Introduction
Worldwide, hepatocellular carcinoma (HCC) is the
sixth most common cancer and the third most prevalent cause of
cancer-related mortality. HCC is also responsible for over 600,000
deaths annually (1). Although this
disease is most prevalent in Asia and Africa, the incidence of HCC
is increasing in Europe and the United States (2–5). Due
to its asymptomatic nature during the early stages, HCC is often
diagnosed at a late stage with a very poor prognosis. Although
tumor ablation surgery can be successfully performed in patients
with early stages of HCC, the recurrence rate is ∼50% within 3
years (6). Even with relatively
aggressive treatments such as systemic chemotherapy, radiotherapy
and trans-arterial chemoembolization, the 5-year relative survival
rate for patients with HCC is only 7% (7). Moreover, systemic therapy with
classical cytotoxic drugs has been reported to be poorly tolerated
and ineffective (8). In
particular, sorafenib, a compound globally approved for treating
unresectable and advanced cases of HCC, is associated with low
response rates and several unwanted side effects such as
hypertension, diarrhea and skin reactions on the hands and feet
(9,10). Therefore, an effective and
well-tolerated pharmaceutical agent for treating advanced HCC is
needed.
In recent years, studies of signaling pathways that
regulate cancer cell proliferation, angiogenesis, invasion and
metastasis have led to the identification of several possible
therapeutic targets. In particular, many clinical investigations
have indicated that the phosphatidylinositol 3-kinase (PI3K)/Akt
signaling pathway plays an integral role in the regulation of many
key cellular processes such as proliferation, growth, survival,
differentiation and metabolism. Additionally, this pathway is one
of the most commonly activated in various cancers (11–13).
PI3K generates a lipid secondary messenger, phosphatidylinositol
3,4,5-triphosphate (PIP3), from phosphatidylinositol 4,
5-diphosphate (PIP2) and then leads to Akt activation. Akt
subsequently phosphorylates mTOR and several other proteins. mTOR
is a serine/threonine protein kinase that exists as two functional
complexes: mTORC1 and mTORC2 (14,15).
This kinase promotes cell growth and cell cycle progression by
phosphorylating several translational regulators such as P70S6
kinase (P70S6K).
Abnormal activation of the PI3K/Akt pathway is
observed in ∼50% of patients with HCC (16). Additionally, Akt phosphorylation is
involved in early HCC recurrence and poor prognosis (17) and 23% of HCC patients have elevated
levels of Akt phosphorylation on Ser473 (18). Concurrently, total mTOR expression
has been found to be increased in 5% of HCC cases and elevated
levels of phosphorylated mTOR have been reported in 15% of HCC
patients. In addition, phosphorylated (p)-mTOR positively
correlates with increased expression of S6K, which is found in 45%
of HCC cases (19). For this
reason, the PI3K/Akt/mTOR pathway has emerged as a key therapeutic
target and seems to offer a promising strategy for treating patient
with HCC.
Although several PI3K/Akt pathway inhibitors have
been identified, clinical trials for these agents are only in the
early stages. In vivo applications have also been limited
due to problems with inhibitor stability, solubility and toxicity
(20–23). To discover a new structural class
of PI3K inhibitors, we used a pharmacophore-directed design. As a
result, we recently identified a novel scaffold belonging to the
imidazopyridine family that acts as a PI3K/mTOR inhibitor (24). Based on this finding, we designed,
synthesized and screened a new series of imidazo[1,2-a]pyridine
derivatives. Among these, a new compound,
N-(5-(3-(3-cyanophenyl)imidazo[1,2-a]pyridin-6-yl)pyridin-3-yl)benzenesulfonamide
(HS-159) was found to be the most potent PI3K inhibitor. In the
present study, we determined whether HS-159 has anticancer effects
against HCC and explored the molecular mechanism involved in this
process.
Materials and methods
In vitro PI3K activity assay
Active PI3Kα (100 ng) was pre-incubated with various
doses of HS-159 or LY294002 for 5 min in kinase reaction buffer (25
mM MOPS, pH 7.0; 5 mM MgCl2 and 1 mM EGTA) and 10
μg L-α-phosphatidylinositol. The L-α-phosphatidylinositol
was sonicated in water for 20 min before being added to the
reaction to facilitate micelle formation. The reaction was
initiated by adding 10 μM ATP and allowed to run for 180 min
at room temperature. To terminate the kinase reaction, an equal
volume of Kinase-Glo® buffer (Promega, Madison, WI) was
added. After 10 min, luminescence of the plates was measured with a
GloMax plate reader.
Cell lines
Human liver cancer (HepG2, Huh-7 and Hep3B) cells
were purchased from the Korean Cell Line Bank (KCLB, Seoul, Korea).
Huh-7 cells were cultured in Roswell Park Memorial Institute 1640
(RPMI-1640) medium with 10% fetal bovine serum (FBS) and 1%
penicillin/streptomycin. HepG2 and Hep3B cells were cultured in
Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10%
fetal bovine serum (FBS) and 1% penicillin/streptomycin. Human
umbilical vein endothelial cells (HUVECs) were grown in a
gelatin-coated 75-cm2 flask with M199 medium containing
20 ng/ml basic fibroblast growth factor (bFGF), 100 U/ml heparin
and 20% FBS. All cell cultures were maintained at 37°C in a
CO2 incubator with a controlled humidified atmosphere
composed of 95% air and 5% CO2.
Cell viability assay
Cell viability was evaluated using a
3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT)
assay. Briefly, cells were seeded at a density of 1×104
cells/well in a 96-well plate and incubated for 24 h. The medium
was then removed and the cells were treated with either DMSO as a
control or various concentrations (0.1–10 μM) of HS-159 for
48 h at 37°C. Next, 10 μl of MTT solution (2 mg/ml) was
added to each well and the plate was incubated for another 4 h at
37°C. The medium was then removed and the formazan crystals that
had formed were dissolved in DMSO (200 μl/well) with
constant shaking for 5 min. Absorbance of the solution was then
measured with a microplate reader at 540 nm. This assay was
conducted in triplicate.
