Introduction
Selective estrogen receptor modulators (SERMs) such
as tamoxifen and raloxifene have proven to be successful in the
treatment of breast cancer. Raloxifene, a second generation SERM,
has been approved for the prevention of osteoporosis and the
reduction of the risk of invasive breast cancer in postmenopausal
women (1). In breast tissue, SERMs
are thought to prevent proliferation of cancer cells by binding
competitively to the estrogen receptor (ER) and blocking the
mitogenic effect of estradiol (2).
Although SERMs have a widespread clinical use, it is not
established whether their therapeutic effects are solely mediated
through the ER. Several studies have demonstrated that SERMs are
effective against tumors that do not express ER such as lung cancer
(3), brain cancer (4), melanoma (5) and breast cancer (6,7).
Furthermore, SERMs have been demonstrated in vitro to
trigger multiple signaling pathways that lead to ER-independent
mediated cell death (reviewed in ref. 8).
Based on these earlier findings, we investigated the
suppressive effects of raloxifene on triple-negative breast cancer
(TNBC) growth. By definition, TNBC do not express ERα, progesterone
receptor (PR) and human epidermal growth factor receptor 2 (Her2,
ErbB2). They account for 10–17% of all breast cancers and represent
85% of the basal- like subtype (9). TNBCs generally have a higher
prevalence in African-American women and an increased occurrence in
premenopausal women (9). TNBCs are
clinically aggressive and generally associated with a poor
prognosis. Currently, chemotherapy remains the only systemic
treatment option available for patients with TNBC (10).
In the present study, a daily oral dose of
raloxifene not only suppressed tumor growth in two TNBC xenograft
mouse models but also promoted tumor regression. The underlying
therapeutic mechanism of raloxifene was associated with a decreased
expression of EGFR which concurred with a decrease in cell
proliferation, an increased incidence of apoptosis and a consequent
decrease in blood vessels count within the tumors. In vitro
experiments demonstrated that raloxifene decreased EGFR expression
by promoting its endocytosis and translocation to small cytoplasmic
vesicles akin to those of the endosomal pathway. In addition,
raloxifene treatment reduced the migration, invasion and
tumorigenicity of MDA-MB-231, a highly metastatic TNBC. Overall
these data clearly showed that exploiting the ER-independent
mechanisms of raloxifene can promote new therapeutic approaches
against TNBC.
Materials and methods
Cell culture
MDA-MB-231, MDA-MB-468 and MCF-7 were obtained from
American Type Culture Collection (ATCC) (Manassas, VA, USA). The
cells were grown in complete growth media composed of DMEM/Ham’s
F12 supplemented with 5% fetal bovine serum, 2 mM L-glutamine, 100
U/ml streptomycin, 100 U/ml penicillin and 2.2 g/l
NaHCO3.
Animals and treatments
All animal protocols were approved by the University
of Otago Animal Ethics Committee (#91/07×2). Female CD1 athymic
nude mice (5–6-week-old) were purchased from Hercus Taieri Resource
Unit (Dunedin, New Zealand). Mice were inoculated subcutaneously
into the right rear flank with triple-negative cells either
MDA-MB-231 (2×106 cells/0.1 ml Matrigel) or MDA-MB-468
(8×106 cells/0.2 ml Matrigel). When tumors reached a
size of ~100 mm3 (MDA-MB-231 cells), 200 mm3
(MDA-MB-468 cells) or 400–500 mm3 (MDA-MB-468 cells for
analyzing tumor regression), six animals were randomly assigned per
treatment groups. Daily for 8–10 weeks as specified, mice received
either raloxifene (0.5 mg/kg), raloxifene (0.85 mg/kg), raloxifene
(12.5 mg/kg) or a vehicle control (0.25% DMSO). Two independent
measurements of tumor volume (length × width × height) were
performed weekly using electronic calipers.
Western blot analysis,
immunohistochemistry and indirect immunofluorescence
Tissues and MDA-MB-468 cells were processed as
described (11) and western blot
analysis was performed either with EGFR antibody (Cell Signaling,
Danvers, MA, USA), β-actin (Sigma-Aldrich, Auckland, New Zealand)
or ERα (Abcam, Cambridge, MA, USA). MCF-7 cell lysates were used as
a positive control for ERα expression. For immunohistochemistry,
frozen sections, obtained from tumors were embedded in OCT, then
incubated overnight with either rat anti-mouse CD105 (BD
Pharmingen, Auckland, New Zealand) or rabbit Ki67 (Epitomics,
Burlingame, CA, USA). Slides were incubated with the appropriate
biotinylated secondary antibody either goat anti-rat (BD
Pharmingen) or goat anti-rabbit (Dako, Campbellfield, Australia).
