Introduction
Head and neck squamous cell carcinoma (HNSCC) arises
at the oral cavity, oropharynx, larynx or hypopharynx and is the
sixth leading cancer by incidence worldwide (1). A subgroup of HNSCCs, particularly
oropharyngeal carcinoma is caused by infection with high-risk types
of human papillomavirus (HPV) (2,3).
Compared with the HPV-negative tumors caused by heavy tobacco and
alcohol use, the incidence of HPV-positive tumors has been recently
reported to be strongly associated with sexual behavior, which is
the predominant means of HPV transmission (4–6). The
incidence of HPV-related oropharyngeal tumors has been increasing
since the early 1990s in the United States and Western Europe, but
the underlying reasons for this rapid increase are unclear
(6,7). Human papillomavirus is a circular and
double-stranded DNA virus. The viral genome encodes two regulatory
proteins (E1 and E2), three oncoproteins (E5, E6 and E7) and two
structural capsid proteins (L1 and L2) (8). The E6 oncoprotein forms complexes
with a cellular E3 ubiquitin ligase (E6-associated protein; E6AP)
and p53 protein, resulting in p53 degradation (9,10).
The E7 oncoprotein binds to pRb family members and disrupts their
ability to form complexes with E2F, increased expression of
E2F-responsive genes, many of which are required for cell cycle
progression (11,12). The E5 oncoprotein cooperates with
E6 and E7 to promote proliferation of infected cells and is likely
to facilitate malignant progression (13). However, the E5 coding sequence is
frequently deleted from the episomal viral DNA during integration
into the host genome (14).
Several recent studies have demonstrated that there
are two distinct HNSCC etiologic risk groups: those who develop
cancer in association with tobacco and alcohol and those who
develop HNC as a result of HPV infection (2–4,15).
The clinical outcomes after treatment with cisplatin and radiation
therapy were significantly better in the patients with HPV-positive
oropharyngeal carcinoma compared with those with HPV-negative
carcinomas (16–18). Chemicals found in smoke, the major
carcinogens responsible for HNSCC, are known to produce specific
types of guanine nucleotide trans-version in crucial genes, such as
the p53 tumor suppressor gene involved in the development of HNSCC
(19). The p53 gene is mutated in
up to half of HNSCCs (20,21), which are not infrequently found in
HPV-positive oropharyngeal cancers (OPCs) (2,22).
A subset of the HPV-positive OPC patients with a
history of extensive smoking have worse clinical outcomes than most
HPV-positive OPC patients, resembling the clinical course in
HPV-negative OPC patients. Considering that these patients
overexpress EGFR and Bcl-xL and have a higher rate of
TP53 mutation, it was proposed that HPV status alone is not an
adequate prognostic marker for classifying patient groups (23). Based on these conflicting findings,
the influence of tobacco in the development of HPV-associated HNSCC
should be elucidated.
Given that the available research data was obtained
from in vitro and in vivo HPV-positive tumor models
unrelated to smoking history, it is necessary to apply the
appropriate experimental models in order to understand a role of
HPV in OPCs coexisting of HPV16 and p53 mutation. In this study, we
investigated how tumor biology and molecular genetic mechanisms
change when HPV-negative OPC cell lines bearing two different
subtypes of TP53 mutations are transfected with HPV E6 and E7
oncogenes in vitro.
Materials and methods
Cell lines and culture conditions
Two HPV16-negative human squamous tongue cancer cell
lines (YD8 and YD10B) and Caski cells (an HPV16-positive human
squamous cervical cancer cell line) were obtained from the Korean
Cell Line Bank (KCLB, Korean Cell Line Bank, Seoul, Korea). Both of
the tongue cancer cell lines harbor p53 mutations, whereas the YD8
cell line has non-disruptive mutation that causes histidine to be
substituted for by arginine at codon 273 in exon 8, the YD10B cell
line has disruptive mutation that causes stop codon to be
substitutes for tyrosine at codon 236 in exon 7 (27). The cell lines were cultured in
RPMI-1640 (Welgene, Seoul, Korea) supplemented with 10%
heat-inactivated fetal bovine serum (FBS; Gibco BRL, Grand Island,
NY, USA), 1% penicillin/streptomycin (Gibco BRL), 10 mmol/l HEPES
(Amresco Inc., Solon, OH, USA) at 37°C in a humidified incubator
with 5% CO2.
Transfection with HPV16 E6 and E7
oncogenes
We used both the HPV16 E6 and E7 oncogenes coding
regions based on the sequences available from the HPV type 16
complete genome (GenBank: K02718.1). Both of the HPV16 E6 and HPV16
E7 oncogenes were amplified by polymer chain reaction (PCR). Primer
sequences used to amplify a 776-bp PCR product were
5′-ATGCACCAAAAGAGAACTGC-3′ (sense) and 5′-TTCTGGTTTCTGAGAACAGAT-3′
(anti-sense). The PCR product was resolved in a 1.5% agarose gel
and observed under ultraviolet light by staining with ethidium
bromide after electrophoresis. We isolated a 776-bp DNA fragment
containing HPV16 E6 and HPV16 E7 sequences and then cloned this
in-frame within the CT-GFP Fusion TOPO®
(pcDNA3.1/CT-GFP-TOPO) of the mammalian expression vector pGFP
(Invitrogen, Carlsbad, CA, USA), producing plasmid HPV16 E6E7. The
resulting plasmids were purified by using a Plasmid Midi kit
(Qiagen, Valencia, CA, USA) according to the manufacturer’s
instructions and the presence of the correct inserts was confirmed
by DNA sequencing (Cocmogenetech, Seoul, Korea).
For transfections, the YD8 and YD10B cells were
plated in 6-well plates at a density of 1×103 cells per
well and allowed to grow overnight to 80–90% confluency. The
following day, the cells were transfected with the mixture of 5
μg plasmid DNA (the target sequence inserted plasmid
HPV16-E6E7 and negative control plasmid) and 1.5 μg of Xfect
polymer nanoparticle (Clontech, Mountain View, CA, USA) in 2 ml of
serum-free medium according to the manufacturer’s instructions.
Four hours later, the medium was replaced by fresh growth medium.
Cells were then incubated at 37°C in 5% CO2 in
humidified chambers for 24 h. Transfectants were then selected
using G418 antibiotic (Abm, BC, Canada), added dropwise to the
culture medium to final concentrations ranging from 100 to 400
μg/ml (YD8-E6E7, YD10B-E6E7). As negative controls, we used
cells transfected with CT-GFP Fusion TOPO vector alone (YD8-V,
YD10B-V).
Western blotting
Cells were harvested with trypsin/EDTA, washed twice
with PBS and lysed with RIPA cell lysis buffer (Gibco BRL) that
contained a protease inhibitor cocktail (Amresco). Protein (30
μg) from each cell type was used in Bio-Rad
detergent-compatible protein assays (Bio-Rad Laboratories Inc.,
Hercules, CA, USA); proteins were resolved on 8–12% polyacrylamide
gels using standard sodium dodecyl sulfate polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred onto polyvinylidene
difluoride (PVDF) membranes (0.45 μm; Millipore Corp.,
Billerica, MA, USA). Membranes were blocked with 5% skim milk
(Becton-Dickinson, NJ, USA). Blots were then probed with the
following antibodies; total Rb (Cell Signaling Technology Inc.,
Danvers, MA, USA), phospho-Rb (Ser807/811) (Cell
Signaling Technology Inc.), E2F-1 (Cell Signaling Technology Inc.),
p16 INK4A (Cell Signaling Technology Inc.), CDK4 (Cell Signaling
Technology Inc.), phospho-p53(Ser392) (Epitomics Inc.,
Burlingame, CA, USA), cyclin D1 (Cell Signaling Technology),
phospho-PTEN(Ser380) (Millipore Corp.), STAT-1
(Epitomics) and GAPDH (Abcam, Cambridge, MA, USA). Horseradish
peroxidase (HRP)-conjugated secondary antibodies were purchased
from Santa Cruz (Santa Cruz Biotechnology Inc., Santa Cruz, CA,
USA). Blots were developed with an enhanced chemiluminescence
reagent (Amersham Pharmacia Biotech Inc., Piscataway, NJ, USA) and
detected using the LAS 3000 Image analyzer system (Fujifilm, Tokyo,
Japan).
For western blot analysis, cells were harvested and
analyzed for the expression of STAT-1 and IGF-1R. Total protein
lysates were obtained and western blotting was performed as
described previously. The antibodies recognized rabbit monoclonal
STAT-1 (Epitomics) and rabbit polyclonal IGF-1R antibody
(Epitomics). Protein expression was normalized against GAPDH
expression (Abcam). Images were acquired with the LAS 3000 Image
analyzer system (Fujifilm) and analyzed using the software provided
by the manufacturer.
