of minimum essential medium (α-MEM),
Opti-MEM, and pre-stained molecular weight markers were purchased
from Gibco BRL (Grand Island, NY, USA). FuGene HD was from Roche
(Indianapolis, IN, USA). Fetal bovine serum (FBS) was obtained from
Equitech-Bio (Kerrville, TX, USA). Anti-phospho-eIF-2α (119A11)
antibody was from Cell Signaling (Danvers, MA, USA).
Anti-phospho-IκBα (Thr291), anti-PKR (M-515) and anti-NF-κB p65
(C-20) antibodies were from Santa Cruz Biotechnology (Santa Cruz,
CA, USA). Antibody for IκBα (MAD-3) were obtained from BD
Biosciences (San Jose, CA, USA). Plastic dishes were from Iwaki
(Chiba, Japan). OA was purchased from Wako (Osaka, Japan).
Cell culture and establishment of the
PKR-K/R mutant MG63 cells
Human PKR cDNA and a PKR-K/R mutant cDNA (carrying a
mutation of amino acid K→R at position 296) and their expression
vector were kindly provided by Dr A. Hovanessian (Institute
Pasteur, Paris, France) (32) and
Dr T. Takizawa (Aichi Human Service Center, Aichi, Japan) (33), respectively. Human osteoblastic
osteosarcoma cell line MG63 cells were obtained from the American
Type Culture Collection (Rockville, MD, USA). The cells were
cultured in α-MEM containing 10% (v/v) FBS and were maintained at
37°C in a humidified atmosphere of 5% CO2 and 95%
air.
PKR-K/R cDNA was subcloned into pcDNA3.1-Flag
(modified pcDNA3.1, Invitrogen, Carlsbad, CA, USA). Transfection of
pcDNA3.1-Falg-PKR-K/R into MG63 cells was performed using FuGene HD
reagents. Two μg of pc-Flag and pc-Flag-PKR-K/R in 100
μl Opti-MEM were mixed with 8 μl FuGene HD reagent
for 15 min at ambient temperature. The DNA-FuGene HD complex was
then added into 35-mm dishes containing 4×105 cells. The
media were replaced at 24 h after transfection and the cells were
subcultured in medium containing G418 Geneticin at a final
concentration of 500 μg/ml for 2 weeks. The media were
replenished every 3 days. The drag-resistant colonies were selected
and cloned. Cell modification was monitored by an Olympus IMT-2
phase-contrast microscope.
SDS-PAGE and western blot analysis
MG63 cells and their PKR-K/R mutant cells were
washed twice with PBS, scraped into lysate buffer containing 1 mM
DTT, 1 mM PMSF, 1 μg/ml leupeptin, 2 μg/ml aprotinin,
5 mM EGTA and protein phosphatase inhibitor cocktail
(Sigma-Aldrich) in phosphate-buffered saline (PBS). The lysates
containing equal amounts of proteins were separated by 10% SDS-PAGE
and transferred to PVDF membranes (Millipore, Bedford, MA, USA).
The membranes were incubated for 2 h at ambient temperature in a
blocking solution consisting of 5% non-fat skim milk in PBS
containing 0.1% Tween-20 (PBS-Tween) and incubated overnight at 4°C
with specific antibodies in PBS-Tween (diluted at 1:500 to 10,000).
After the membranes had been washed 4 times within 30 min in
PBS-Tween, they were incubated for 2 h at ambient temperature in
PBS-Tween containing horseradish peroxidase-conjugated second
antibodies (diluted at 1:5,000). The membranes were washed again as
described above and the proteins recognized by the antibodies were
visualized with an ECL detection kit (Pharmacia Biotech, Uppsala,
Sweden) according to the manufacturer’s instuctions. To strip off
the antibody the membrane was treated for 30 min at 50°C with 2%
SDS and 0.35% 2-mercaptoehanol in 62.5 mM Tris-HCl (pH 6.8). The
antibody-stripped membrane was then blocked again and re-incubated
with other antibodies.
RNA preparation, real-time PCR, and
RT-PCR
MG63 cells and the PKR-K/R cells were cultured in
35-mm dishes (1.0×104 cells/dish). Quantitative
real-time PCR analysis was performed using SYBER Premix Ex taq
Perfect Real-time (Takara Bio, Kyoto, Japan). The sequences of the
primers used are as follows: IκBα forward,
CACACGTGTCTACACTTAGCCTCTA; IκBα reverse, AATAGCCCTGGTAGGTAACTCTGTT;
GAPDH forward, GACCCCTTCATTGACCTCAAC; GAPDH reverse,
CTTCTCCATGGTGGTGAAGA. DNA amplification and detection was performed
in the ABI PRISM 7500 (Perkin-Elmer Applied Biosystems, Foster
City, CA, USA). PCR amplification (40 cycles) was performed
following conditions: 95°C for 10 sec and 60°C for 34 sec. Standard
curves were generated using 10-fold serial dilutions of genomic
DNA. The concentrations of unknown samples were calculated by
extrapolation from this standard curve and expression levels were
normalized with GAPDH expression. The data were analyzed by
Sequence Detection Software (SDS) vo1. 4 (Perkin-Elmer).
For RT-PCR analysis, total cellular RNA was
extracted by using TRIzol (Invitrogen) and subjected to PCR using
RT-PCR kit (Takara). The primers used for PCR were as indicated
above. PCR reactions at 94°C for 30 sec, at 57°C for 30 sec and at
72°C for 30 sec were carried out for 32 cycles. The PCR products
were separated by electrophoresis on 1.5% agarose gels and
visualized by ethidium bromide staining with UV light
illumination.