Western blotting
Total cellular proteins were extracted with lysis
buffer containing 1% Triton X-100, 1% Nonidet P-40 and a 1%
protease and phosphatase inhibitor cocktail containing aprotinin
(10 mg/ml), leupeptin (10 mg/ml; ICN Biomedicals, Asse-Relegem,
Belgium), phenylmethylsulfonyl fluoride (1.72 mM), NaF (100 mM),
NaVO3 (500 mM) and
Na4P2O7 (500 mg/ml; Sigma-Aldrich,
St. Louis, MO). The proteins were separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto nitro-cellulose membranes. The blots were
incubated with the appropriate primary antibodies followed by
incubation with secondary antibodies conjugated to horseradish
peroxidase. Antibody binding was detected with an enhanced
chemiluminescence reagent (Amersham Biosciences). Antibodies
against p-mTOR (Ser2448), mTOR, p-Akt
(Ser473), Akt, p-P70S6K (Thr389), P70S6K,
PARP, cleaved caspase-3, hypoxia inducible factor-1α (HIF-1α),
vascular endothelial growth factor (VEGF) and β-actin were
purchased from Cell Signaling Technology Inc. (Danvers, MA).
Immunofluorescence microscopy
Huh-7 cells were plated on 18-mm cover glasses in
RPMI-1640 medium and incubated for 24 h at 37°C so that ∼70%
confluence was reached. The cells were then incubated in the
presence or absence of 10 μM HS-159, washed twice with PBS
and fixed in an acetic acid: ethanol solution (1:2, v/v) for 10 min
at −20°C. The cells were blocked in 1.5% horse serum in PBS for 30
min at room temperature and then incubated overnight at 4°C with
primary antibodies against p-Akt, p-mTOR and p-P70S6K (Cell
Signaling) in a humidified chamber. After washing twice with PBS,
the cells were then incubated with rabbit TRITC secondary antibody
(1:100, Dianova, Germany) for 1 h at room temperature. The cells
were also stained with 4, 6-diamidino-2-phenylindole (DAPI) to
visualize the nuclei. The slides were then washed twice with PBS
and covered with fluorescent mounting medium (Dako North America
Inc., Carpinteria, CA) before viewing with a confocal laser
scanning microscope (Olympus, Tokyo, Japan).
Cell cycle analysis
Huh-7 cells were plated in 100-mm culture dishes
with RPMI-1640 medium at ∼70% confluence and incubated for 24 h at
37°C. The next day, the cells were treated with either DMSO as a
control or various concentrations (0.01–10 μM) of HS-159.
Floating and adherent cells were collected and fixed overnight in
cold 70% ethanol at 4°C. After washing with PBS, the cells were
subsequently stained with 50 μg/ml propidium iodide (PI) and
100 μg/ml RNase A for 1 h at room temperature in the dark.
Flow cytometry was then performed with a FACSCalibur flow cytometer
(Becton-Dickinson, San Jose, CA) to determine the percentage of
cells in specific phases of the cell cycle (sub-G1,
G0/G1, S and G2/M). The data were
analyzed using FlowJo software (Tree Star, Inc.). All the
experiments were performed in triplicate.
DAPI staining and terminal
deoxynucleotidyltransferase-mediated nick end labeling (TUNEL)
Huh-7 cells were plated on an 18-mm cover glass with
RPMI-1640 medium at ∼70% confluence and incubated for 24 h at 37°C.
The cells were then incubated in the presence or absence of 10
μM HS-159 for 24 h. The cells were then fixed in cold 2%
para-formaldehyde (PFA), washed with PBS and stained with 2
μg/ml of DAPI (Sigma-Aldrich) for 20 min at 37°C. The
stained cells were examined under a fluorescence microscope for
evidence of nuclear fragmentation. A TUNEL assay was subsequently
performed using a commercial kit (Chemicon, Temecula, CA) according
to the manufacturer’s instructions.
Measurement of mitochondrial membrane
potential (ψm)
Mitochondrial membrane potential was assessed with a
mitochondria staining kit (MitoPT, Immunohistochemistry
Technologies, Bloomington, MN) using 5, 5’, 6, 6’-tetrachloro-1,
1’, 3, 3’-tetraethylbenzimidazol-carbocyanine iodide (JC-1), which
indicates potential-dependent accumulation in mitochondria. Huh-7
cells were plated on 18-mm cover glass in RPMI-1640 medium and
incubated for 24 h at 3°C. When confluence reached ∼70%, the cells
were incubated in the presence or absence of 10 μM HS-159 at
37°C for 6 h. Next, 100 μl of a JC-1 solution (12.5
μg/ml final concentration) was added to each well and the
plate was incubated for 30 min at 3°C. After washing twice with
PBS, the cells were stained with DAPI to visualize the nuclei. The
slides were then washed twice with PBS and covered with DABCO
before viewing with a confocal laser scanning microscope (Olympus).
The results [green/red (G/R) fluorescence ratio] were expressed as
a percentage of the control.
Capillary tube formation assay
Matrigel (10 mg/ml, 200 μl; BD Biosciences)
was polymerized for 30 min at 37°C. HUVECs were suspended at a
density of 2.5×105 cells/well in M199 medium
supplemented with 5% FBS. Aliquots (0.2 ml) of the cell suspension
were added to each well coated with Matrigel and incubated with or
without different concentrations of HS-159 for 24 h at 37°C.
Morphological changes of the cells and tube formation were observed
with a phase-contrast microscope. The cells were captured at
magnification ×200.
Wound migration assay
HUVECs were plated on 60-mm culture dishes at ∼90%
confluence. A razor blade was then used to create a wound 2 mm in
width in the cell mono-layer and the injury line was marked. After
wounding, the detached cells were removed with serum-free medium
and the remaining HUVECs were further incubated in M199 medium
supplemented with 5% FBS, 1 mM thymidine (Sigma-Aldrich) and HS-159
(0.5 μM). The cells were allowed to migrate for 18 h before
being rinsed with serum-free medium and fixed in absolute methanol.
Cell migration was quantitated by counting the number of cells that
had moved beyond the injury line.
Statistical analysis
Data are expressed as the mean ± standard deviation
(SD). Statistical analysis was performed using an ANOVA and
unpaired Student’s t-test. P-values ≤0.05 were considered
statistically significant. All calculations were performed using
SPSS software for the MS Windows operating system (version 10.0;
SPSS, Chicago, IL).