Cell proliferation and invasion
Cell proliferation was measured using the
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) assay. Cells were plated in a 96-well plate at
2×103 cells per well. For 5 days, cells were incubated
with 10 μl of MTS/phenazine methosulfate (PMS) reagent for 4
h at 37°C in a 5% CO2 incubator. Following incubation in
MTS, viable cells were counted every day by reading the absorbance
at 490 nm using enzyme-linked immunosorbent assay (ELISA) reader
(Spectra Max 250; Molecular Devices, Sunnyvale, CA, USA). Cell
viability was calculated using Excel (Microsoft, Albuquerque, NM,
USA) and expressed as the percentage of MTS absorption: % survival
= (mean experimental absorbance/mean control absorbance) × 100.
Data represent the mean ± SD.
The migratory potential of cells was evaluated using
a 24-well format insert with 8-μm pores (Becton-Dickinson,
Franklin Lakes, NJ, USA). For the invasion assay, 1×105
cells in serum-free medium were added to each upper insert
pre-coated with matrigel matrix (BD, NJ, USA) and 750 μl 10%
FBS medium was added to the matched lower chamber. After 24-h
incubation, cells that remained in the upper chamber were removed
from the upper surface of the transwell membrane with a cotton swab
and migrated to the bottom of the upper membrane surface were fixed
in methanol, stained with Diff-Quik™, captured and counted. For
migration assay, the procedures were similar, except that
1×105 cells were added into the inserts without matrix
gel pre-coated. Five random fields at ×200 magnification for each
insert were counted. Inserts were conducted in triplicate in three
separate experiments. The percentage of invasion was calculated as
the mean number of cells invading through matrigel insert
membrane/mean of cell migrating through control insert membrane x
100. Invasion was expressed as the invasion index, which was
calculated as % invasion by HPV16 E6E7 transfected cells/% invasion
by vector-alone cells.
Cell cycle analysis
Cells were seeded at 5×105 in 100-mm
plastic dishes (Techno Plastic Products AG) and incubated for 72 h.
The cells were trypsinized, washed twice with PBS and harvested by
centrifugation. Briefly, cells were fixed with ice cold 70% ethanol
for ≥1 h, centrifuged, washed twice in cold PBS, resuspended in 1
ml PBS and stained with propidium iodide (PI) solution (0.05 mg/ml
PI, 10 mg/ml RNase A) for 20 min at 37°C in the dark. The
fluorescence intensity was measured using a flow cytometer
(FACSCalibur; Becton-Dickinson Biosciences, San Jose, CA, USA); at
least 1×104 cells were counted and DNA contents were
analyzed using CellQuest software (Becton-Dickinson, Franklin
Lakes, NJ, USA). All experiments were performed in triplicate. Data
represent the mean ± SD. Statistically significant differences
between the control and treatment groups were accepted at
P<0.01.
RNA isolation and cDNA microarray
analysis
Total RNA was isolated from all the cells grown to
90% confluency using the TRIzol reagent (Invitrogen) according to
the manufacturer’s instructions. Briefly, total RNA was extracted
from cell lysate by phase separation with chloroform and RNA
precipitation with isopropanol. After washing with 70% alcohol, the
RNA was eluted in RNase-free water. Total RNA was quantified using
a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies Inc.,
Wilmington, DE, USA). A quality control test of total RNA was
performed using the Experion™ system (Bio-Rad). Total RNA was
cleaned up using Ambion columns (Illumina Total-Prep RNA
Amplification kit, Ambion). Microarray analysis was performed using
an Illumina HumanHT-12 v4 Sentrix Expression BeadChip (Illumina,
San Diego, CA, USA). After hybridization of the biotinylated cRNA
to the chips, the chips were scanned according to the standard
protocol (Illumina). The arrays were scanned on the Illumina
BeadArray reader, a confocal-type imaging system with 532 (Cy3) nm
laser illumination. Data from each sample was extracted with Genome
Studio software (Illumina) using default parameters.
Quantitative real-time PCR (qRT-PCR)
array
For the qRT-PCR array, we selected the Human p53
Signaling Pathway RT2 Profiler™ PCR Array (PAHS-027;
Qiagen) for 84 genes relative cell proliferation, cell cycle,
apoptosis. Total RNA was isolated from YD8-HPV and YD8-V cells
grown to 90% confluency using the RNeasy Mini kit (Qiagen)
according to the manufacturer’s instructions. Total RNA was
quantified using a NanoDrop ND-1000 Spectrophotometer (NanoDrop
Technologies Inc.). Reverse transcription was performed using the
RT2 First Strand kit (Qiagen) as described by the
manufacturer and carried out with RT2 Fast SYBR® Green
qPCR Mastermix (Qiagen) using a Bio-Rad CFX96 system (Bio-Rad). The
cycling conditions comprised 10-min enzyme activation at 95°C,
followed by 40 cycles at 95°C for 15 sec, 55°C for 30 sec and 72°C
for 30 sec. The complete data set obtained from the array analysis
upload Excel Spreadsheet at http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php
(SABiosciences, Qiagen) and threshold cycle (Ct) value for each
gene was used to calculate the fold-change in levels. Five
housekeeping genes were included on the array to normalize the cDNA
amounts: β-actin (ACTB), β-glucuronidase (GUSB),
glyceraldehyde-3 phosphate dehydrogenase (GAPDH), heat shock
protein 90 kDa α class B member 1 (Hsp90ab1) and
hypoxanthine guanine phosphoribosyl transferase 1 (HPRT1).
The formula used to calculate the relative gene expression level
was (2−ΔCt). ΔCt = Ct (GOI) - avg. [Ct (HKG)], where GOI
is the abundance of each gene and HKG are the housekeeping genes
chosen from the ‘YD8-E6E7 Gene - YD8-V Gene’ worksheet. With the
use of appropriate cut-off criteria, a 2-fold induction or
repression of expression was considered to represent significantly
up- or downregulated gene expression.
Quantitative real-time PCR (qRT-PCR)
Quantitative real-time reverse transcriptase-PCR
(qRT-PCR) was employed to validate genes that were differentially
expressed by cDNA Microarray and qRT-PCR array. Synthesis of cDNA
was performed with the Maxime RT PreMix kit (iNtRON Biotechnology,
Korea) using 1 μg of RNA in the reaction. FastStart
Universal SYBR Green master mix (Roche, Mannheim, Germany) was
added to the RT products and PCR was performed using a Bio-Rad
CFX96 system (Bio-Rad). The primer pairs used for STAT-1 were
5′-CAAAGTCATGGCTGCTGAGA-3′ (forward) and 5′-AGGAAAACTGTCGCCAGAGA-3′
(reverse), whereas those for IGF-1R were 5-TGGAGTGCTGTATGCCTCTG-3′
(forward) and 5′-TGATGACCAGTGTTGGCTGG-3′ (reverse). The
amplification program for all primer sets was 95°C for 5 min,
followed by 40 cycles at 95°C for 30 sec, 60°C for 30 sec and 72°C
for 1 min.
Assays were performed in accordance with the
manufacturer’s instructions and the mRNA levels were normalized
relative to levels of GAPDH transcripts. Relative expression
levels of the mRNAs were calculated using the 2−ΔΔCT
values. Statistical analyses were performed using
Microsoft® Excel®. The average of triplicate real-time
PCR measurements was used to calculate the mean induction ratio ±
SD for each gene.
Immunohistochemistry
Detection of HPV16-DNA was previously reported by
in situ hybridization (ISH) methods (48). Samples were collected from 139
patients who underwent curative surgery for squamous cell carcinoma
of the head and neck (HNSCC) in Seoul St. Mary’s Hospital between
1994 and 2009. The sites of HNSCC tumors included buccal mucosa (4
cases; 3%), tongue (66 cases; 47%), floor of the mouth (4 cases;
3%), soft palate (3 cases; 2%), tonsil (59 cases; 42%), oropharynx
(2 cases; 1%) and uvula (1 case; 1%). To construct the tissue
microarray block, tissue cylinders with a diameter of 2.0 mm, were
taken from non-necrotic, morphologically representative areas of
paraffin-embedded tumor tissues. Tissue cores from each specimen
were assembled on a recipient paraffin block using a manual tissue
arrayer (Quick-Ray Manual Tissue Microarrayer, Unitma Co. Ltd.,
Seoul, Korea). After construction, 4-μm sections were cut
and stained with hematoxylin-eosin staining on the initial slide
for histological verification. Rabbit monoclonal anti-STAT1
(Epitomics) and rabbit polyclonal anti-IGF-1R (Epitomics) were used
for immunohistochemical staining. Paraffin sections (4 μm)
from samples were deparaffinized in 100% xylene and re-hydrated in
an ethanol series of decreasing concentrations of aqueous ethanol
using standard protocols. Antigen was performed using the heat
induced epitope retrieval method (HIER) in 0.01 M citrate buffer
(pH 6.0). Endogenous peroxidase activity was blocked by immersion
in 3% hydrogen peroxide in methanol for 10 min, followed by
overnight incubation with rabbit monoclonal anti-STAT1 (1:200) and
rabbit polyclonal anti-IGF-1R (1:100) at 4°C. After washing, the
sections were incubated with polymer-conjugated horseradish
peroxidase (HRP) for 10 min at room temperature. The peroxidase
reaction was developed using 3,3-diaminobenzidine chromogen
solution in diaminobenzidine (DAB) buffer substrate using polink-2
plus DAB detection kit (Two-step polymer-HRP detection system,
biotin-free) (D43-15; Life Science Division, Mukiteo, WA, USA).