DNA isolation and agarose gel
electrophoresis
Purification of DNA from cultured cells was started
by lysis of the cells in cold 10 mM Tris-HCl, pH 7.5, containing 1
mM EDTA and 0.5% Triton X-100. After lysis, debris was removed by
centrifugation at 15,000 g for 20 min. DNAse-free RNAse
(Sigma-Aldrich) was added to the lysates at a final concentration
of 40 μg/ml, and the lysates were then incubated with gentle
shaking for 1 h at 37°C. Proteinase K (Sigma-Aldrich) was added to
the RNAse-treated lysates at a final concentration of 40
μg/ml. The lysates were further incubated for 1 h at 37°C
with gentle shaking. DNA was precipitated with 2-propanol and
sodium chloride overnight at −20°C. After centrifugation and
drying, the DNA was dissolved in TE-buffer (10 mM Tris, pH 8.0,
containing 1 mM EDTA). Agarose gel electrophoresis of DNA was
performed by using 2.0% agarose gel containing 0.5 μg/ml
ethidium bromide. DNA markers (100 bp) (New England BioLabs,
Ipswich, MA, USA) were run in the same gels. To visualize apoptotic
alterations to DNA integrity, we observed the DNA bands on a UV
transilluminator. Images were taken with a Polaroid DS-300
camera.
Statistical analysis
All data are presented as mean ± SEM. Statistical
analysis was performed using Student’s t-test. Results are
representative examples of three or more independent
experiments.
Results
Okadaic acid stimulated the expression of
IκBα in MG63 cells
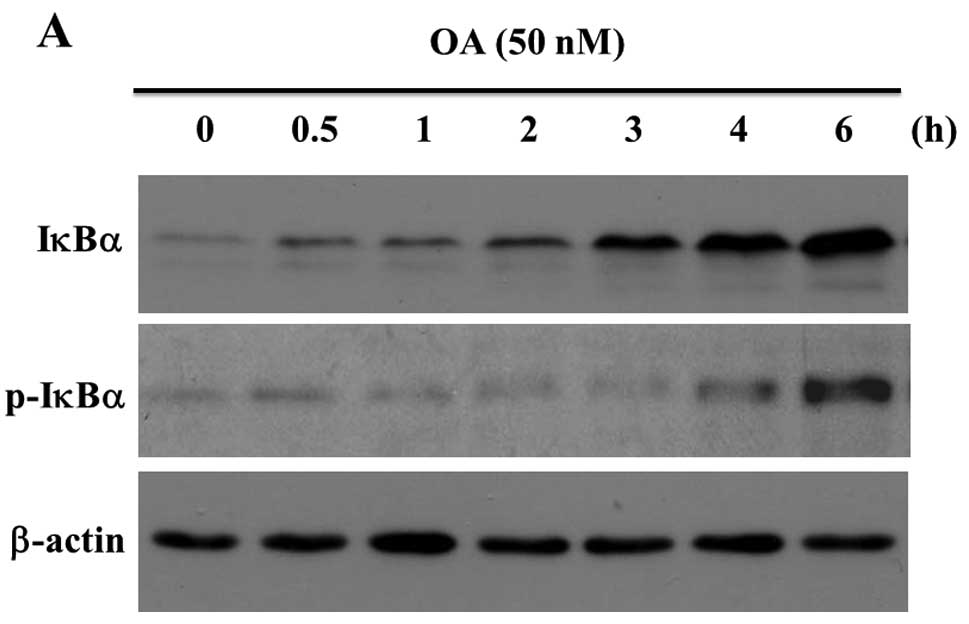
Fig. 1A shows that
the anti-IκBα antibody recognized a band corresponding to IκBα (36
kDa) in a sample prepared from the unstimulated MG63 cells. OA at
50 nM increased the expression of IκBα protein in a time-dependent
manner (Fig. 1A, upper panel). The
anti-phospho-IκBα (S32) antibody interacted with a band
corresponding to IκBα and 50 nM OA increased the staining intensity
of phosphorylated IκBα in MG63 cells (Fig. 1A, middle panel). The bound
antibodies were stripped off the membranes and re-incubated with
the anti-β-actin antibody as loading controls (Fig. 1A, lower panel). A band
corresponding to the position of IκBα was not detected in the blots
of the extracts incubated with the same dilution of normal rabbit
serum (data not shown). We also analyzed the expression of IκBα
mRNA. The result of RT-PCR shows that OA increased the expression
of IκBα mRNA in MG63 cells (Fig.
1B).
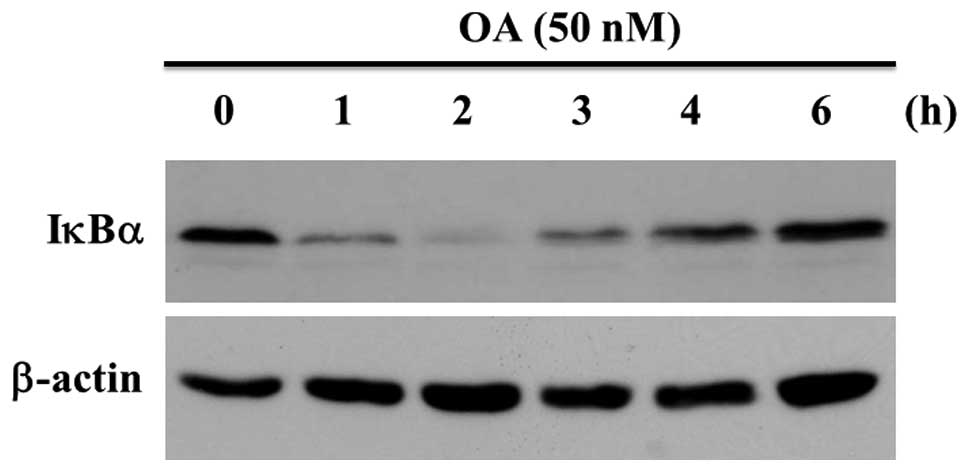
New protein synthesis in MG63 cells was inhibited by
the treatment of 100 ng/ml of CHX for 30 min. The expression of
IκBα protein was evaluated by western blot analysis. OA-treatment
decreased the staining intensity of IκBα ≤2 h, at which time the
staining level was minimum. After that the staining level increased
in a time-dependent manner up to 6 h (Fig. 2). The amounts of IκBα decreased in
MG63 cells treated with OA for 2 h in a dose-dependent fashion up
to 100 nM (data not shown). These findings indicate that IκBα was
degraded in MG63 cells with OA-treatment. The expression of β-actin
was not changed with OA-treatment (Fig. 2).