Results
HS-159 inhibits PI3K activity
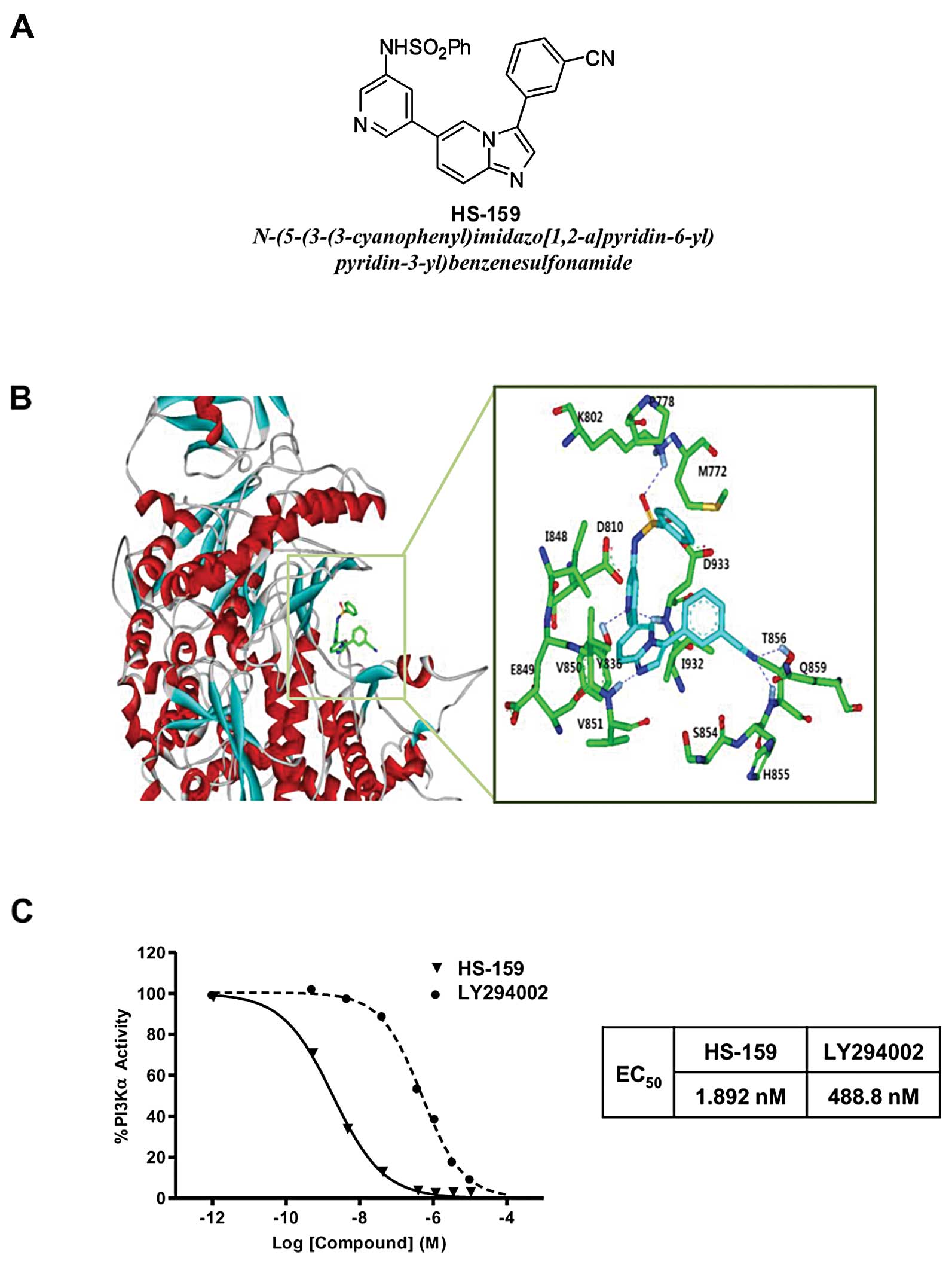
A new PI3Kα inhibitor, HS-159
(N-(5-(3-(3-cyanophenyl)imidazo[1,2-a]pyridin-6-yl)pyridin-3-yl)benzenesulfonamide)
was synthesized as described in our previous study (Fig. 1A) (25). Three-dimensional (3D) coordinates
in the X-ray crystal structure of PI3Kα in the resting form (PDB
code: 2RD0) were selected as the receptor model for docking
simulations. After removing the solvent molecules, hydrogen atoms
were added to each protein atom. We used the Auto Dock program for
studying docking between PI3Kα and HS-159 because the
outperformance of its scoring function over those of the others had
been shown in several target proteins. The predicted binding mode
of HS-159 in the ATP-binding site of PI3Kα is shown in Fig. 1B. The imidazopyridine of HS-159
maintained stable hydrogen-bonding in the hinge region (Val851).
The pyridyl group was anchored by hydrogen bonds to Asp933 and
interacted with gatekeeper residue Tyr836. Two stable hydrogen
bonds were established between the cyano group of C3 and Thr856.
These hydrogen bonds were received from the enzyme and seemed to
play a crucial role as an anchor for HS-159 binding. HS-159 could
be further stabilized in the ATP-binding site by forming
hydrophobic interactions with Met772, Pro778, Lys802, Val850,
Ser854, Gln859 and Ile932. Judging from the overall structural
features derived from our docking simulations, the inhibitory
activity of HS-159 is likely to stem from multiple hydrogen bonds
and hydrophobic interactions simultaneously established in the
ATP-binding site of PI3Kα. We also found that HS-159 more strongly
inhibited PI3Kα activity (EC50 1.892 nM) compared to
LY294002, a conventional specific PI3K inhibitor (EC50
488.8 nM, Fig. 1C).