Following incubation, the sections were visualized with DAB and
counterstained with hematoxylin, mounted in neutral gum and
analyzed using a bright field microscope.
The results were interpreted by a pathologist who
was blinded to the specific diagnosis and prognosis for each case.
The percentage of positive tumor cells was scored as follows: 0, no
tumor cells stained; 1, 1–5% of cells stained; 2, 5–20% of cells
stained; 3, 21–50% of cells stained; 4, 51–75% of cells stained;
and 5, >75% of cells stained. The intensity of staining was
scored as follows: 0, no staining; 1, low staining; 2, moderate
staining; and 3, high staining. The immunoreactive score was
calculated by multiplying the percentage of positive cells (scored
0–5) by staining intensity (scored 0–3). Tumors with an
immunoreactive score were considered positive for STAT and IGF-1R
expression. The total score was calculated by summing the
percentage of positive cells and staining intensity values. For
statistical analysis, a final staining was scored as follows: 0,
negative; 1–4, low expression; 5 and 6, 8 and 9, intermediate
expression; 10 and 12 and 15, high expression.
Statistical analysis
Data were evaluated for statistical significance by
analysis using Student’s t-test. A statistically significant
difference was considered to be significant at P<0.05 or
P<0.01. All experiments were performed independently at least
three times and the data presented are from a representative
experiment. The results are presented as mean ± SD.
Results
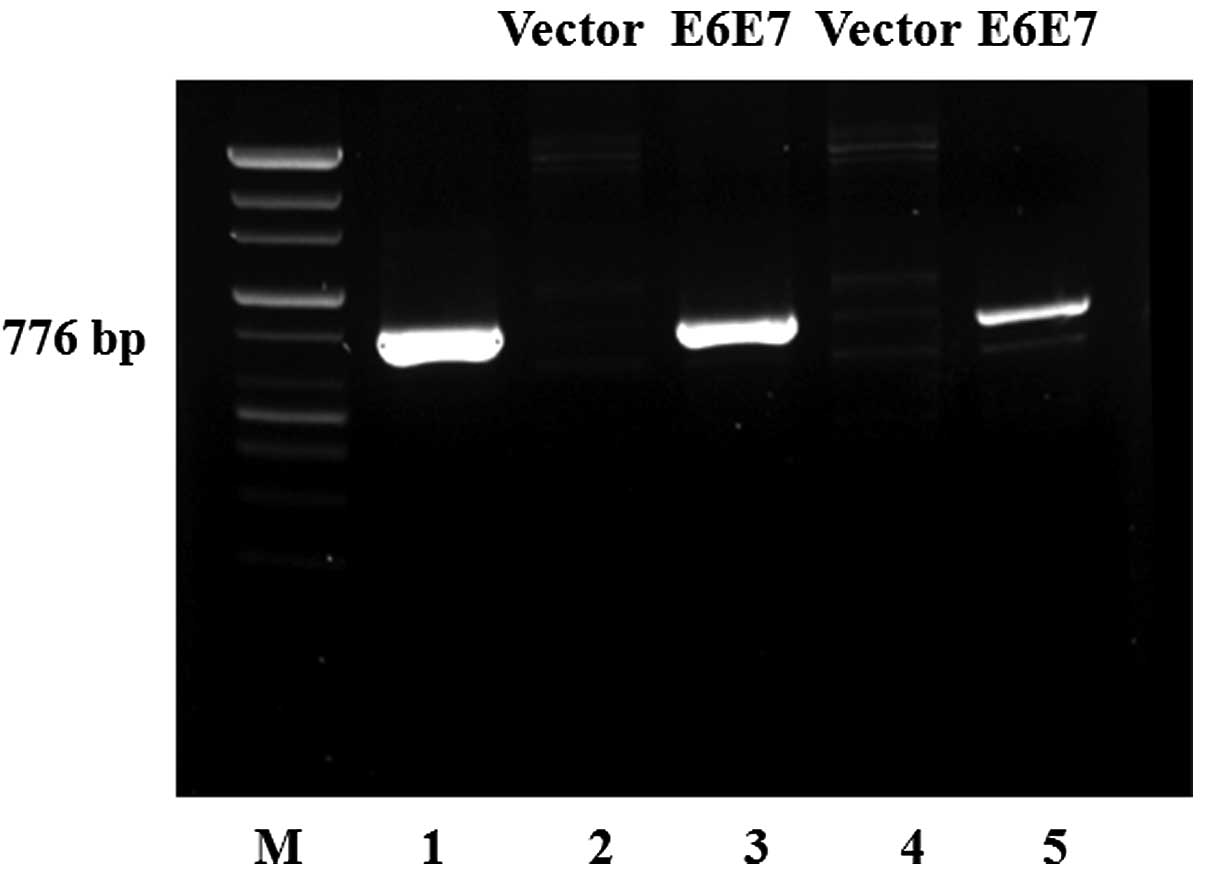
Expression of the E6E7 gene in two cancer
cell lines transfected with HPV16 E6 and E7 oncogenes
We confirmed the E6E7 DNA amplifications of the
stable YD8 and YD10B cell lines transfected with HPV-16 E6 and E7
oncogenes. We performed PCR analysis using one primer pair specific
to the HPV16 E6 and E7 oncogenes. Both YD8- and YD10B-E6E7 cells
expressed the E6E7 gene. Neither YD8- nor YD10B-V expressed the
E6E7 gene. We used Caski cell line as positive control (Fig. 1).
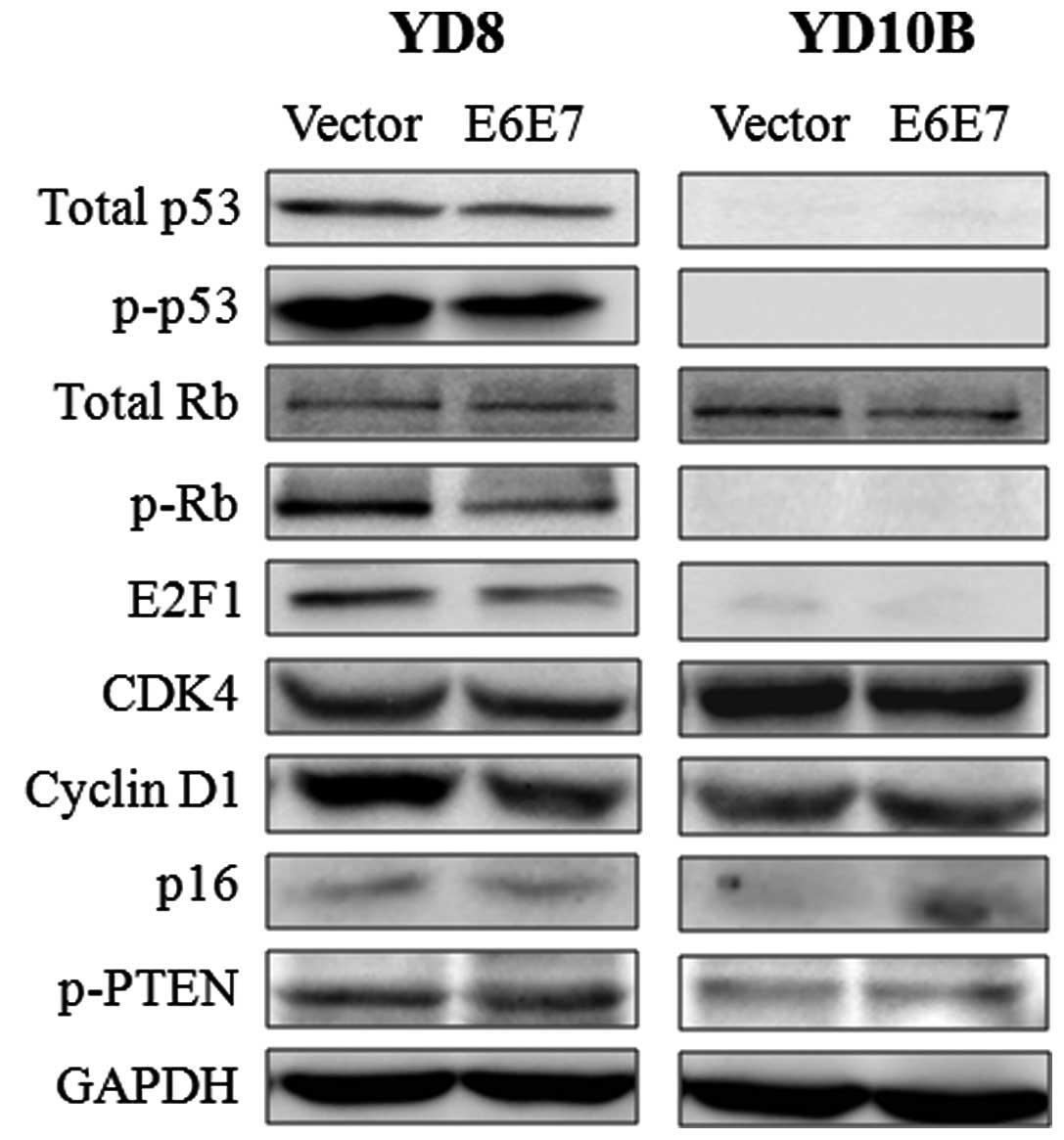
Expression of p53- and Rb-related
proteins
We next examined the biochemical responses of genes
related to the p53 and Rb pathways in two cancer cell lines
transfected with HPV16 E6 and E7 oncogenes. Although p53 protein
was abundant in YD8 cells, levels were lower in YD8-E6E7 cells than
YD8-V cells (40.6%). However, no expression of total p53 and p53
was observed in YD10B cells. Additionally, the levels of expression
of pRb and E2F-1 were substantially lower in YD8-E6E7 cells than in
YD8-V cells (38 and 33.7%) and were barely evident in either
YD10B-E6E7 or YD10B-V cells. The level of expression of cyclin D1
was lower in YD8-E6E7 than in YD8-V cells (25.6%), but there was no
difference in expression of cyclin D1 between YD10B-E6E7 and
YD10B-V. The protein level of p-PTEN, total Rb and CDK4 were not
considerably different in HPV16 E6E7 transfected cells and vector
alone cells (Fig. 2).