The sections were then incubated with streptavidin (BD Pharmingen)
before development with 3,3′-diaminobenzidine tetrahydrochloride
(DAB) (BD Pharmingen) and counterstained with hematoxylin QS
(Vector Laboratories). In situ labeling of fragmented DNA,
TUNEL assay (terminal deoxynucleotidyl transferase-mediated
2′-deoxyuridine 5′-triphosphate nick-end labeling) was carried out
using the apotag peroxidase In Situ Apoptosis Detection kit
(Millipore, North Ryde, Australia) according to the manufacturer’s
instructions. Indirect immunofluorescence microscopy was carried
out as described previously (12).
Briefly, cells were incubated with raloxifene for 48 h, fixed with
4% paraformaldehyde and incubated with EGFR antibody alone or in
combination with EEA1 or caveolin-1 antibodies (Cell Signalling).
Secondary antibodies, conjugated to fluorescein or Texas Red
(Dako), were used for co-localization. Nuclei were visualized using
4′,6-diamidino-2-phenylindole (DAPI) staining.
Cell migration
Migration of MDA-MB-231 cells was measured with the
in vitro cell scratch assay. Confluent cells were scratched
with a pipette tip and cellular debris were removed by extensive
washing with serum-free medium. Raloxifene (10 μM) or DMSO as
control was then added. Cells were allowed to migrate into the
scrapped area for up to 20 h at 37°C and were captured at indicated
intervals.
Invasion assay
MDA-MB-231 (5×105 cells/ml) were seeded
onto growth factor-reduced Matrigel invasion chambers (8-μm pore;
BD Biosciences) with or without raloxifene 10 μM for 20 h. Lower
chambers contained DMEM/Ham’s F12 supplemented with 5% FBS, a
chemoattractant. Filters were fixed in methanol and stained using
Diff-Quick staining solutions. Cells were counted in four fields of
each well under an inverted microscope at magnification, ×20. Their
migration towards FBS was calculated as a percentage of the
control. Data were collected from three independent experiments,
each done in triplicate. Migrated cells were counted and the mean
(± SE) between groups were analyzed using a Student’s t-test.
Soft-agar assay
The base layer consisted of 0.6% ultra-pure agarose
(Invitrogen, Auckland, New Zealand) in DMEM/Ham’s F12 supplemented
with 5% FBS medium. Soft agar composed of 0.3% ultra-pure agarose
in DMEM/Ham’s F12 supplemented with 5% FBS medium was mixed with
15×104 MDA-MB-231 cells and plated on top of the
solidified base layer in a 6-well-plate. Soft agar cultures were
maintained at 37°C for an additional 21 days and treated with
raloxifene at the indicated concentration (5, 10 or 15 μM) or DMSO
(0.1%). Formed colonies were stained with 0.2% (w/v) crystal violet
(Sigma-Aldrich) solution in 6% (v/v) paraformaldehyde solution
(Sigma-Aldrich). Colonies were counted in images taken in four
fields in each well. The assay was repeated three times with
duplicate samples.
Statistical analysis
Before statistical analysis, data were
log-transformed if parameters showed significantly different
variances between control and treated mice (namely, tumor volume
and tumor weight). Tumor growth experiments were analyzed using a
repeated measures two-way ANOVA coupled with a Student-Newman-Keuls
post hoc test, where p<0.05 is required for statistical
significance. Analyses that were independent of time (i.e., tumor
weight and protein expression) were analyzed using a one-way ANOVA
coupled with a Student-Newman-Keuls post hoc test, where p<0.05
is required for statistical significance.