Expression of eIF-2α and PKR in the
okadaic acid-treated cells
PKR functions via phosphorylation of IκBα (34,35)
and OA increased the amounts of phosphorylated form of IκBα
(Fig. 1). We considered that PKR
might play an essential role in the phosphorylation of IκBα. We
transfected MG63 cells with a catalytically inactive mutant of
human PKR obtained by substituting Lys at 296 with Arg and
established cells stably expressing dominant-negative PKR gene
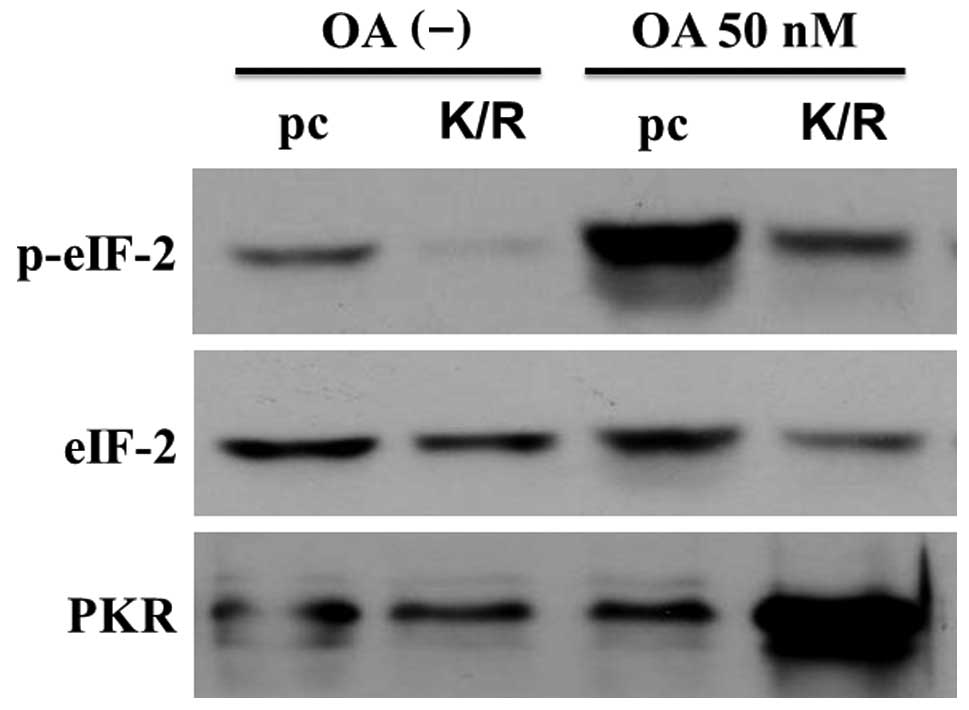
(PKR-K/R). To verify the PKR mutation, we analyzed the
phosphorylation status of eIF-2α, the best-characterized substrate
of PKR, by western blotting using an anti-phospho-eIF-2α antibody.
Fig. 3 shows that the intensity of
phosphorylated form of eIF-2α increased in the 50 nM OA-treated
pcDNA-transfected MG63 cells (pc cells). However, the level of this
form was low in the PKR-K/R cells treated with OA at the same
conditions (Fig. 3). The amounts
of eIF-2α did not differ between the control and PKR-K/R cells. OA
increased the amount of PKR in the PKR-K/R cells whereas the
expression levels of PKR were low in the OA-treated pc cells
(Fig. 3). These data indicate that
although PKR was overexpressed in PKR-K/R cells, functional
PKR/eIF-2α pathway was inactivated by the transfection with the
plasmid bearing the PKR dominant-negative mutation.
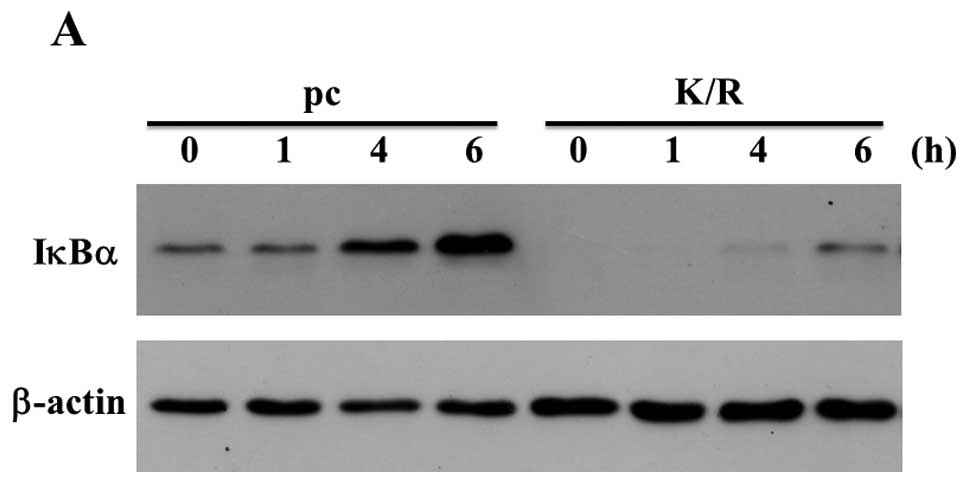
We treated pc and PKR-K/R cells with 50 nM OA for
various time-points. Expression of IκBα was detected in the samples
of the unstimulated pc cells whereas the expression levels of IκBα
were low in the PKR-K/R cells (Fig.