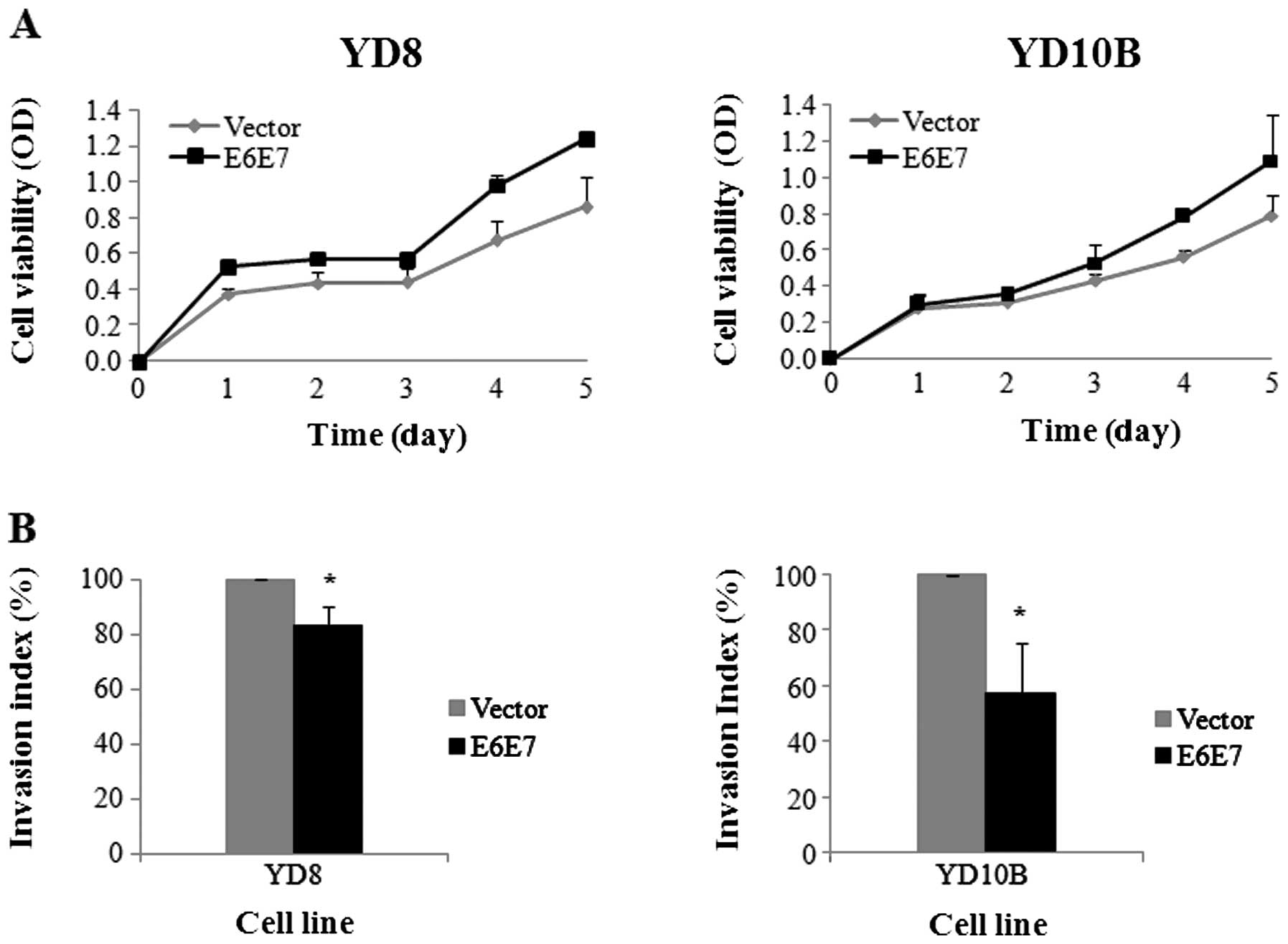
Differences in cell viability and
invasive capacities of the two classes of transfected cells
To test our hypothesis that HPV infection alters the
proliferative potential of oropharyngeal cancer cells, we compared
cell proliferation activity in HPV16 E6E7 transfected cells and
cells transfected with vector alone using the
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
(MTS) assay, which was carried out on days 0, 1, 2, 3, 4 and 5. The
rate of proliferation of YD8-E6E7 cells was higher than that of
YD8-V cells (43.9%, P=0.09) and YD10B-E6E7 cell proliferated at a
rate higher than YD10B-V (38.1%, P=0.26).
Proliferation of cells transfected with HPV16 E6E7
was higher than in cells transfected with vector alone. Especially,
in YD8-E6E7 cells, cell proliferation increased rapidly from day 1
and then, gradually increased from day 2 to day 3 in YD10B cells
(Fig. 3A).
Cells were seeded in the upper parts of the
transwells. Invasion activity was expressed as an invasion index,
which was calculated as the percentage of initial cell numbers
attached to the bottom of a matrigel-coated membrane after 24 h. As
shown in Fig. 3, we observed that
invasion activity was significantly reduced in transfected cells
with E6E7 compared with cells transfected with vector alone. The
invasion activity of YD8-E6E7 cells was lower than that of YD8-V
cells (17.1%, P=0.02) and invasion activity of YD10B-E6E7 was lower
than that of YD10B-V cells (42.7%, P=0.05) (Fig. 3B).
Changes in cell cycle distributions
Flow cytometry was used to show that the cell cycle
distribution of cells transfected with HPV16 E6E7 was significantly
different from that of cells transfected with vector alone.
Compared with YD8-V values, the proportion of YD8-E6E7 cells in the
G0/G1 phase was lower (66.1±31.5 vs. 71.9±1.2%; P=0.006) and the
proportion in the G2/M was higher (18.3±1.2 vs. 13.9±1.4%,
P=0.003). In contrast, the fraction of YD10B-E6E7 cells in the
G0/G1 phase increased (79.4±2.8 vs. 74.5±2.8%, P=0.098) and the
fraction in the G2/M phase decreased (12.3±0.7 vs. 16.4±0.4%,
P=0.003) relative to YD10B-V values (Table I).
 | Table ICell cycle distributions between
HPV16 E6E7 transfected cells and vector alone cells. |
Table I
Cell cycle distributions between
HPV16 E6E7 transfected cells and vector alone cells.