Results
A daily oral dose of raloxifene
suppresses tumor growth
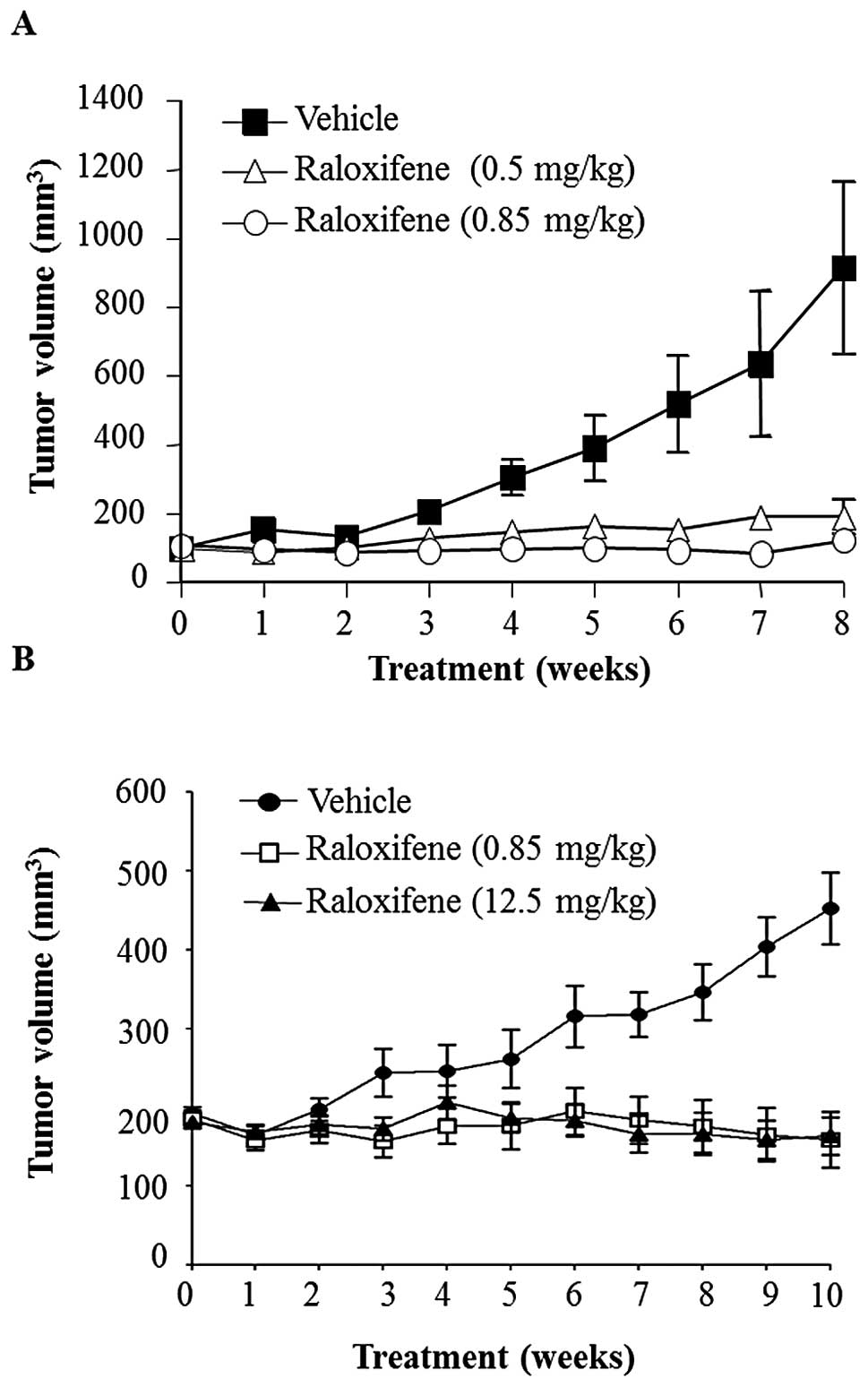
Tumor growth was abolished in two mouse models by a
daily administration of raloxifene with optimal dose being 0.85
mg/kg (Fig. 1). In the first
model, MDA-MB-231 xenograft tumors were seeded in mice until
reaching a size of 100±12 mm3. The doses of raloxifene
used in this experiment were 25 and 15 times lower than the human
equivalent dose of 60 mg when calculated based on body surface area
(13). The results showed that
after 4 weeks, the mice receiving raloxifene daily showed a
significant reduction in tumor growth compared to vehicle control
(Fig. 1A). At 8 weeks, these
raloxifene treated groups showed tumors sized at ~100
mm3, 10 times lower than those of the vehicle group
(~1,000 mm3) (p<0.001) and a similar size comparable
to those before the start of the treatment. In the second model,
the rear flanks of mice were implanted with MDA-MB-468 cells. The
doses of raloxifene used were either 15 times lower or equivalent
to the human dose of 60 mg. The results showed that after 10 weeks
of treatment, tumor size for both raloxifene treated groups were
three times lower than that of the vehicle treated group (450
mm3) (p<0.001). Interestingly, the low dose of
raloxifene (0.85 mg/kg) was as effective at suppressing MDA-MB-468
xenograft tumor growth as the higher (12.5 mg/kg) human equivalent
dose (Fig. 1B). Moreover, in both
tumorigenic models, the body weights of animals receiving
raloxifene did not change in comparison to controls.