HS-159 inhibits the PI3K/Akt signaling
pathway in Huh-7 cells
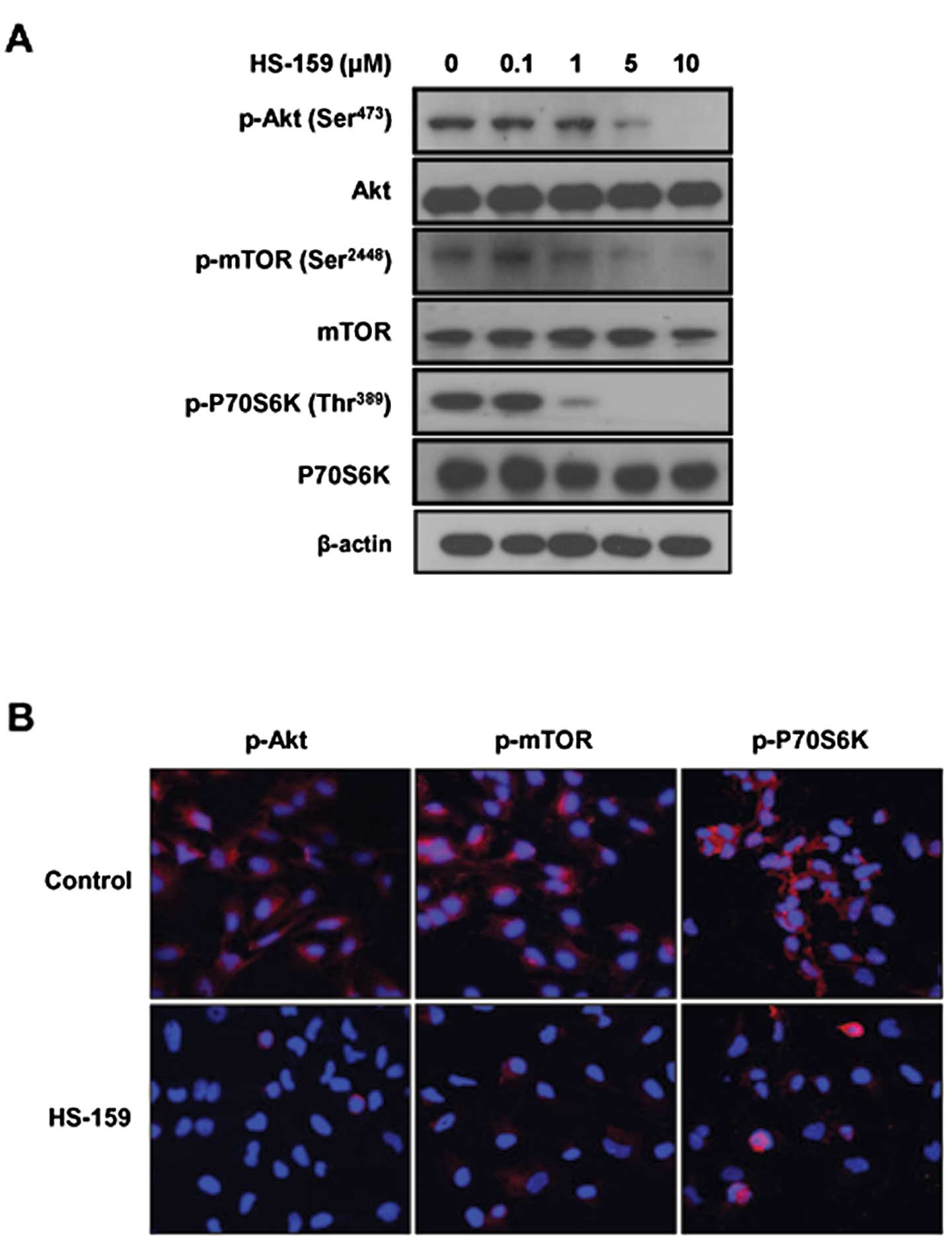
Since HS-159 inhibited PI3Kα activity, we next
determined whether this drug also affected downstream signal
transduction factors that promote PI3K-mediated cell survival. When
Huh-7 cells were treated with various concentrations of HS-159 for
6 h, the phosphorylation of Akt and its downstream components such
as mTOR and P70S6K was effectively suppressed in a dose-dependent
manner (Fig. 2A). We further
assessed the effect of HS-159 on Akt, mTOR and P70S6K using a
fluorescent imaging system. Results from this assay again showed
that HS-159 strongly suppressed phosphorylation of these proteins
in Huh-7 cells, similar to the western blotting results (Fig. 2B).
HS-159 inhibits the growth of HCC
cells
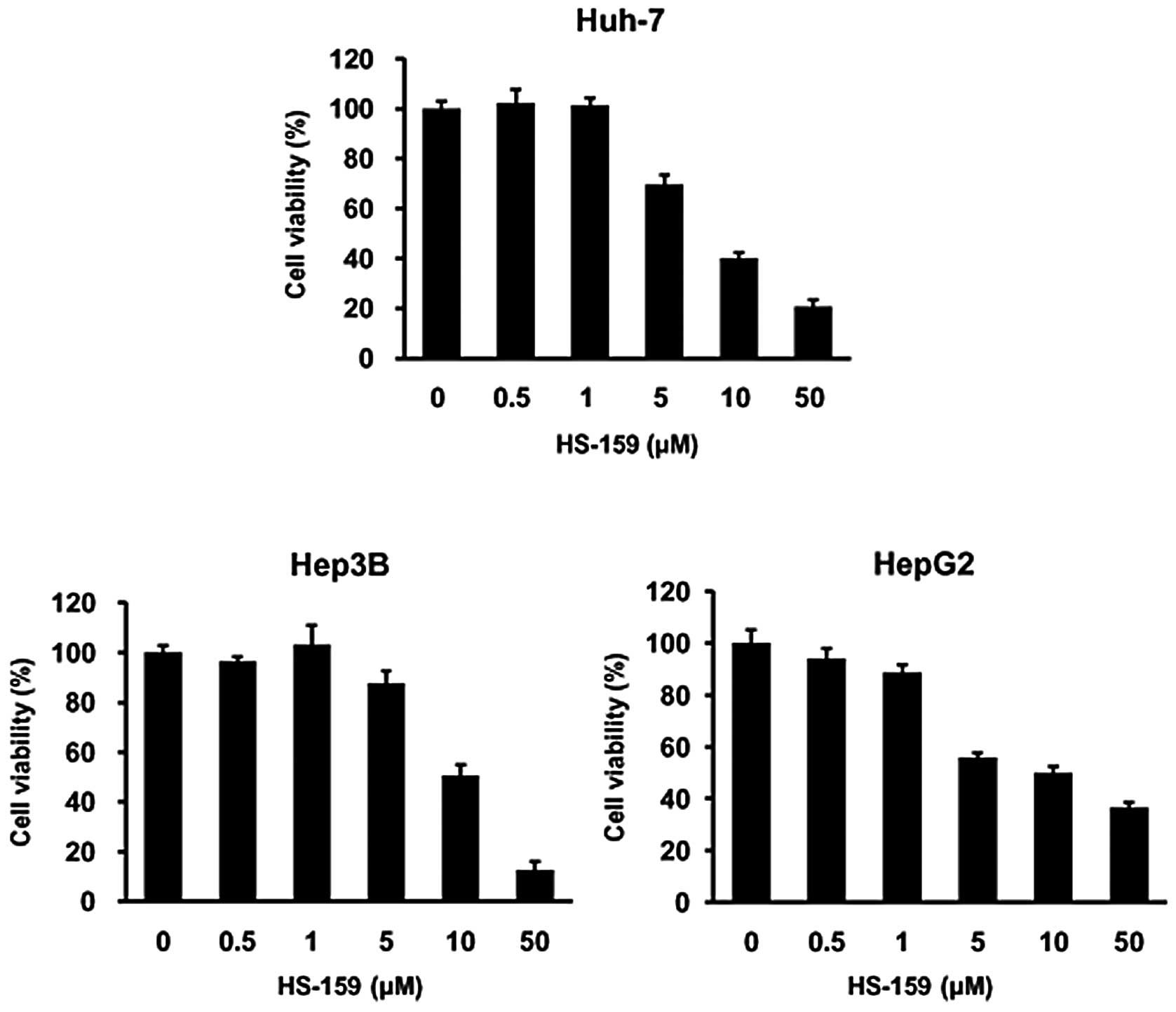
Next, we further evaluated the anti-proliferative
effects of HS-159 on HCC cells using an MTT assay. Huh-7, HepG2 and
Hep3B cells were exposed to various concentrations (0.1–10
μM) of HS-159 for 48 h. HS-159 markedly reduced viability of
the three HCC cell lines in a dose-dependent manner (Fig. 3). These results indicated that
HS-159 exerts potent anti-proliferative effects on HCC cells.