| Cells | Cell cycle
distributions
|
|---|
| G0/G1 (%) | S (%) | G2/M (%) | Sub-G1 (%) |
|---|
| YD8 | | | | |
| E6E7 | 66.1±1.5a | 6.3±0.6 | 18.3±1.2a | 10.2±1.5 |
| Vector | 71.9±1.2 | 7.6±1.4 | 13.9±1.4 | 6.8±0.6 |
| YD10B | | | | |
| E6E7 | 79.4±2.8 | 5.6±1.8 | 12.3±0.7a | 1.8±0.4 |
| Vector | 74.5±2.8 | 4.5±1.2 | 16.4±0.4 | 3.4±0.6 |
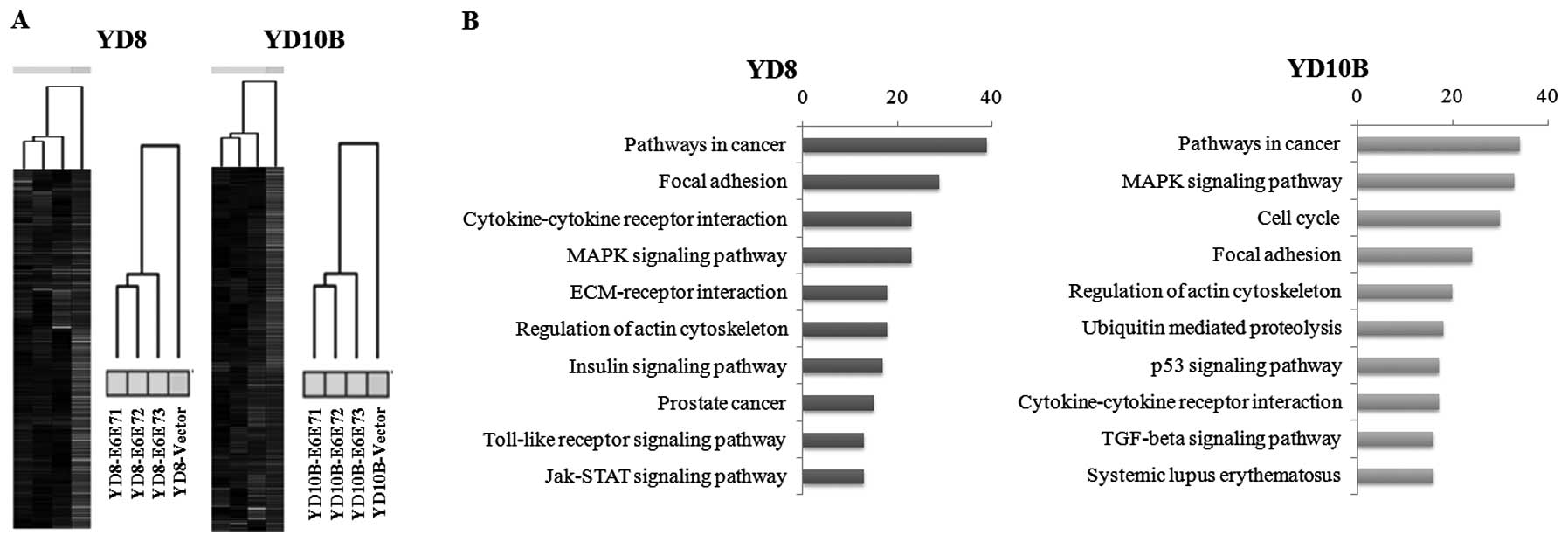
Gene expression profiles of HPV E6E7
transfected cells
We found a significant difference in gene
expressions between HPV16 E6E7 transfected cells and cells
transfected with vector alone. A total of 1,079 genes were
differentially expressed between YD8-V and YD8-E6E7, with 2,414
genes differentially expressed between YD10B-V and YD10B-E6E7
(Fig. 4A). We next sought to
identify the molecular mechanisms responsible for these differences
in expression by using the pathway mining tool of the Kyoto
Encyclopedia of Genes and the Genomes (KEGG) pathway database
(http://www.genome.jp/kegg/). This tool
maps genes to known pathways and provides a summary of the
biological processes affected. Based on this database analysis, we
identified 10 pathways that containing ≥10 genetic elements mapped
in pathways from 1,079 genes of the molecular signature in YD8
cells and from 2,414 genes of the molecular signature in YD10B
cells (Fig. 4B).
As shown in Fig.
4B, the major signaling pathways affected in HPV16 E6E7
transformed cells were identified as focal adhesion, the
cytokine-cytokine receptor interaction MAPK signaling pathway,
extracellular matrix (ECM)-receptor interaction, the JAK-STAT
signaling pathway, the cell cycle and the p53 signaling pathway.
The majority of genes involved in focal adhesion, the
cytokine-cytokine receptor interaction MAPK signaling pathway and
the ECM-receptor interaction, were downregulated in HPV16 E6E7
transfected cells compared with cells transfected with vector
alone.
Most of these genes were downregulated in HPV16 E6E7
transfected cells compared with vector alone cells. However,
RAC1, VAV3, GSK3B, THBS3, ITGB4, LAMA3, STAT1, IFI44L, FITM1,
IFIH1, SOCS2 and CDC25B were expressed at higher levels
in HPV16 E6E7 transfected cells compared with vector-alone cells
(Table II).
| Table IIThe classification of differentially
expressed genes according to signaling pathways in HPV16 E6E7
transfected cells compared with vector alone cells. |
Table II
The classification of differentially
expressed genes according to signaling pathways in HPV16 E6E7
transfected cells compared with vector alone cells.
| KEGG pathway | Regulation
|
|---|
| Upregulation | Downregulation |
|---|
| Focal adhesion | RAC1, VAV3, GSK3B,
THBS3, ITGB4, LAMA3 | ITGA11, LAMB1,
ITGA2, IGF1R, THBS2, PDGFC, CRKL, COL1A1, TNC, CAV2, BIRC2, PRKCA,
COL3A1, COL6A2, COL1A2, PDGFRB, MAPK1 |
| Cytokine-cytokine
receptor interaction | CCL26, CXCL16,
TNFSF9, CD70, TNFSF10, KITLG, IL28RA | IL4R, IL8,
TNFRSF11B, PDGFC, IL6, IL1RAP, IL7R, TNFRSF19, IL1B, CXCL1, IL11,
TGFB2, TNFRSF9, PDGFRB |
| MAPK signaling
pathway | RAC1, MKNK2, IKBKG,
CD14, BDNF, HSPA2, CDC25B | CRKL, NGF, PPM1B,
PRKCA, IL1B, PPP3CB, EVI1, TGFB2, PDGFRB, MAPK1 |
| ECM-receptor
interaction | THBS3, ITGB4,
LAMA3 | ITGA11, LAMB1,
ITGA2, THBS2, COL1A1, TNC, COL3A1, COL6A2, COL1A2, CD47 |
| Regulation of actin
cytoskeleton | RAC1, VAV3, BAIAP2,
CD14, ITGB4 | ITGA11, ITGB2,
ITGA2, PDGFC, CRKL, DIAPH3, PDGFRB, MAPK1 |
| Jak-STAT signaling
pathway | STAT1, IRF7, STAT4,
SOCS2, IL28RA, IFI30, IFI35, IFI44L, IFIH1, IFIT1, IFIT2, IFIT3,
IFITM1, IKBKG | IL4R, JAK2, IL7R,
IL6, IL11, SPRY2 |
| Cell cycle | GSK3B, CDC25B | CDKN2C, CHEK1,
CDC45L, CDC2, CDKN1B, MAD2L1, TGFB2, CCNE2 |
| p53 signaling
pathway | | CHEK1, CDC2,
SERPINE1, STEAP3, CCNE2 |
Apoptosis, cell growth and cell
cycle-related gene expression in HPV E6E7 transfected cells
To gain further insight into the molecular
mechanisms responsible for differential expression of the markers
identified in HPV16 E6E7 transfected cells after their comparison
with cells transfected with vector alone, we used qRT-PCR array
technology to examine the pattern of expression of 84 genes
associated with p53-mediated signal transduction. The array
includes p53-related genes involved in the processes of apoptosis,
cell cycle progression, cell growth, cell proliferation and cell
differentiation and DNA repair. We found significant differences in
gene expression between YD8-vector and YD8-E6E7 cells. Four genes
were upregulated (i.e., STAT1, TP73, WT1 and BCL2),
but seven genes were downregulated (i.e., ESR1, PRKCA, IGF-1R,
EGR1, MSH2, CDKN1A and JUN). The expression of STAT1 was
upregulated by 6.47-fold (P<0.05). IGF-1R was downregulated
2.40-fold in YD8-vector compared to YD8-E6E7 (P<0.01) (Table III).
| Table IIIApoptosis, cell growth and cell
cycle-related genes where were differentially expressed in YD8-E6E7
cells. |
Table III
Apoptosis, cell growth and cell
cycle-related genes where were differentially expressed in YD8-E6E7
cells.