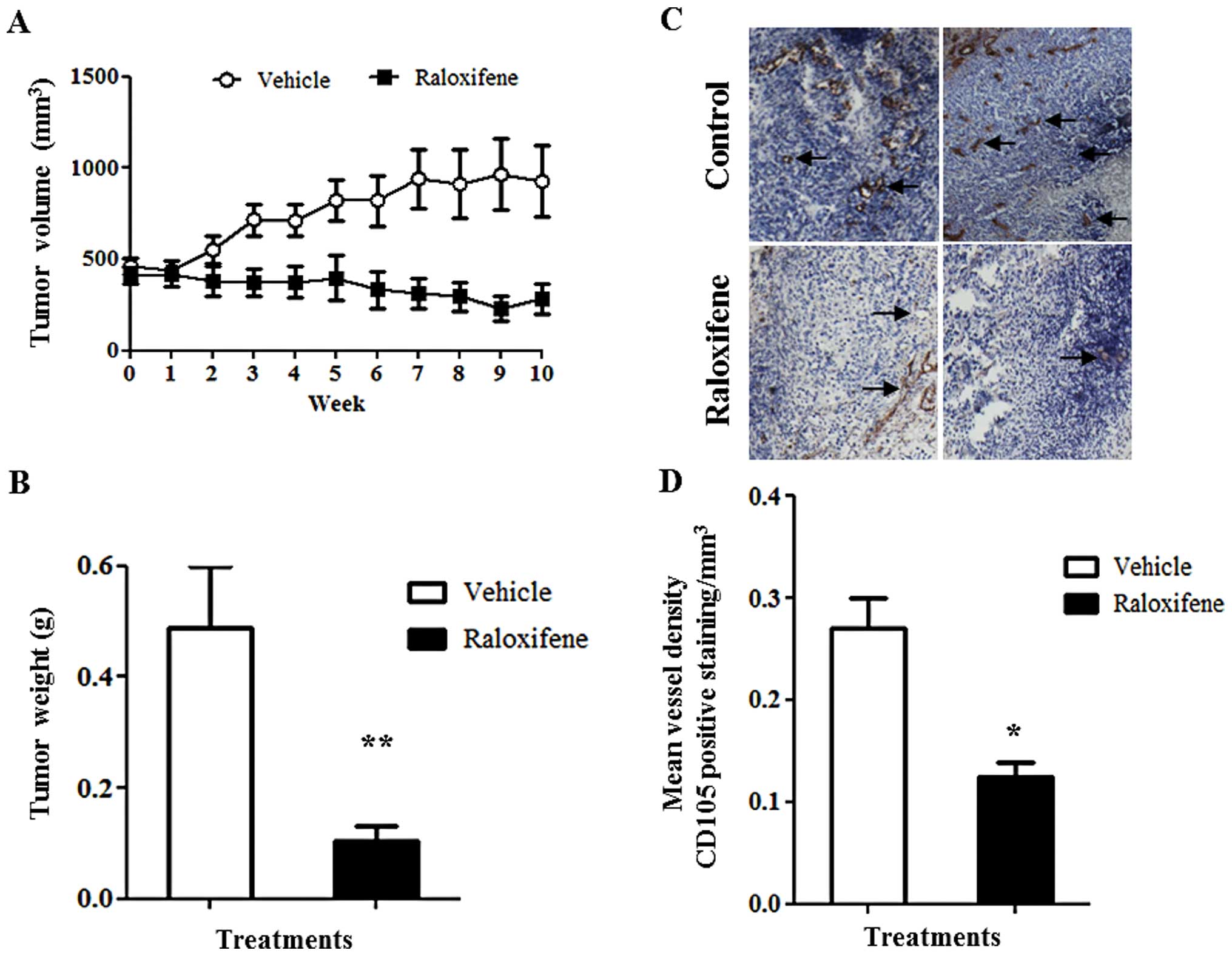
A daily oral dose of raloxifene caused
regression of TNBC xenograft tumors
The effective low dose of raloxifene (0.85 mg/kg)
promoted tumor volume regression. Female athymic nude mice were
implanted with MDA-MB-468 cells. In this model after tumors reached
an average volume of 400–500 mm3, the mice were treated
daily for a period of 10 weeks with an oral dose of 0.85 mg/kg of
raloxifene, or vehicle control (DMSO (0.25%) (Fig. 2). After 5 weeks, tumors of
raloxifene treated mice were 52% smaller (p<0.05) than those of
vehicle controls (Fig. 2A). After
10 weeks of treatment, tumor size had decreased to 283
mm3, a 39% reduction from their initial size and a 70%
volume reduction compared to vehicle controls (p<0.001)
(Fig. 2A). Consequently, tumor
weight was 5 times lower in raloxifene treated mice compared to
vehicle controls (Fig. 2B).