HS-159 induces apoptosis in Huh-7
cells
Cell apoptosis and growth are correlated with cell
cycle progression (26). In the
present study, HS-159 (0.1-10 μM) inhibited the growth of
HCC cells in a dose-dependent manner (Fig. 3). Thus, we performed flow cytometry
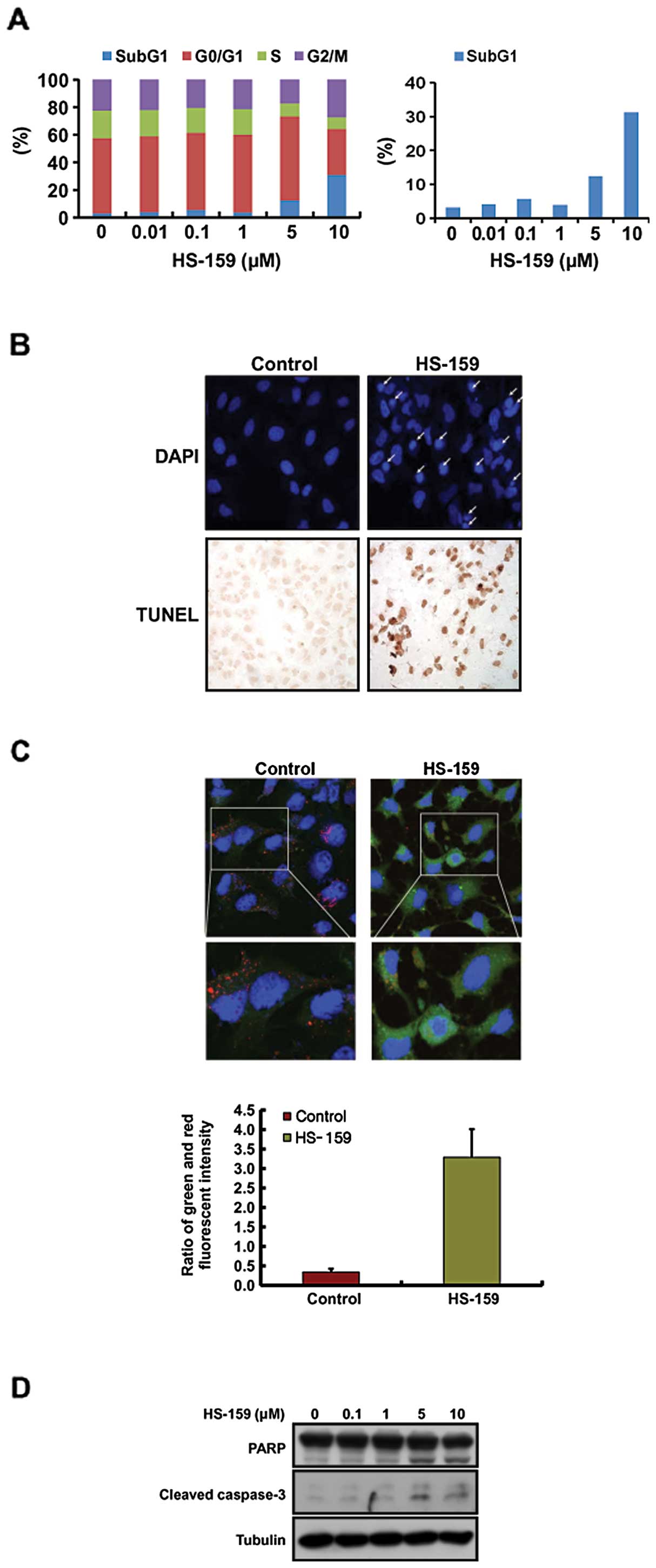
to monitor changes of the cell cycle profile induced by HS-159.
Data from this analysis revealed that treatment with 5 and 10
μM HS-159 increased the number of cells in a
sub-G1 stage, indicative of apoptosis and decreased the
fraction of cells in the G0/G1 phase in Huh-7
cells (Fig. 4A).
HS-159-induced apoptosis was also evaluated by DAPI
and TUNEL staining to characterize nuclear morphology. As shown in
Fig. 4B, cells treated with 10
μM HS-159 had morphological features characteristic of
apoptotic cells such as bright nuclear condensation and perinuclear
apoptotic bodies observed with DAPI staining. HS-159-induced
apoptosis was also confirmed by detecting DNA fragmentation using a
TUNEL assay. In addition, we performed JC-1 staining to measure the
effect of HS-159 on changes in mitochondrial membrane potential
that correlate with the intrinsic pathway of apoptosis. As shown in
Fig. 4C, the cytoplasm of control
cells showed heterogeneous staining with both red and green
fluorescence coexisting in the same cells. Consistent with
mitochondrial localization, red fluorescence (corresponding to high
mitochondrial membrane potential, ψm) was mostly found
in granular structures distributed throughout the cytoplasm.
Treatment with 10 μM HS-159 decreased the red fluorescence
while frequent clusters of mitochondria were still observed. HS-159
induced marked changes in ψm as evident from the
disappearance of red fluorescence or increased green fluorescence
in most cells. We next performed western blotting to evaluate the
activation of caspase-3 and PARP after treatment with HS-159. As
expected, HS-159 increased the levels of cleaved caspase-3 and PARP
in Huh-7 cells in a dose-dependent manner (Fig. 4D). Taken together, these results
showed that HS-159 induces apoptotic cell death in Huh-7 cells.