| Gene symbol | Fold change | P-value |
|---|
| Overexpression | | |
| STAT1 | 6.47 | 0.046a |
| TP73 | 2.72 | 0.416 |
| WT1 | 2.22 | 0.621 |
| BCL2 | 2.03 | 0.450 |
|
Underexpression | | |
| ESR1 | −3.98 | 0.454 |
| PRKCA | −2.71 | 0.129 |
| IGF-1R | −2.40 | 0.009b |
| EGR1 | −2.35 | 0.337 |
| MSH2 | −2.28 | 0.349 |
| CDKN1A | −2.10 | 0.091 |
| JUN | −2.10 | 0.383 |
STAT1 and IGF-1R expression in YD8
cells
We analyzed similarities in differential gene
expression revealed in data from cDNA microarray and qRT-PCR array
experiments that compared HPV16 E6E7 transformed cells and cells
transfected with vector alone. This analysis revealed that STAT1
and IGF-1R displayed the most significantly differential expression
when gene expression was compared in YD8-E6E7 and YD8-V. In order
to validate gene expression data obtained using the microarray and
qRT-PCR array technologies, we compared the data generated using
qRT-PCR and western blot analyses for two genes that are
differentially expressed in cells transfected with HPV16 E6E7 or
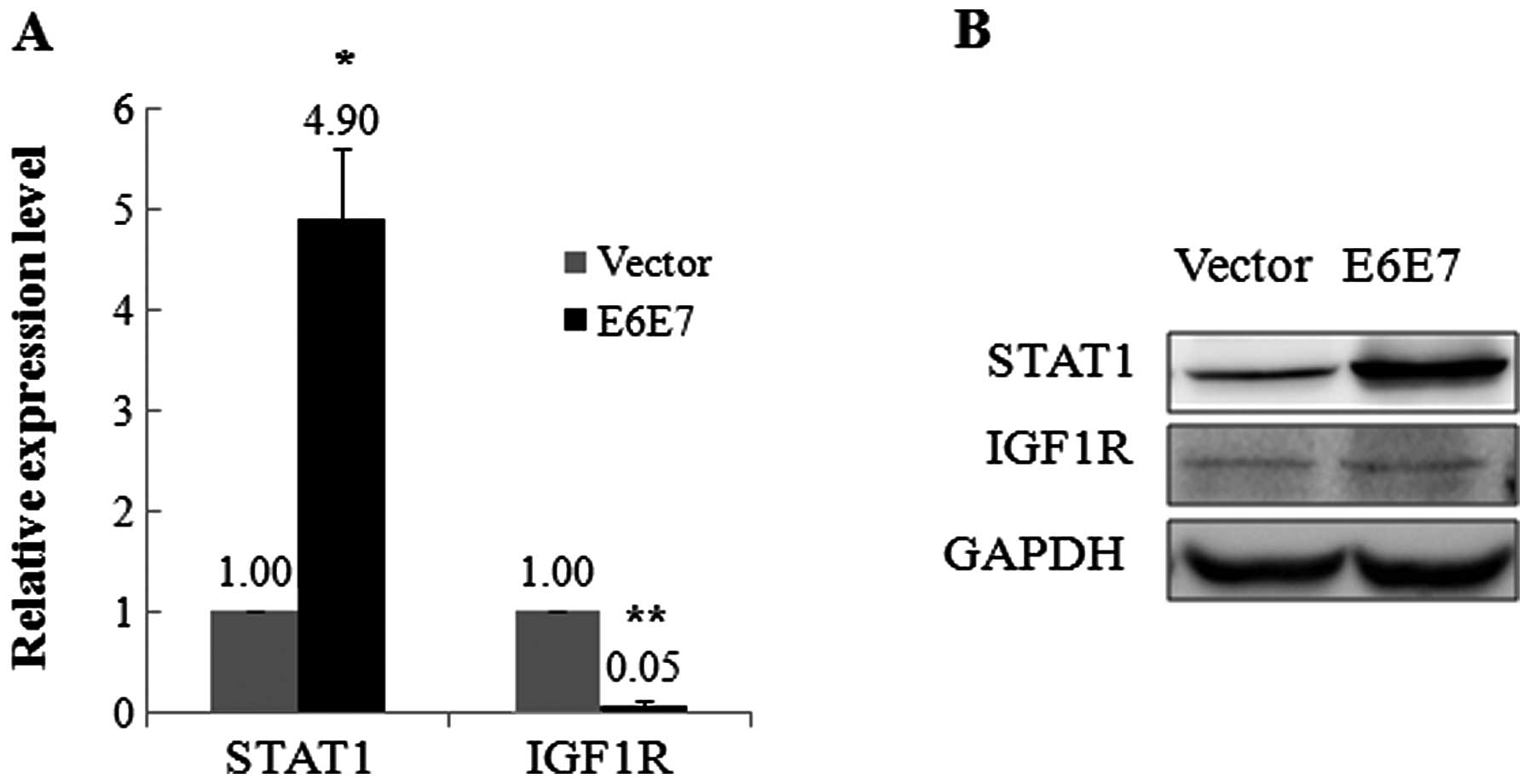
vector alone. As shown in Fig. 5,
transcription of STAT1 was expressed at a higher level in
YD8-E6E7 cells compared with YD8-V cells. In contrast, levels of
IGF-1R transcripts were less abundant in YD8-E6E7 cells
compared with YD8-V cells (Fig.
5A). The level of STAT1 protein was also higher in YD8-E6E7
cells than in YD8-V cells. Nonetheless, level of IGF-1R protein was
not differentially expressed in YD8-E6E7 and YD8-V cells (Fig. 5B).
STAT1 and IGF-1R expression in
oropharyngeal tumors
Representative examples for the immunohistochemical
staining of tumors with low, intermediate and high STAT1 activation
are shown in Table IV. STAT1
expression was assessed by immunohistochemistry as described in
Materials and methods through evaluation of the percentage of cells
with nuclear STAT1 and cytoplasmic STAT1 in HPV-positive/negative
cancer. As a result, high-level expression of nuclear STAT1 was
slightly higher in HPV-positive than HPV-negative tumors (84 and
88%, respectively) (P=0.18). However, the high-level expression of
cytoplasmic STAT1 was significantly lower in HPV-positive tumors
than in HPV-negative tumors (27 and 19%, respectively)
(P=0.01).
| Table IVImmunohistochemical staining for
STAT1. |
Table IV
Immunohistochemical staining for
STAT1.
| HPV-negative | HPV-positive |
|---|
|
|
|---|
| Nucleus | Cytoplasm | Nucleus | Cytoplasm |
|---|
| STAT1 | | | | |
| Negative | 0 (0) | 17 (15) | 0 (0) | 5 (19) |
| Low | 2 (2) | 26 (23) | 0 (0) | 4 (15) |
| Intermediate | 16 (14) | 40 (35) | 3 (12) | 12 (47) |
| High | 95 (84) | 30 (27) | 23 (88) | 5 (19) |
| No. of patients
(%) | 113 (100) | 113 (100) | 26 (100) | 26 (100) |
IGF-1R expression was evaluated by determining the
percentage of cells with cytoplasmic IGF-1R in
HPV-positive/negative cancer. The high-level expression of
cytoplasmic IGF-1R was expressed at a low level in HPV-positive
tumors compared with HPV-negative tumors (46 and 64%, respectively)
(P=0.03) (Table V).
| Table VImmunohistochemical staining for
IGF-1R. |
Table V
Immunohistochemical staining for
IGF-1R.
| HPV-negative | HPV-positive |
|---|
| IGF-1R | | |
| Negative | 11 (10) | 1 (4) |
| Low | 9 (8) | 4 (15) |
| Intermediate | 11 (19) | 9 (35) |
| High | 72 (64) | 12 (46) |
| No. of patients
(%) | 113 (100) | 26 (100) |
Discussion
Patients with HPV-positive oropharyngeal cancer show
better tumor response to radiation or chemotherapy than patients
with HPV-negative cancer (16–18).
However, HPV oncoprotein E6 binds and degrades a typically
wild-type p53 protein product (24,25).
HPV16 infection and p53 mutation may infrequently coexist in a
subset of HNSCC, but there is an inverse correlation between HPV16
and disruptive p53 mutation (26).
Even if this information has mostly been based upon clinical
studies, little is known about the molecular genetics and tumor
biology of HPV-positive oropharyngeal cancer characterized as two
different subtypes of TP53 mutations.
To address this deficiency, we investigated the
biological and molecular changes in two HPV16-negative tongue
cancer cell lines: YD8 cells bearing non-disruptive p53 mutation
and YD10B cells bearing disruptive p53 mutation (26,27),
which had been transfected with the HPV16 E6E7 oncogene. We
confirmed the existence of E6E7 DNA amplifications in these stably
transfected YD8 and YD10B cells (Fig.
1). Then, we evaluated the expression of the protein products
of the tumor suppressor genes, such as p-p53, p-Rb and cell
cycle-related genes before and after E6E7 transformation in
YD8-E6E7 cells. We observed that most of the proteins were less
abundant in YD8-E6E7 cells than in YD8-V cells (Fig. 2). Several studies have suggested
that downregulation of p53, pRb and cyclin D1 and upregulation of
p16INK4A in HPV-positive head and neck cancer patients
are the consequence of functional inactivation of two key tumor
suppressor proteins, p-p53 and p-Rb, by the HPV E6 and E7
oncoproteins (18,23,28).
Increased cell proliferative activity in cells
transformed with the E6E7 gene may be attributed to the degradation
of pRb and p53. E6-mediated degradation of p53 results in the
abrogation of the G2/M cell cycle checkpoint upon DNA damage
(29). However, in our data, the
significantly increased cell population in the G2/M phase in
YD8-E6E7 cells may be attributed to p53-mediated growth arrest
(Fig. 3A and Table I).