Immunohistological analysis using CD105 antibody revealed that
raloxifene treatment reduced CD105 positive microvascular density
by 50% (Fig. 2C and D). This
reduction in microvasculature correlated with the observed decrease
in tumor volume following the 10 weeks of treatment.
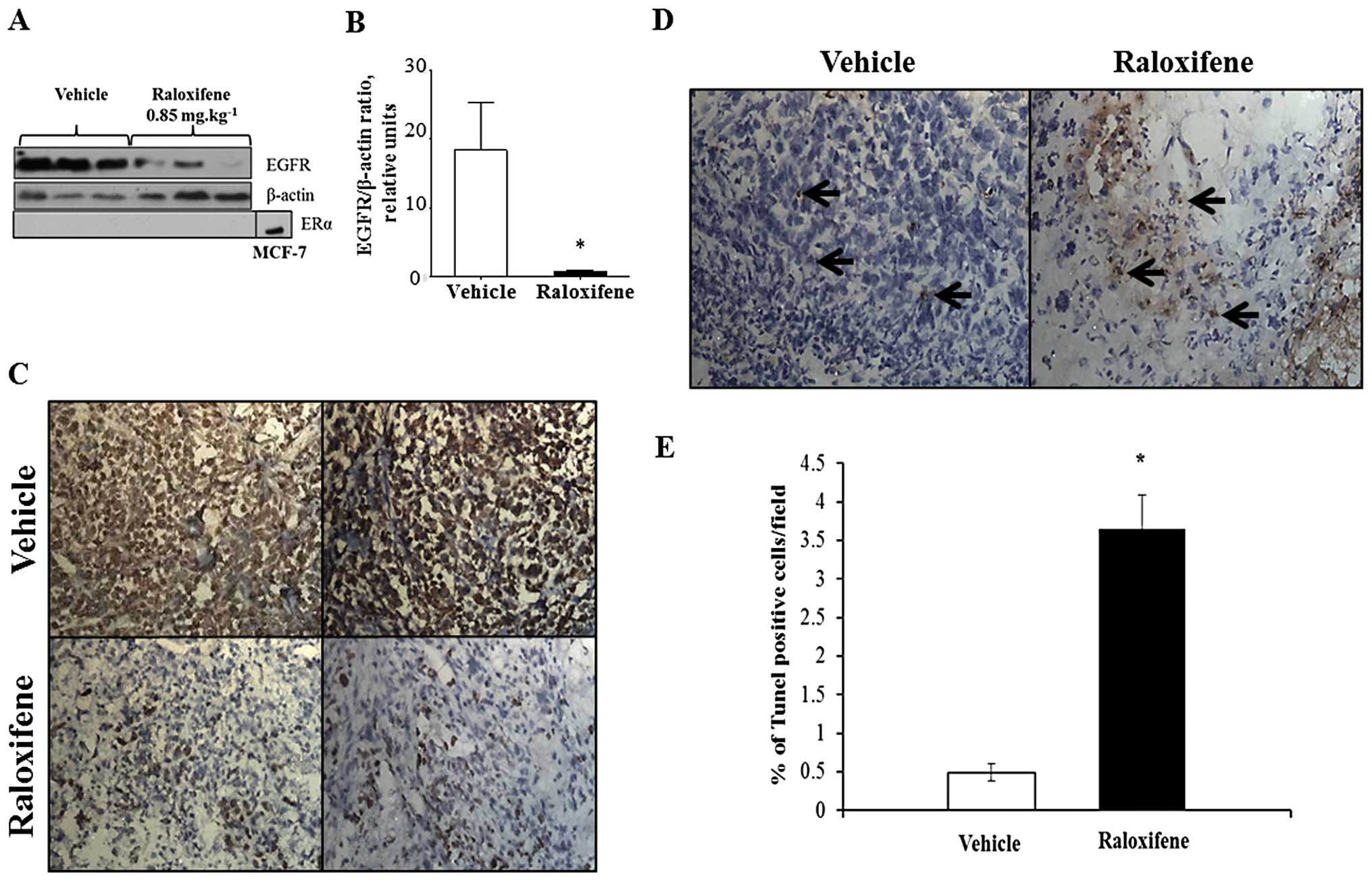
Raloxifene treatment reduces EGFR
expression, decreases cell proliferation and increases apoptosis in
tumors
The EGFR is among the few proteins that can
characterize basal-like subtype and TNBC by immunohistology and is
one of the highest concordant markers. It is expressed in 60% of
basal- like tumors (14) and is
highly expressed in MDA-MB-468 cells (15). Furthermore, EGFR stimulates cell
proliferation, motility and invasion of breast cancer cells
(16,17). Western blot analysis showed that
raloxifene (0.85 mg/kg) significantly decreased the expression of
EGFR protein (Fig. 3A and B).