4A). OA increased the IκBα expression in pc cells compared with
that in PKR-K/R cells (Fig. 4A).
The bound anti-IκBα antibody was stripped off and re-incubated with
anti-β-actin antibody as a loading control (Fig. 4A). The result of real-time PCR
shows that the expression of IκBα mRNA in MG63 cells was stimulated
with 50 nM OA in a time-dependent manner (Fig. 4B). The expression of IκBα in the 50
nM OA-stimulated PKR-K/R cells was low compared with that of the
wild-type MG63 cells (Fig.
4B).
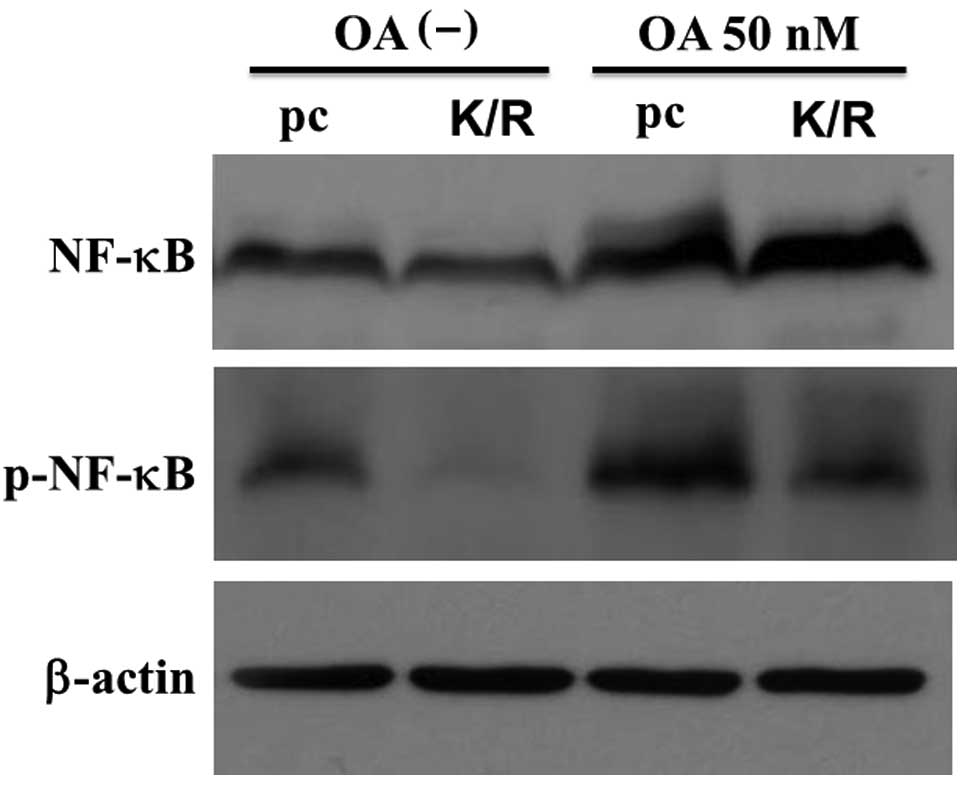
Regulation of NF-κB proteins in the cells
treated with okadaic acid
To determine the expression of NF-κB in pc and
PKR-K/R cells whole cell lysates prepared from the 50 nM
OA-stimulated cells were analyzed by western blotting with an
antibody against p65NF-κB. The anti-p65NF-κB antibody interacted
with a major band having an estimated molecular weight of 65 kDa
(Fig. 5, upper panel). This
antibody also recognized a more slowly migrating band in the
OA-treated pc cells. The levels of slowly migrated bands in the
PKR-K/R cells were low compared with that in the pc cells (Fig. 5). These slower migrated bands were
not detected in the extracts prepared from the unstimulated cells.
The slowly migrated band was a phosphorylated form of NF-κB
(31). Fig. 5 also shows that the
anti-phospho-Ser536 p65NF-κB antibody interacted with a 65-kDa band
and OA increased the staining intensity of the band in pc and
PKR-K/R cells. However, the staining intensity was higher in pc
cells than that in the PKR-K/R cells (Fig. 5, middle panel). The bound antibody
was stripped off the membrane and re-incubated with anti-β-actin
antibody for the loading controls (Fig. 5, lower panel).
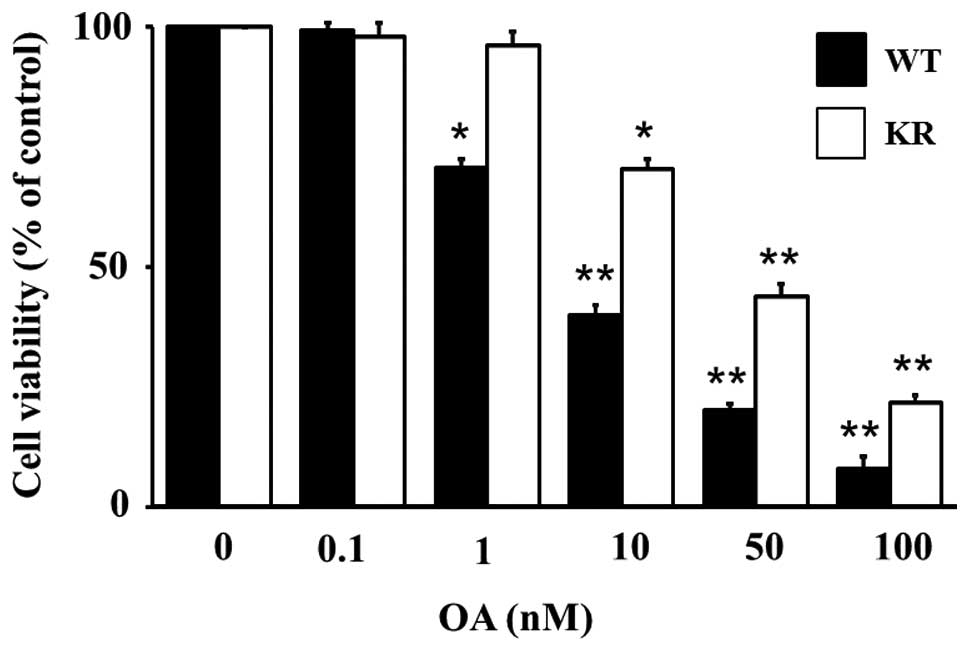
Apoptosis in MG63 and PKR-K/R cells
In our previous study, OA induced apoptosis in MG63
cells (7). We examined whether OA
could induce apoptosis in the PKR-K/R cells. To quantify the
OA-induced cytotoxicity in MG63 and PKR-K/R cells, the cells were
treated with various concentrations of OA for 24 h and the cell
viability was measured by the WST-8 assay. Fig. 6 shows that OA decreased the cell
viability in MG63 cells in a dose-dependent manner up to 100 nM. OA
at 10 nM decreased the cell viability to ∼40% that of the control
cells, whereas the viability of 50 nM OA-treated cells was 20% that
of the control cultures (Fig. 6).