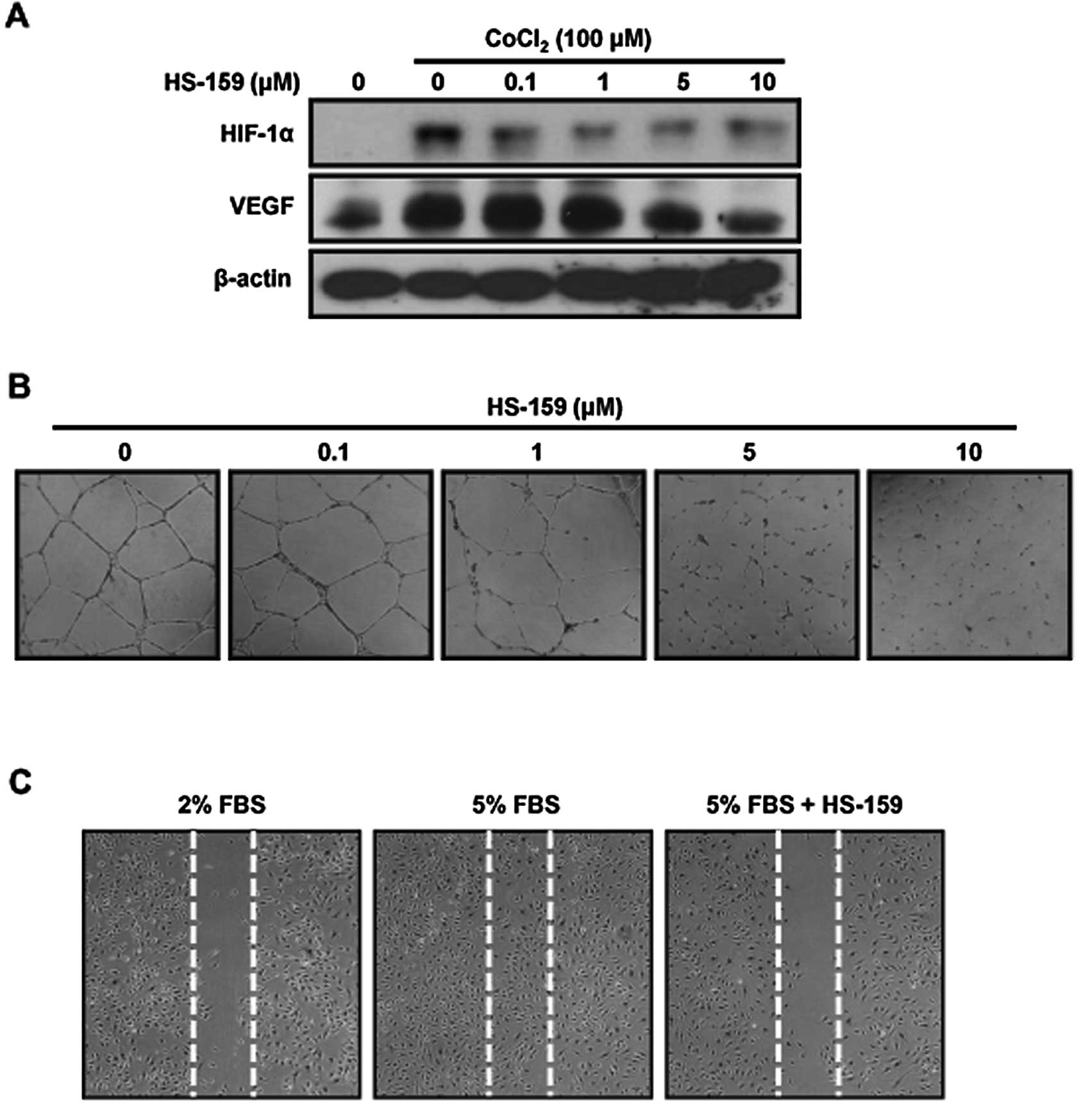
HS-159 inhibits the expression of HIF-1α
and VEGF in Huh-7 cells
A low oxygen level is characteristic of solid tumors
and a negative prognostic factor for cancer patient survival. The
response of cancer cells to hypoxia not only drives
neo-angiogenesis, but also enhances cell survival and malignant
phenotype development. HIF-1α is a major regulator of cellular
adaptive responses to hypoxia as well as a major transcriptional
modulator of angiogenic factors such as VEGF (27). We therefore evaluated the effect of
HS-159 on the expression of HIF-1α and VEGF in Huh-7 cells under
hypoxia conditions. The cells were treated with various
concentrations of HS-159 cultured for 16 h with 100 μM
CoCl2 for mimic hypoxic conditions. As shown in Fig. 5A, the expression of HIF-1α and VEGF
was increased after exposure to hypoxia and subsequently suppressed
by HS-159 treatment, respectively.
HS-159 suppresses angiogenesis
In endothelial cells, PI3K/Akt signaling actively
mediates processes associated with angiogenesis including cell
migration and capillary tube formation (28). Therefore, we performed capillary
tube formation and migration assays to determine whether HS-159
suppresses the angiogenic activity of HUVECs. HS-159 inhibited the
formation of vessel-like structures resulting from HUVEC elongation
and alignment (Fig. 5B). HS-159
also markedly inhibited cell migration (Fig. 5C). These results indicate that
HS-159 is a potential anti-angiogenic reagent that inhibits the
tube formation and migration of HUVECs.
Discussion
Many clinical studies have indicated that
hyper-activation of the PI3K/Akt pathway plays a critical role in
the pathogenesis of various human cancers. Consequently, the
PI3K/Akt pathway has emerged as an important therapeutic target for
anticancer drug development. Several inhibitors targeting this
pathway have been recently developed and some are being evaluated
in clinical trials (29,30). These compounds have a wide variety
of targets and, include pan-PI3K, isoform-specific PI3K, dual
PI3K-mTOR, mTOR catalytic site and Akt inhibitors (23,31–33).
Nevertheless, an optimal therapeutic strategy for targeting the
PI3K/Akt pathways in cases of HCC has not been identified. In an
effort to develop potent PI3K inhibitors, we screened a large
number of novel imidazopyridine derivatives. Among them HS-159
acted as a specific PI3K inhibitor, which more strongly inhibited
PI3Kα activity (EC50 1.892 nM) than LY294002, a
conventional PI3K-specific inhibitor (EC50 488.8 nM). In
addition it significantly suppressed proliferation and angiogenesis
as well as induced apoptosis by blocking the PI3K/Akt signaling
pathway in HCC cells.
Akt is considered the most important signaling
factor in the PI3K pathway as it regulates various substrates
affecting processes that control cell growth and survival (34). It was also reported that Akt
phosphorylation on Ser473 is associated with poor
prognosis in patients with different types of solid tumors
including those in the pancreas, liver, prostate and breast
(35). In our western blotting and
immunofluorescent studies, HS-159 was found to effectively suppress
the phosphorylation of downstream PI3K effectors including Akt,
mTOR and P70S6K dose-dependently in HCC cells. Considering that the
PI3K/Akt pathway is important for cell growth and progression, we
also tested whether HS-159 inhibited the survival of three HCC cell
lines. HS-159 inhibited the growth of all three cell lines in a
dose-dependent manner. Thus, we suggest that HS-159 prevents cell
growth by blocking the PI3K/Akt pathway in HCC cells.