A large body of evidence demonstrated differences in
the expression of DNA replication, DNA repair and cell
cycle-related genes between HPV-positive and HPV-negative head and
neck cancer patients (30–32). Our gene expression profiling study
revealed significant changes in the expression of genes not only
related to the cell cycle but also to focal adhesion,
cytokine-cytokine receptor interaction, MAPK signaling and JAK-STAT
signaling (Table II). In addition,
a previous study reported that gene expression changes associated
with cytokines, growth factors and JAK-STAT signaling pathways as
part of an in vitro study, which involved HaCaT cells
(immortalized human keratinocytes) that had been transfected with
the HPV16 genome (33).
Given that the levels of the mRNA and protein
products of the cell cycle-related genes, including p53,
expressed by YD8-E6E7 cells were shown to be altered by cDNA
microarray and western blot analyses, we focused on the mRNA
expressions of p53 signal pathway-related genes in non-disruptive
p53 mutant YD8-E6E7 cells. RT-PCR array analysis revealed that HPV
significantly increased the levels of STAT1, TP73, WT1 and
BCL-2 transcripts and significantly decreased the levels of
IGF-1R, CDKN1A, FGR1, ESR1 and JUN (Table III).
Analysis of the results from both cDNA microarray
and qRT-PCR array experiments enabled us to select the STAT1
and IGF-1R genes for further analysis. We also verified the
higher expression of STAT1 and the lower expression of
IGF-1R at the mRNA in the E6E7 transformed cells than in the
cells transfected with the vector alone, but the level of IGF-1R
protein was not differentially expressed in those cells.
The JAK-STAT pathway is known to be activated in
many solid tumors, HNSCC, non-small cell lung cancer (NSCLC) and
small cell lung cancer (SCLC) (34). Dysregulation of the JAK-STAT
pathway is implicated in tumor formation and progression (35). A recent study has reported that
STAT1 expression was decreased in human foreskin keratinocytes
(HFK) transfected with wild-type HPV16/31 genomes and E6/E7
(36). In contrast, our data
showed that the level of STAT1 mRNA was higher in YD8-E6E7
cells than in YD8-V cells. We concluded that these conflicting
results of STAT1 expression may have originated from the
differences between the two target cell lines.
Most of all, previous experiments have been
conducted using normal keratinocytes transfected with the HPV viral
genome or E6 and E7 oncogenes, these transformed keratinocytes were
substantially influence the expression of numerous intracellular
target genes, via degradation of tumor suppressor genes such as p53
and pRb (37,38). However, we observed slight
decreases in the levels of mRNA and protein that encode p53 when
p53-mutated YD8 cells were transformed with the E6E7 gene. Our
results were quite different from the previous results showing a
significant decrease in the wild-type p53 protein by E6 (24). These data suggest that there is
less possibility that E6/E7 oncoproteins control intracellular
target genes, including STAT1, through the inhibition of p53
expression in the case of HPV-positive oropharyngeal cancer cells
bearing non-disruptive p53 mutation.
Analysis of mRNA expression profiling by cDNA
micro-array revealed significant changes in the Toll-like receptor
genes and genes related to the JAK-STAT signaling pathway-related
genes, especially a 2–4-fold upregulation of interferon regulatory
factor 7 (IRF7) and interferon-induced genes in the E6E7
transformed cells than in the cells transfected with the vector
alone. Considering that a remarkable increase in STAT1 mRNA
was commonly identified in both cDNA microarray and qRT-PCR array
when YD8-E6E7 cells were compared with YD8-V cells, we can
postulate that the interferon response element in the promoter of
the transfected E6 gene binds the interferon regulatory factor,
which activates the interferon signal that subsequently accelerates
interferon production. As part of a positive feedback loop,
extracellular interferon binds to the INF-α receptor and then
ultimately activates the JAK-STAT signaling pathway.
Immunohistochemical analysis showed that the
expression of STAT1 protein was slightly higher in HPV-positive
than in HPV-negative oropharyngeal cases (P=0.18); however,
cytoplasmic STAT1 was significantly lower in HPV-positive cases
(P=0.03) (Table IV).
This result suggests that STAT1 may be translocated
more from the cytoplasm to the nucleus in HPV-positive than in
HPV-negative oropharyngeal cancers.
STAT1, STAT3 and STAT5 proteins are frequently
overexpressed in head and neck cancer cell lines (39). While STAT1 increases the rates of
apoptosis, improves functioning of the immune system and functions
as a tumor suppressor by reducing cancer proliferation (40,41),
STAT3 maintains the malignant transformation by increasing the
proliferation of tumors (39,42).
Levels of IGF-1R mRNA were lower in YD8-E6E7
cells than in YD8-V cells and their migration and invasion were
also significantly decreased relative to YD8-V cells (Fig. 3B). In addition, immunohistochemical
staining of IGF-1R revealed that its abundance in the cytoplasm was
remarkably lower in HPV-positive tumors than in HPV-negative tumors
when compared with oropharyngeal carcinomas with a high level of
IGF-1R expression (Table V).
The IGF-1R protein has been implicated in
controlling cellular adhesion, cytoskeletal organization and
migration of various solid tumors, including HNSCC, via two major
signal pathways: the PI3-K/AKT and RAS/RAF/MAPK pathways (43,44).
The early oncoproteins of HPV-16 (E5, E6 and E7) enhance
trophoblastic growth by impairing cell adhesion, leading to
increased cellular motility and invasive properties (45) and HPV16 E6 increasing the ability
of human keratinocytes to adhere on poly(HEME) (46). Our data suggest that decreased
invasion activity might be caused by downregulated IGF-1R mRNA.
During the early stage of HPV infection into
oropharyngeal mucosal cells, STAT1 expression is suppressed and
viral replication is activated with evasion of the immune
surveillance (36,47). However, infection of non-disruptive
p53-mutated oropharyngeal cancer cells with HPV activates
interferon signaling associated with the immune response, which
increases rates of STAT1 phosphorylation and apoptosis while
reducing the rates of cell proliferation.
In conclusion, we propose that the molecular changes
of INF-related and JAK-STAT signals that are triggered by HPV
infection might account substantially for the increased sensitivity
to chemotherapy or radiotherapy that improves the outcome in
HPV-positive oropharyngeal carcinoma cases. Although we did not
clearly identify the downstream signals and role of STAT1, our data
suggest that activated STAT1 and interferon signals by HPV16 E6 and
E7 may play a major role in the relatively favorable prognosis for
patients with a non-disruptive p53 mutation, HPV-positive
oropharyngeal squamous cell carcinomas. Therefore, upregulated
INF-related and JAK-STAT signals likely play a pivotal role in
mediating the immune surveillance of HPV-related oropharyngeal
cancers and strategies designed to upregulate the immune response
hold promise for further improving patient outcomes.
Acknowledgements
This study was supported by BK21
Project for Biomedical Science and a grant of the Disease-Oriented
Translational Research Project of Ministry of Education & Human
Resources Development in Korea.
References
|
1.
|
Kamangar F, Dores GM and Anderson WF:
Patterns of cancer incidence, mortality and prevalence across five
continents: defining priorities to reduce cancer disparities in
different geographic regions of the world. J Clin Oncol.
24:2137–2150. 2006. View Article : Google Scholar
|
|
2.
|
Gillison ML, Koch WM, Capone RB, et al:
Evidence for a causal association between human papillomavirus and
a subset of head and neck cancers. J Natl Cancer Inst. 92:709–720.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
D’Souza G, Kreimer AR, Viscidi R, et al:
Case-control study of human papillomavirus and oropharyngeal
cancer. N Engl J Med. 356:1944–1956. 2007.PubMed/NCBI
|
|
4.
|
Applebaum KM, Furniss CS, Zeka A, et al:
Lack of association of alcohol and tobacco with HPV16-associated
head and neck cancer. J Natl Cancer Inst. 99:1801–1810. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Gillison ML, D’Souza G, Westra W, et al:
Distinct risk factor profiles for human papillomavirus type
16-positive and human papillomavirus type 16-negative head and neck
cancers. J Natl Cancer Inst. 100:407–420. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Smith EM, Ritchie JM, Summersgill KF, et
al: Age, sexual behavior and human papillomavirus infection in oral
cavity and oropharyngeal cancers. Int J Cancer. 108:766–772. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Näsman A, Attner P, Hammarstedt L, et al:
Incidence of human papillomavirus (HPV) positive tonsillar
carcinoma in Stockholm, Sweden: an epidemic of viral-induced
carcinoma. Int J Cancer. 125:362–366. 2009.PubMed/NCBI
|
|
8.
|
McLaughlin-Drubin ME and Munger K:
Oncogenic activities of human papillomaviruses. Virus Res.
143:195–208. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Li X and Coffino P: High-risk human
papillomavirus E6 protein has two distinct binding sites within
p53, of which only one determines degradation. J Virol.