OA also decreased the cell viability in PKR-K/R cells (Fig. 6). However, the level of cell
viability was higher in the PKR-K/R cells compared with that in the
MG63 cells (Fig. 6). The cell
viability of PKR-K/R cells treated with 50 nM OA was 40% that of
the control cells (Fig. 6).
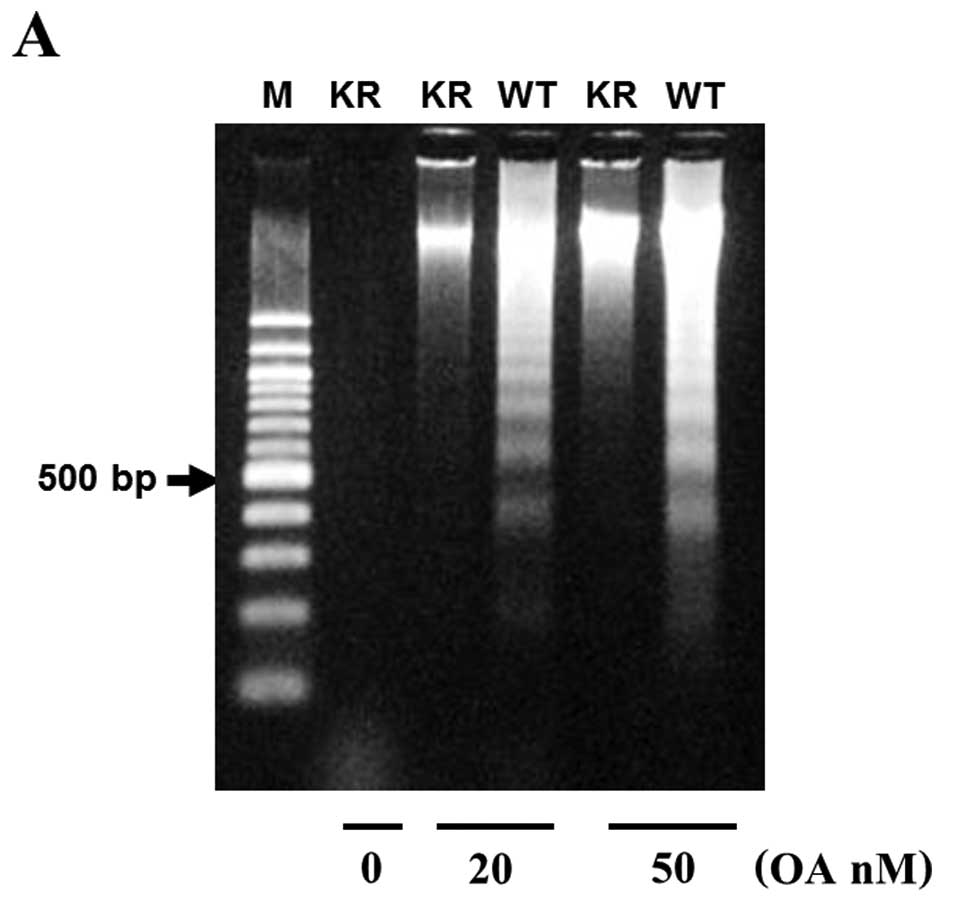
To determine if the OA-induced cell viability was
due to apoptosis, we looked for the presence of nuclear
fragmentation in MG63 and PKR-K/R cells treated with a low (20 nM)
or higher (50 nM) concentrations of OA for 48 h. The extracted DNA
was analyzed by agarose gel electrophoresis and stained with
ethidium bromide. In the 50 nM OA-treated MG63 cells, a DNA
fragmentation pattern forming a ladder of multiples of 185–200 bp
was observed (Fig. 7A). However,
DNA laddering pattern was minimum in the PKR-K/R cells treated with
the same concentration of OA. OA at 20 nM also stimulated the DNA
laddering pattern in MG63 cells however the same concentration of
OA did not induce DNA laddering in the PKR-K/R cells (Fig. 7A). We also evaluated the nuclear
fragmentation and condensation of chromatin in MG63 and PKR-K/R
cells by Hoechst staining. The control cells did not show any
apoptotic features in MG63 cells and PKR-K/R cells (Fig. 7Ba and c). In the 50 nM OA-treated
MG63 cells nucleic acid staining with Hoechst 33342 exhibited
typical apoptotic nuclei, which had highly fluorescent condensed
chromatin structures (Fig. 7Bb).
However, in the PKR-K/R cells, the number of the apoptotic cells
significantly decreased, although the cells still manifested
apoptotic features (Fig. 7Bd).
Discussion
We transfected a human cDNA having the amino acid
lysine at 296 replaced with Arginine in the catalytic domain of PKR
into human osteoblastic MG63 cells and established the stable cell
lines that express mutant gene construct (PKRK/R cells).