Treatment of MDA-MB-468 cells with raloxifene (10 μM) for 24 h
in vitro also triggered a decreased expression of EGFR by
>30% (data not shown). We confirmed that long-term in
vivo raloxifene treatment did not induce ERα expression in
MDA-MB-468 tumors (Fig. 3A).
Raloxifene therapy significantly inhibited tumor cell
proliferation, as shown by a 70% decrease in cells positive for
Ki67 compared to vehicle controls (Fig. 3C). Moreover, raloxifene treatment
induced an 8-fold elevation of the number of TUNEL-positive cells
compared to the vehicle group (Fig. 3D
and E).
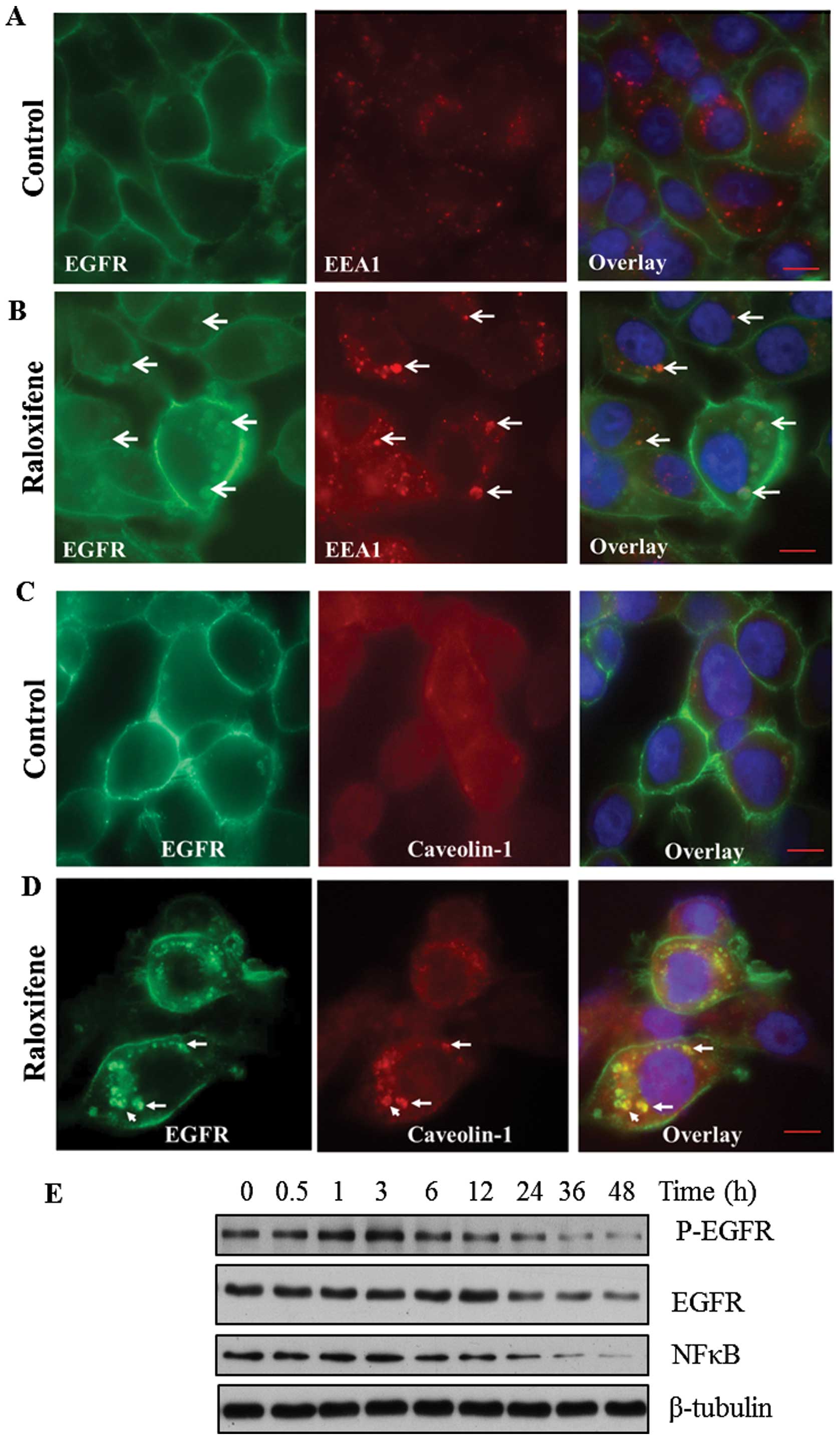
Raloxifene treatment promotes EGFR
endocytosis
Raloxifene treatment of MDA-MB-468 cells in
vitro changes EGFR localization and promotes its transit
towards small cytoplasmic vesicles. In untreated MDA-MB-468 cells,
EGFR is highly expressed and localizes at the membrane but also
shows a diffuse punctuate cytoplasmic staining (Fig. 4A). Whereas, after 48-h exposure to
raloxifene (10 μM) triggers the accumulation of EGFR in small
cytoplasmic vesicles (Fig. 4B). To
establish the origin of the cytoplasmic vesicles containing EGFR,
we probed two protein markers: early endosome antigen-1 (EEA1),
which is essential for early endosome formation and trafficking
(18) and caveolin-1, a marker of
caveolae and endosome formation (19). In control cells, dual labeling with
EEA1 and EGFR showed that these proteins distinctively
compartmentalize (Fig. 4A) where
EEA1 is cytoplasmic but can also cluster within small vesicles
while EGFR is mainly expressed at the cell membrane. However, in
raloxifene treated cells, the two proteins colocalized within a few
larger vesicles (Fig. 4B), while
some vesicles contained only EGFR protein (Fig. 4B). When probing raloxifene treated
cells with EGFR and caveolae marker, we found that caveolin-1 and
EGFR antibodies colocalized both proteins within a few vesicles
(Fig. 4D) while in the untreated
control cells, the caveolin-1 staining diffuse (Fig. 4C). Western blot analysis of
MDA-MB-468 cells showed that raloxifene treatment decreased EGFR
phosphorylation and protein expression after a 24-h incubation and
this was associated with a reduced expression of downstream
effectors such as NFκB (Fig. 4E).
Overall, these experiments show that the EGFR is internalized into
cytoplasmic vesicles related to endosome formation. This suggests
that the raloxifene mediated decrease in proliferation of cancer
cells may be mediated through endocytosis of the EGFR which
decreases proliferative signaling pathways.