Cell cycle arrest in tumor cells is a major
indicator of anticancer activity. Additionally, cell cycle
dysregulation has been implicated in tumor development and
proliferation characteristic of different cancers, including HCC
(30). Thus, we analyzed cell
cycle changes following treatment with HS-159. We found that HS-159
induced cell cycle arrest during the G0/G1
phase. We also observed that the anti-proliferative activity of
HS-159 was associated with the induction of apoptosis in Huh-7
cells. This was indicated by elevated levels of cleaved caspase-3
and PARP as well as increased number of TUNEL-positive cells. The
alteration of mitochondrial membrane potential induces the release
of cytochrome c from mitochondria into the cytosol and is
associated with caspase-3 activation (36). We therefore monitored changes of
mitochondrial membrane potential following treatment with HS-159.
Our results showed that treatment with HS-159 disrupted the
mitochondrial membrane potential in Huh-7 cells. Overall, these
data suggest that HS-159 induced apoptotic cell death through
activation of the intrinsic apoptotic pathway.
Rapid tumor cell proliferation in a stable vascular
system leads to the development of hypoxia in almost all solid
tumors (26). Because the liver is
a highly vascular organ that depends on angiogenesis for cellular
regeneration, angiogenesis may be specifically targeted as a novel
therapeutic approach for treating HCC (37). Hypoxia in many solid tumors
increases the expression of major angiogenic factors such as HIF-1α
and VEGF to promote neo-angiogenesis (38,39).
Additionally, it has been reported that PI3K signaling regulates
HIF-1α and VEGF expression (40,41).
We thus assessed the effect of HS-159 on expression of these two
angiogenic factors. As expected, HS-159 significantly reduced the
expression of both HIF-1α and VEGF under hypoxic conditions induced
by CoCl2 in Huh-7 cells. The activation of endothelial
cells by angiogenic processes results in cell proliferation and
migration as well as cord tube formation for the creation of new
microvessels (42). We performed
tube formation and migration assays to study the effect of HS-159
on HUVECs. HS-159 significantly inhibited HUVEC tube formation as
well as migration. Overall, our data suggest that HS-159 may be a
potent anti-angiogenic agent.
In conclusion, our study demonstrated that HS-159 is
a specific PI3K inhibitor and exerts potent anticancer effects
against HCC cells. Additionally, blocking the PI3K/Akt pathway with
HS-159 inhibited cell proliferation and angiogenesis and induced
apoptosis of HCC cells. These finding suggest that HS-159 is a
novel anticancer agent with the potential for treating patients
with HCC and other diseases associated with dyregulated PI3K
signaling.
Acknowledgements
This study was supported by the Korean
Health Technology R&D Project (A110862, A120266), Ministry of
Health and Welfare, the National Research Foundation of Korea (NRF)
funded by the Ministry of Education, Science and Technology
(NRF-2012-0002988, 2012R1A2A2A01045602, 2011-0016436, 2011-0020322,
2012-0003009).
References
|
1.
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar
|
|
2.
|
Poon RT, Fan ST, Lo CM, Liu CL and Wong J:
Intrahepatic recurrence after curative resection of hepatocellular
carcinoma: long-term results of treatment and prognostic factors.
Ann Surg. 229:216–222. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
El-Serag HB and Mason AC: Rising incidence
of hepatocellular carcinoma in the United States. N Engl J Med.
340:745–750. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Llovet JM and Bruix J: Novel advancements
in the management of hepatocellular carcinoma in 2008. J Hepatol.
48(Suppl 1): S20–S37. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Bruix J and Sherman M: Management of
hepatocellular carcinoma. Hepatology. 42:1208–1236. 2005.
View Article : Google Scholar
|
|
6.
|
Mulcahy MF: Management of hepatocellular
cancer. Curr Treat Options Oncol. 6:423–435. 2005. View Article : Google Scholar
|
|
7.
|
Bosch FX, Ribes J, Diaz M and Cleries R:
Primary liver cancer: worldwide incidence and trends.
Gastroenterology. 127:S5–S16. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Verslype C, Van Cutsem E, Dicato M, Arber
N, Berlin JD, Cunningham D, De Gramont A, Diaz-Rubio E, Ducreux M,
Gruenberger T, Haller D, Haustermans K, Hoff P, Kerr D, Labianca R,
Moore M, Nordlinger B, Ohtsu A, Rougier P, Scheithauer W, Schmoll
HJ, Sobrero A, Tabernero J and van de Velde C: The management of
hepatocellular carcinoma. Current expert opinion and
recommendations derived from the 10th World Congress on
Gastrointestinal Cancer, Barcelona, 2008. Ann Oncol. 20(Suppl 7):
vii1–vii6. 2009. View Article : Google Scholar
|
|
9.
|
Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S,
Kim JS, Luo R, Feng J, Ye S, Yang TS, Xu J, Sun Y, Liang H, Liu J,
Wang J, Tak WY, Pan H, Burock K, Zou J, Voliotis D and Guan Z:
Efficacy and safety of sorafenib in patients in the Asia-Pacific
region with advanced hepatocellular carcinoma: a phase III
randomised, double-blind, placebo-controlled trial. Lancet Oncol.
10:25–34. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Kim R, Byrne MT, Tan A and Aucejo F: What
is the indication for sorafenib in hepatocellular carcinoma? A
clinical challenge. Oncology. 25:283–295. 2011.PubMed/NCBI
|
|
11.
|
Yap TA, Garrett MD, Walton MI, Raynaud F,
de Bono JS and Workman P: Targeting the PI3K-AKT-mTOR pathway:
progress, pitfalls and promises. Curr Opin Pharmacol. 8:393–412.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Ward CS, Venkatesh HS, Chaumeil MM,
Brandes AH, Vancriekinge M, Dafni H, Sukumar S, Nelson SJ, Vigneron
DB, Kurhanewicz J, James CD, Haas-Kogan DA and Ronen SM:
Noninvasive detection of target modulation following
phosphatidylinositol 3-kinase inhibition using hyperpolarized 13C
magnetic resonance spectroscopy. Cancer Res. 70:1296–1305. 2010.
View Article : Google Scholar
|
|
13.
|
Fry MJ: Phosphoinositide 3-kinase
signalling in breast cancer: how big a role might it play? Breast
Cancer Res. 3:304–312. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Dunlop EA and Tee AR: Mammalian target of
rapamycin complex 1: signalling inputs, substrates and feedback
mechanisms. Cell Signal. 21:827–835. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Sparks CA and Guertin DA: Targeting mTOR:
prospects for mTOR complex 2 inhibitors in cancer therapy.