Phosphorylation of eIF-2α was not detected in the PKR-K/R cells
whereas strong phosphorylation of eIF-2α occurred in
pcDNA-transfected MG63 cells. Because eIF-2α is a substrate for PKR
(6,7), the mutant cells we established have a
PKR dominant-negative characteristics. OA stimulated the
phosphorylation of eIF-2α in MG63 cells, however, phosphorylation
of eIF-2α in PKR-K/R cells was not stimulated with the
OA-treatment.
We explored the effects of OA on the expression and
phosphorylation of IκBα and p65NF-κB in MG63 and PKR-K/R cells.
During the OA-treatment, the expression and phosphorylation of IκBα
increased in MG63 cells. IκBα was degraded upon OA treatment in
MG63 cells. We previously demonstrated that IκBα was phosphorylated
on tyrosine residues by the OA-treatment (36). In the present study IκBα was
phosphorylated at least on serine residues at 32 position, because
the anti-phospho IκBα (Ser32) recognized the phosphorylated form of
IκBα. We also detected the phosphorylation at Ser36 in MG63 cells
(data not shown).
OA is one of the many stimuli activating NF-κB in
the cultured cells. It has been reported that OA increased the
phosphorylation of cellular proteins (24). We previously reported that OA
induced activation of PKR/eIF-2α, nuclear translocation of p65NF-κB
and apoptosis in MG63 cells (5,7,31).
In human neutrophils and HL-60 cells, OA and orthovanadate, an
inhibitor of tyrosine phosphatase, stimulated the activation of
NF-κB and rapid degradation of IκBα (37). The NF-κB activation was caused by
the OA-induced inhibition of PKCδ and IKK phosphatases or by the
OA-induced activation of ERK1, a member of the MAP kinase family
(37). These reports indicate that
the phosphorylation and degradation of IκBα was influenced by
OA-sensitive phosphatases. However, it was reported that OA-induced
activation of NF-κB did not depend on the inhibitor properties of
OA but rather on the production of reactive oxygen intermediates
(38). The phosphorylation of IκBα
preceding its degradation occurs on the Ser32 and 36 residues,
making it a subject to degradation by proteosomes. These findings
consist with our present results. Degradation of IκBα liberates the
NF-κB complex, which is able to migrate to the nucleus and to
activate gene expression (39). In
our previous study we demonstrated that 100 nM OA-treatment induced
IκBα phosphorylation without causing its degradation. This
phosphorylation appears to occur on a tyrosine residue because
anti-phospho-tyrosine antibody bound to the samples
immunoprecipitated with the anti-IκBα antibody (36). This finding confirms an earlier one
that tyrosine phosphorylation of IκBα induced NF-κB activation
without its degradation (40). In
the present study, we demonstrated that 50 nM OA-treatment induced
phosphorylation of IκBα on Ser32 residue and degradation of IκBα.
The discrepancy of the results derived from the use of CHX to block
the new protein synthesis in the present study.
PKR is an interferon-induced protein, initially
identified and characterized as a translational inhibitor in an
antiviral pathway regulated by interferon (1,2). It
was reported that PKR could function as a signal transducer for
mediating transcriptional activation in response to dsRNA via its
ability to phosphorylate IκBα, resulting in the activation of NF-κB
(41). The relationships between
PKR and IκBα has not been reported previously. To provide a further
insight into the role of phosphorylation of IκBα in PKR pathway, we
used PKR-K/R cells stably expressing the dominant-negative PKR. PKR
was earlier found to be associated with IKK complex, where its
major contribution appears to be activation of NF-κB (35,41).
In our previous study, we demonstrated that the mutation of PKR
kinase resulted in the basal translocation of p65NF-κB into the
nucleus in the unstimulated cells (7,31).
This translocation was accompanied by the degradation of IκBα.
These results indicate that functional PKR is necessary for the
cytosolic localization of NF-κB and for the phosphorylation of IκBα
in the OA-stimulated cells. Expression of the PKR mutation is
associated with enhanced levels of NF-κB DNA-binding and
transcriptional activities compared with those of the control cells
(42), further supporting our
present results. Serine phosphorylation of IκBα and its degradation
did not require PKR kinase activity (41). We demonstrated that IκBα was
degraded without phosphorylation in PKR-K/R cells suggesting that
the kinase activity of PKR is required for the phosphorylation of
IκBα. PKR has Ser/Thr kinase activity; however, it also
phosphorylates tyrosine residue in place of Ser51 in eIF-2α
(42). The relationship between
kinase activity of PKR and other kinases, such as c-Src, remains to
be examined.
PKR is required to osteoblast differentiation,
osteoclast formation and chondrogenesis (43–46).
Phosphorylation of IκBα plays important roles in
osteoclastogenesis, osteoclast recruitment and osteolysis (45,47,48).
We previously reported that OA induced apoptosis in osteoblastic
cells (7,27–31).
During OA-induced apoptosis, the PKR/eIF-2α pathway was activated,
that activation was followed by the inhibition of protein synthesis
(7). Although the detailed
functions of PKR activity in osteoblastic apoptosis remain unclear,
it might be possible that PKR mediates the OA-induced apoptosis in
MG63 cells by phosphorylating IκBα and causing p65NF-κB
translocation. It still remains to be determined whether an
OA-sensitive phosphatase could regulate this pathway.
Acknowledgements
We thank Mrs. Eiko Sasaki for her
skillful technical assistance. This study was supported in part by
grants from the Grant-in-Aid for Scientific Research from the
Ministry of Education, Science, Sports and Culture of Japan (T.H.
and H.M.).
References
|
1
|
Sadler AJ and Williams BRG:
Interferon-inducible antiviral effectors. Nat Rev Immunol.