70:4509–4516. 1996.PubMed/NCBI
|
|
10.
|
Talis AL, Huibregtse JM and Howley PM: The
role of E6AP in the regulation of p53 protein levels in human
papillomavirus (HPV)-positive and HPV-negative cells. J Biol Chem.
273:6439–6445. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Dyson N: The regulation of E2F by
pRB-family proteins. Genes Dev. 12:2245–2262. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Münger K, Basile JR, Duensing S, et al:
Biological activities and molecular targets of the human
papillomavirus E7 oncoprotein. Oncogene. 20:7888–7898.
2001.PubMed/NCBI
|
|
13.
|
Bouvard V, Matlashewski G, Gu ZM, Storey A
and Banks L: The human papillomavirus type 16 E5 gene cooperates
with the E7 gene to stimulate proliferation of primary cells and
increases viral gene expression. Virology. 203:73–80. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Schwarz E, Freese UK, Gissmann L, et al:
Structure and transcription of human papillomavirus sequences in
cervical carcinoma cells. Nature. 314:111–114. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Smith EM, Rubenstein LM, Haugen TH,
Pawlita M and Turek LP: Complex etiology underlies risk and
survival in head and neck cancer human papillomavirus, tobacco and
alcohol: a case for multifactor disease. J Oncol.
5718622012.PubMed/NCBI
|
|
16.
|
Fakhry C, Westra WH, Li S, et al: Improved
survival of patients with human papillomavirus positive head and
neck squamous cell carcinoma in a prospective clinical trial. J
Natl Cancer Inst. 100:261–269. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Gillison ML, Harris J, Westra W, et al:
Survival outcomes by tumor human papillomavirus (HPV) status in
stage III–IV oropharyngeal cancer (OPC) in RTOG 0129. Proc Am Soc
Clin Oncol. 27:60032009.
|
|
18.
|
Chung CH and Gillison ML: Human
papillomavirus in head and neck cancer: its role in pathogenesis
and clinical implications. Clin Cancer Res. 15:6758–6762. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Brennan JA, Boyle JO, Koch WM, et al:
Association between cigarette smoking and mutation of the p53 gene
in squamous-cell carcinoma of the head and neck. N Engl J Med.
332:712–717. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Carlos de Vicente J, Junquera Gutiérrez
LM, Zapatero AH, Fresno Forcelledo MF, Hernández-Vallejo G and
López Arranz JS: Prognostic significance of p53 expression in oral
squamous cell carcinoma without neck node metastases. Head Neck.
26:22–30. 2004.PubMed/NCBI
|
|
21.
|
Khademi B, Shirazi FM, Vasei M, et al: The
expression of p53, c-erbB-1 and c-erbB-2 molecules and their
correlation with prognostic markers in patients with head and neck
tumors. Cancer Lett. 184:223–230. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Licitra L, Perrone F, Bossi P, et al:
High-risk human papillomavirus affects prognosis in patients with
surgically treated oropharyngeal squamous cell carcinoma. J Clin
Oncol. 24:5630–5636. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Kumar B, Cordell KG, Lee JS, Worden FP, et
al: EGFR, p16, HPV Titer, Bcl-xL and p53, sex and smoking as
indicators of response to therapy and survival in oropharyngeal
cancer. J Clin Oncol. 26:3128–3137. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Scheffner M, Werness BA, Huibregtse JM,
Levine AJ and Howley PM: The E6 oncoprotein encoded by human
papillomavirus types 16 and 18 promotes the degradation of p53.
Cell. 63:1129–1136. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Wiest T, Schwarz E, Enders C,
Flechtenmacher C and Bosch FX: Involvement of intact HPV16 E6/E7
gene expression in head and neck cancers with unaltered p53 status
and perturbed pRb cell cycle control. Oncogene. 21:1510–1517. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Westra WH, Taube JM, Poeta ML, Begum S,
Sidransky D and Koch WM: Inverse relationship between human
papillomavirus-16 infection and disruptive p53 gene mutations in
squamous cell carcinoma of the head and neck. Clin Cancer Res.
14:366–369. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Lee EJ, Kim J, Lee SA, et al:
Characterization of newly established oral cancer cell lines
derived from six squamous cell carcinoma and two mucoepidermoid
carcinoma cells. Exp Mol Med. 37:379–390. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Hafkamp HC, Mooren JJ, Claessen SM, et al:
P21Cip1/WAF1expression is strongly associated with
HPV-positive tonsillar carcinoma and a favorable prognosis. Mod
Pathol. 22:686–698. 2009.
|
|
29.
|
Kessis TD, Slebos RJ, Nelson WG, et al:
Human papillomavirus 16 E6 expression disrupts the p53-mediated
cellular response to DNA damage. Proc Natl Acad Sci USA.
90:3988–3992. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Martinez I, Wang J, Hobson KF, Ferris RL
and Khan SA: Identification of differentially expressed genes in
HPV-positive and HPV-negative oropharyngeal squamous cell
carcinomas. Eur J Cancer. 43:415–432. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Pyeon D, Newton MA, Lambert PF, et al:
Fundamental differences in cell cycle deregulation in human
papillomavirus-positive and human papillomavirus-negative head/neck
and cervical cancers. Cancer Res. 67:4605–4619. 2007. View Article : Google Scholar
|
|
32.
|
Lohavanichbutr P, Houck J, Fan W, et al:
Genomewide gene expression profiles of HPV-positive and
HPV-negative oropharyngeal cancer: potential implications for
treatment choices. Arch Otolaryngol Head Neck Surg. 135:180–188.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Kaczkowski B, Rossing M, Andersen DK, et
al: Integrative analyses reveal novel strategies in HPV11, -16 and
-45 early infection. Sci Rep. 2:5152012. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Lai SY and Johnson FM: Defining the role
of the JAK-STAT pathway in head and neck and thoracic malignancies:
Implications for future therapeutic approaches. Drug Resist Updat.
13:67–78. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Yu H and Jove R: The STATs of cancer - new
molecular targets come of age. Nat Rev Cancer. 4:97–105. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Hong S, Mehta KP and Laimins LA:
Suppression of STAT-1 expression by human papillomaviruses is
necessary for differentiation-dependent genome amplification and
plasmid maintenance. J Virol. 85:9486–9494. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Boccardo E, Manzini Baldi CV, Carvalho AF,
et al: Expression of human papillomavirus type 16 E7 oncoprotein
alters keratinocytes expression profile in response to tumor
necrosis factor-α. Carcinogenesis. 31:521–531. 2010.PubMed/NCBI
|
|
38.
|
Smeets SJ, van der Plas M, Schaaij-Visser
TB, van Veen EA, et al: Immortalization of oral keratinocytes by
functional inactivation of the p53 and pRb pathways. Int J Cancer.
128:1596–1605. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Shim SH, Sung MW, Park SW and Heo DS:
Absence of STAT1 disturbs the anticancer effect induced by STAT3
inhibition in head and neck carcinoma cell lines. Int J Mol Med.
23:805–810. 2009.PubMed/NCBI
|
|
40.
|
Xi S, Dyer KF, Kimak M, et al: Decreased
STAT1 expression by promoter methylation in squamous cell
carcinogenesis. J Natl Cancer Inst. 98:181–189. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Laimer K, Spizzo G, Obrist P, et al: STAT1
activation in squamous cell cancer of the oral cavity: a potential
predictive marker of response to adjuvant chemotherapy. Cancer.
110:326–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Riebe C, Pries R, Schroeder KN and
Wollenberg B: Phosphorylation of STAT3 in head and neck cancer
requires p38 MAPKinase, whereas phosphorylation of STAT1 occurs via
a different signaling pathway. Anticancer Res. 31:3819–3825.
2011.PubMed/NCBI
|
|
43.
|
Barnes CJ, Ohshiro K, Rayala SK, El-Naggar
AK and Kumar R: Insulin-like growth factor receptor as a
therapeutic target in head and neck cancer. Clin Cancer Res.
13:4291–4299. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Bähr C and Groner B: The IGF-1 receptor
and its contributions to metastatic tumor growth-novel approaches
to the inhibition of IGF-1R function. Growth Factors. 23:1–14.
2005.PubMed/NCBI
|
|
45.
|
Boulenouar S, Weyn C, Van Noppen M, et al:
Effects of HPV-16 E5, E6 and E7 proteins on survival, adhesion,
migration and invasion of trophoblastic cells. Carcinogenesis.
31:473–480. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Epshtein A, Jackman A, Gonen P and Sherman
L: HPV16 E6 oncoprotein increases cell adhesion in human
keratinocytes. Arch Virol. 154:55–63. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Einstein MH, Schiller JT, Viscidi RP, et
al: Clinician’s guide to human papillomavirus immunology: knowns
and unknowns. Lancet Infect Dis. 9:347–356. 2009.
|
|
48.
|
Won HS, Jung CK, Chun SH, et al:
Difference in expression of EGFR, pAkt and PTEN between
oropharyngeal and oral cavity squamous cell carcinoma. Oral Oncol.
48:985–990. 2012. View Article : Google Scholar : PubMed/NCBI
|