Oncogene. 29:3733–3744. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Nakanishi K, Sakamoto M, Yamasaki S, Todo
S and Hirohashi S: Akt phosphorylation is a risk factor for early
disease recurrence and poor prognosis in hepatocellular carcinoma.
Cancer. 103:307–312. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Boyault S, Rickman DS, de Reyniès A,
Balabaud C, Rebouissou S, Jeannot E, Hérault A, Saric J, Belghiti
J, Franco D, Bioulac-Sage P, Laurent-Puig P and Zucman-Rossi J:
Transcriptome classification of HCC is related to gene alterations
and to new therapeutic targets. Hepatology. 45:42–52. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Sahin F, Kannangai R, Adegbola O, Wang J,
Su G and Torbenson M: mTOR and P70 S6 kinase expression in primary
liver neoplasms. Clin Cancer Res. 10:8421–8425. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Powis G, Bonjouklian R, Berggren MM,
Gallegos A, Abraham R, Ashendel C, Zalkow L, Matter WF, Dodge J,
Grindey G and Vlahos CJ: Wortmannin, a potent and selective
inhibitor of phosphatidylinositol-3-kinase. Cancer Res.
54:2419–2423. 1994.PubMed/NCBI
|
|
21.
|
Vlahos CJ, Matter WF, Hui KY and Brown RF:
A specific inhibitor of phosphatidylinositol 3-kinase,
2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol
Chem. 269:5241–5248. 1994.PubMed/NCBI
|
|
22.
|
West KA, Castillo SS and Dennis PA:
Activation of the PI3K/Akt pathway and chemotherapeutic resistance.
Drug Resist Updat. 5:234–248. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Workman P: Inhibiting the phosphoinositide
3-kinase pathway for cancer treatment. Biochem Soc Trans.
32:393–396. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Hu XT and Owens MA: Multiplexed protein
quantification in maize leaves by liquid chromatography coupled
with tandem mass spectrometry: an alternative tool to immunoassays
for target protein analysis in genetically engineered crops. J
Agric Food Chem. 59:3551–3558. 2011. View Article : Google Scholar
|
|
25.
|
Kim O, Jeong Y, Lee H, Hong SS and Hong S:
Design and synthesis of imidazopyridine analogues as inhibitors of
phosphoinositide 3-kinase signaling and angiogenesis. J Med Chem.
54:2455–2466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Malumbres M, Pevarello P, Barbacid M and
Bischoff JR: CDK inhibitors in cancer therapy: what is next? Trends
Pharmacol Sci. 29:16–21. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Semenza GL: Expression of
hypoxia-inducible factor 1: mechanisms and consequences. Biochem
Pharmacol. 59:47–53. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Shiojima I and Walsh K: Role of Akt
signaling in vascular homeostasis and angiogenesis. Circ Res.
90:1243–1250. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Sommers I and Baskin D: Sex, race, age and
violent offending. Violence Vict. 7:191–201. 1992.PubMed/NCBI
|
|
30.
|
O’Brien C, Wallin JJ, Sampath D,
GuhaThakurta D, Savage H, Punnoose EA, Guan J, Berry L, Prior WW,
Amler LC, Belvin M, Friedman LS and Lackner MR: Predictive
biomarkers of sensitivity to the phosphatidylinositol 3’ kinase
inhibitor GDC-0941 in breast cancer preclinical models. Clin Cancer
Res. 16:3670–3683. 2010.
|
|
31.
|
Yang L, Dan HC, Sun M, Liu Q, Sun XM,
Feldman RI, Hamilton AD, Polokoff M, Nicosia SV, Herlyn M, Sebti SM
and Cheng JQ: Akt/protein kinase B signaling inhibitor-2, a
selective small molecule inhibitor of Akt signaling with antitumor
activity in cancer cells overexpressing Akt. Cancer Res.
64:4394–4399. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Schultz RM, Merriman RL, Andis SL,
Bonjouklian R, Grindey GB, Rutherford PG, Gallegos A, Massey K and
Powis G: In vitro and in vivo antitumor activity of the
phosphatidylinositol-3-kinase inhibitor, wortmannin. Anticancer
Res. 15:1135–1139. 1995.PubMed/NCBI
|
|
33.
|
Rowinsky EK: Targeting the molecular
target of rapamycin (mTOR). Curr Opin Oncol. 16:564–575. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
LoPiccolo J, Granville CA, Gills JJ and
Dennis PA: Targeting Akt in cancer therapy. Anticancer Drugs.
18:861–874. 2007.
|
|
35.
|
Ashe PC and Berry MD: Apoptotic signaling
cascades. Prog Neuropsychopharmacol Biol Psychiatry. 27:199–214.
2003. View Article : Google Scholar
|
|
36.
|
Belozerov VE and Van Meir EG: Hypoxia
inducible factor-1: a novel target for cancer therapy. Anticancer
Drugs. 16:901–909. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Hamedi M, Wigenius J, Tai FI, Björk P and
Aili D: Polypeptide-guided assembly of conducting polymer
nanocomposites. Nanoscale. 2:2058–2061. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Ben-Shoshan M, Amir S, Dang DT, Dang LH,
Weisman Y and Mabjeesh NJ: 1alpha,25-dihydroxyvitamin D3
(Calcitriol) inhibits hypoxia-inducible factor-1/vascular
endothelial growth factor pathway in human cancer cells. Mol Cancer
Ther. 6:1433–1439. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
López-Lázaro M: Hypoxia-inducible factor 1
as a possible target for cancer chemoprevention. Cancer Epidemiol
Biomarkers Prev. 15:2332–2335. 2006.PubMed/NCBI
|
|
40.
|
Zhong H, Chiles K, Feldser D, Laughner E,
Hanrahan C, Georgescu MM, Simons JW and Semenza GL: Modulation of
hypoxia-inducible factor 1alpha expression by the epidermal growth
factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human
prostate cancer cells: implications for tumor angiogenesis and
therapeutics. Cancer Res. 60:1541–1545. 2000.
|
|
41.
|
Mazure NM, Chen EY, Laderoute KR and
Giaccia AJ: Induction of vascular endothelial growth factor by
hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt
signaling pathway in Ha-ras-transformed cells through a hypoxia
inducible factor-1 transcriptional element. Blood. 90:3322–3331.
1997.
|
|
42.
|
Papetti M and Herman IM: Mechanisms of
normal and tumor-derived angiogenesis. Am J Physiol Cell Physiol.
282:C947–C970. 2002. View Article : Google Scholar : PubMed/NCBI
|