8:559–568. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pindel A and Sadler AJ: The role of
protein kinase R in the interferon responses. J Interferon Cytokine
Res. 31:59–70. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nallagatla SR, Toroney R and Bevilacqua
PC: Regulation of innate immunity through RNA structure and the
protein kinase PKR. Curr Opin Struct Biol. 21:119–127. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pfaller CK, Li Z, George CX and Samuel CE:
Protein kinase PKR and RNA adenosine deaminase ADR1: new roles for
old players as modulators of the interferon response. Curr Opin
Immunol. 23:573–582. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang QL, Zhou LY, Mu YQ, Zhou QX, Luo JY,
Cheng L, Deng ZL, He TC, Haydon RC and He BC: All-trans retinoic
acid inhibits tumor growth of human osteosarcoma by activating Smad
signaling-induced osteogenic differentiation. Int J Oncol.
41:153–160. 2012.PubMed/NCBI
|
|
6
|
de Haro C, Méndez R and Santoyo J: The
eIF-2α kinases and the control of protein synthesis. FASEB J.
10:1378–1387. 1996.
|
|
7
|
Morimoto H, Okamura H, Yoshida K, Kitamura
S and Haneji T: Okadaic acid induces apoptosis through
double-stranded RNA-dependent protein kinase/eukaryotic initiation
factor-2α pathway in human osteoblastic MG63 cells. J Biochem.
136:433–438. 2004.
|
|
8
|
Yeung MC, Liu J and Lau AS: An essential
role for the interferoninducible, double-stranded RNA-activated
protein kinase PKR in the tumor necrosis factor-induced apoptosis
in U937 cells. Proc Natl Acad Sci USA. 93:12451–12455. 1977.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Der SD, Yang YL, Weissmann C and Williams
BRG: A double-stranded RNA-activated protein kinase-dependent
pathway mediating stress-induced apoptosis. Proc Natl Acad Sci USA.
94:3279–3283. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kaufman RJ: Double-stranded RNA-activated
protein kinase mediates virus-induced apoptosis: A new role for an
old actor. Proc Natl Acad Sci USA. 96:11693–11695. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gil J and Esteban M: Induction of
apoptosis by the dsRNA-dependent protein kinase (PKR): Mechanism of
action. Apoptosis. 5:107–114. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Saelens X, Kalai M and Vandenabeele P:
Translation inhibition in apoptosis: caspase-dependent PKR
activation and eIF2-α phosphorylation. J Biol Chem.
276:41620–41628. 2001.
|
|
13
|
Szyszka R, Kudlicki W, Kramer G, Hardesty
B, Galabru J and Hovanessian A: A type 1 phosphoprotein phosphatase
active with phosphorylated Mr = 68,000 initiation factor
2 kinase. J Biol Chem. 264:3827–3831. 1989.PubMed/NCBI
|
|
14
|
Jammi NV and Beal PA: Phosphorylation of
the RNA-dependent protein kinase regulates its RNA-binding
activity. Nucleic Acids Res. 14:3020–3029. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tan SL, Tareen SU, Melville MW, Blakely CM
and Katze MG: The direct binding of the catalytic subunit of
protein phosphatase 1 to the PKR protein kinase is necessary but
not sufficient for inactivation and disruption of enzyme dimer
formation. J Biol Chem. 277:36109–36117. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jacobson MD, Weil M and Raff MC:
Programmed cell death in animal development. Cell. 88:347–354.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Haneji T: Association of protein
phosphatase 1 delta with nucleolin in osteoblastic cells and
cleavage of nucleolin in apoptosis-inducing osteoblastic cells.
Acta Histochem Cytochem. 38:1–8. 2005. View
Article : Google Scholar
|
|
18
|
Tanaka H, Yoshida K, Okamura H, Morimoto
H, Nagata T and Haneji T: Calyculin A induces apoptosis and
stimulates phosphorylation of p65 NF-κB in human osteoblastic
osteosarcoma MG63 cells. Int J Oncol. 31:389–396. 2007.PubMed/NCBI
|
|
19
|
Lin CC, Kuo CL, Lee MH, Lai KC, Lin JP,
Yang JS, Yu CS, Lu CC, Chiang JH, Chueh FS and Chung JG: Wogonin
triggers apoptosis in human osteosarcoma U-2 cells through the
endoplasmic reticulum stress, mitochondrial dysfunction and
caspase-3-dependent signaling pathways. Int J Oncol. 39:217–224.
2011.
|
|
20
|
Li B, Yang Y, Jiang S, Ni B, Chen K and
Jiang L: Adenovirus-mediated overexpression of BMP-9 inhibits human
osteosarcoma cell growth and migration through downregulation of
the PI3K/AKT pathway. Int J Oncol. 41:1809–1819. 2012.PubMed/NCBI
|
|
21
|
Savill J and Fadok V: Corpse clearance
defines the meaning of cell death. Nature. 407:784–788. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wyllie AH: Glucocorticoid-induced
thymocyte apoptosis is associated with endogenous endonuclease
activation. Nature. 284:555–556. 1980. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gong J, Traganos F and Darzynkiewicz Z: A
selected procedure for DNA extraction from apoptotic cells
applicable for gel electrophoresis and flow cytometry. Anal
Biochem. 218:314–319. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fernandez JJ, Candenas ML, Souto ML,
Trujillo MM and Norte M: Okadaic acid, useful tool for studying
cellular processes. Curr Med Chem. 9:229–262. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Goto K, Fukuda J and Haneji T: Okadaic
acid stimulates apoptosis through expression of Fas receptor and
Fas ligand in human oral squamous carcinoma cells. Oral Oncol.
38:16–22. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fujita M, Goto K, Yoshida K, Okamura H,
Morimoto H, Kito S, Fukuda J and Haneji T: Okadaic acid stimulates
expression of Fas receptor and Fas ligand by activation of nuclear
factor kappa-B in human oral squamous carcinoma cells. Oral Oncol.
40:199–206. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Morimoto Y, Ohba T, Kobayashi S and Haneji
T: The protein phosphatase inhibitors okadaic acid and calyculin A
induce apoptosis in human osteoblastic cells. Exp Cell Res.
230:181–186. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Morimoto H, Morimoto Y, Ohba T, Kido H,
Kobayashi S and Haneji T: Inhibitors of protein synthesis and RNA
synthesis protect against okadaic acid-induced apoptosis in human
osteosarcoma cell line MG63 cells but not in Saos-2 cells. J Bone
Miner Metab. 17:266–273. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kito S, Shimizu K, Okamura H, Yoshida K,
Morimoto H, Fujita M, Morimoto Y, Ohba T and Haneji T: Cleavage of
nucleolin and argyrophilic nucleolar organizer region associated
proteins in apoptosis-induced cells. Biochem Biophys Res Commun.
300:950–956. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Haneji T, Teramachi J, Hirashima K, Kimura
K and Morimoto H: Interaction of protein phosphatase 1δ with
nucleophosmin in human osteoblastic cells. Acta Histochem Cytochem.
45:1–7. 2012.
|
|
31
|
Ozaki A, Morimoto H, Tanaka H, Okamura H,
Yoshida K, Amorim BR and Haneji T: Okadaic acid induces
phosphorylation of p65NF-κB on serine 536 and activates NF-κB
transcriptional activity in human osteoblastic MG63 cells. J Cell
Biochem. 99:1275–1284. 2006.
|
|
32
|
Meurs E, Chong K, Galabru J, Thomas NSB,
Kerr IM, Williams BRG and Hovanessian AG: Molecular cloning and
characterization of the human double-stranded RNA-activated protein
kinase induced by interferon. Cell. 62:379–390. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Takizawa T, Ohashi K and Nakanishi Y:
Possible involvement of double-stranded RNA-activated protein
kinase in cell death by influenza virus infection. J Virol.
70:8128–8132. 1996.PubMed/NCBI
|
|
34
|
Ishii T, Kwon H, Hiscott J, Mosialos G and
Koromilas AE: Activation of the IκBα kinase (IKK) complex by
double-stranded RNA-binding defective and catalytic inactive
mutants of the interferon-inducible protein kinase PKR. Oncogene.
20:1900–1912. 2001.
|
|
35
|
Gil J, García MA, Gomez-Puertas P, Guerra
S, Rullas J, Nakano H, Alcamí J and Esteban M: TRAF family proteins
link PKR with NF-κB activation. Mol Cell Biol. 24:4502–4512.
2004.PubMed/NCBI
|
|
36
|
Morimoto H, Ozaki A, Okamura H, Yoshida K,
Kitamura S and Haneji T: Okadaic acid induces tyrosine
phosphorylation of IκBα that mediated by PKR pathway in human
osteoblastic MG63 cells. Mol Cell Biochem. 276:211–217. 2005.
|
|
37
|
Miskolci V, Castro-Alcaraz S, Nguyen P,
Vancura A, Davidson D and Vancurova I: Okadaic acid induces
sustained activation of NFκB and degradation of the nuclear IκBα in
human neutrophils. Arch Biochem Biophys. 417:44–52. 2003.
|
|
38
|
Schmidt KN, Traenckner EBM, Meier B and
Baeuerle PA: Induction of oxidative stress by okadaic acid is
required for activation of transcription factor NF-κB. J Biol Chem.
270:27136–27142. 1995.PubMed/NCBI
|
|
39
|
Karin M and Lin A: NF-κB at the crossroads
of life and death. Nat Immunol. 3:221–227. 2002.
|
|
40
|
Imbert V, Rupec RA, Livolsi A, et al:
Tyrosine phosphorylation of IκB-α activates NF-κB without
proteolytic degradation of IκB-α. Cell. 86:787–798. 1996.
|
|
41
|
Bonnet MC, Weil R, Dam E, Hovanessian AG
and Meurs EF: PKR stimulates NF-κB irrespective of its kinase
function by interacting with the IκB kinase complex. Mol Cell Biol.
20:4532–4542. 2000.
|
|
42
|
Lu J, O’Hara EB, Trieselmann BA, Romano PR
and Dever TE: The interferon-induced double-stranded RNA-activated
protein kinase PKR will phosphorylate serine, threonine, or
tyrosine at residue 51 in eukaryotic initiation factor 2α. J Biol
Chem. 274:32198–32203. 1999.PubMed/NCBI
|
|
43
|
Yoshida K, Okamura H, Amorim BR, Ozaki A,
Tanaka H, Morimoto H and Haneji T: Double-stranded RNA-dependent
protein kinase is required for bone calcification in MC3T3-E1 cells
in vitro. Exp Cell Res. 311:117–125. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yoshida K, Okamura H, Amorim BR, Hinode H,
Yoshida H and Haneji T: PKR-mediated degradation of STAT1 regulates
osteoblast differentiation. Exp Cell Res. 315:2105–2114. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Teramachi J, Morimoto H, Baba R, Doi Y,
Hirashima K and Haneji T: Double stranded RNA-dependent protein
kinase is involved in osteoclast differentiation of RAW264.7 cells
in vitro. Exp Cell Res. 316:3254–3262. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Morimoto H, Baba R, Haneji T and Doi Y:
Double-stranded RNA-dependent protein kinase regulates
insulin-stimulated chondrogenesis in mouse clonal chondrogenic
cells, ATDC-5. Cell Tissue Res. 35:41–47. 2013. View Article : Google Scholar
|
|
47
|
Abu-Amer Y, Dowdy SF, Ross FP, Clohisy JC
and Teitelbaum SL: TAT fusion proteins containing tyrosine
42-deleted IκBα arrest osteoclastogenesis. J Biol Chem.
276:30499–30503. 2001.
|
|
48
|
Clohisy JC, Roy BC, Biondo C, Frazier E,
Willis D, Teitelbaum SL and Abu-Amer Y: Direct inhibition of NF-κB
blocks bone erosion associated with inflammarory arthritis. J
Immunol. 171:5547–5553. 2003.
|