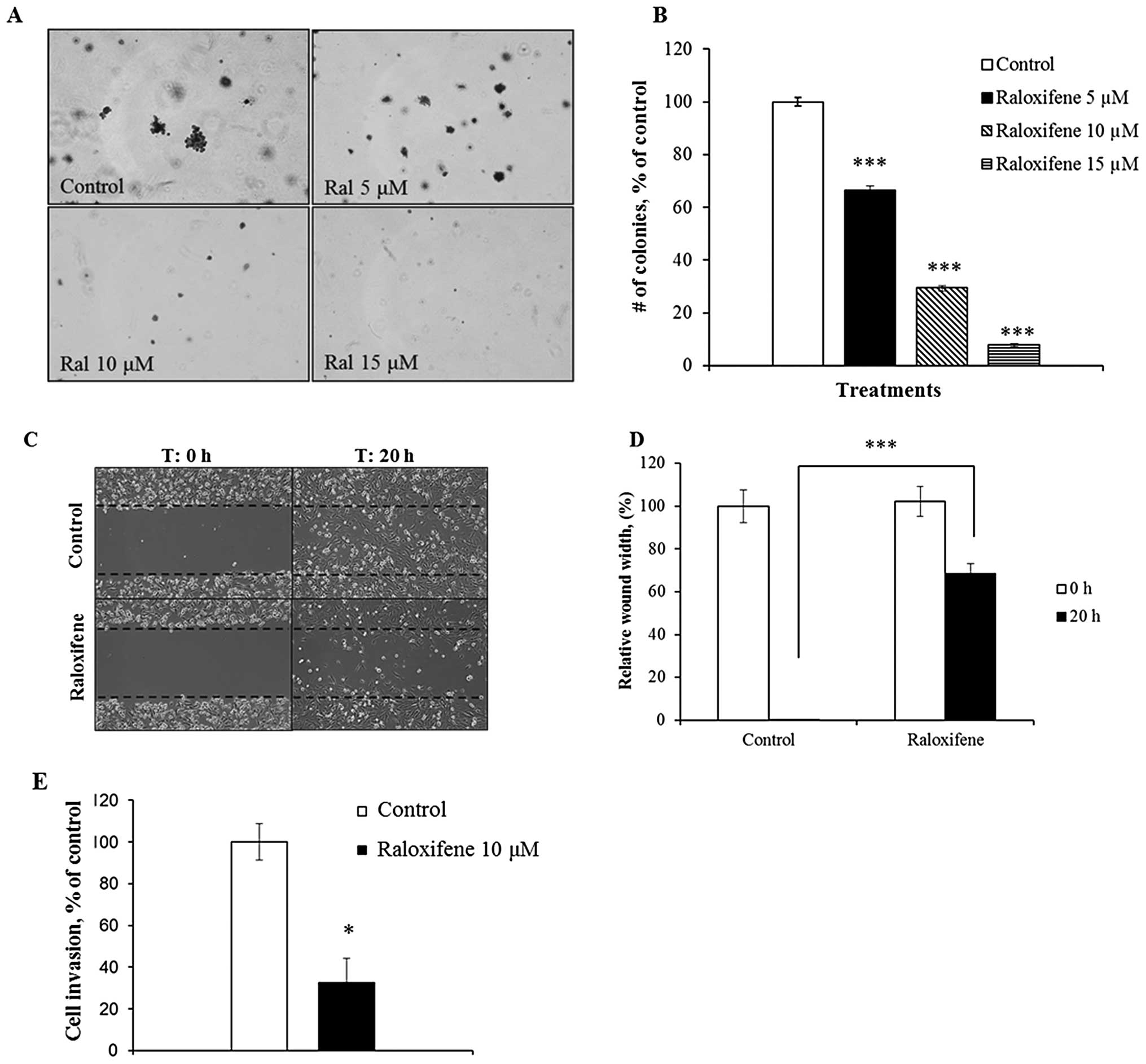
Raloxifene decreases tumorigenicity, cell
migration and cell invasion
Raloxifene treatment significantly decreased
anchorage-independent growth of the highly metastatic MDA-MB-231
cells in soft agar. The assay we used represents an in vitro
transformation phenotype that is highly correlated with in
vivo tumorigenicity (20).
Raloxifene not only dose-dependently suppressed colony formation of
MDA-MB-231 cells (Fig. 5A and B)
but treatment decreased their cell migration and invasion.
Specifically, raloxifene (10 μM) reduced cell migration and delayed
wound closure by 70% (Fig. 5C and
D). The ability of raloxifene to prevent MDA-MB-231 cells to
migrate and invade through Matrigel was then measured in a
transwell chamber. Raloxifene impaired invasion by >70% compared
to control (Fig. 5E). Together
these in vitro results provide further evidence that
raloxifene significantly reduces the metastatic potential of TNBC
cells.
Discussion
Despite the high rate of response to chemotherapy,
patients presenting TNBC have considerably poor prognosis. This
urges the development of novel targeted or combination therapies to
reduce the mortality associated with these cancers. By identifying
the targets and mechanisms behind the efficacy of a drug helps to
carefully adapt treatment conditions to specific pathology of TNBC.
The present study describes four major findings: i) a daily oral
dose of raloxifene suppresses tumor growth in two xenograft mouse
models of TNBC; ii) a daily oral dose of raloxifene promotes tumor
regression in a xenograft model using MDA-MB-468 cells; iii)
raloxifene treatment decreases the expression of EGFR and promotes
its accumulation in endosomes; iv) raloxifene decreases
tumorigenicity, cell migration and invasion of a highly invasive
human TNBC cell line.
The efficacy of SERMs, tamoxifen and raloxifene, has
been attributed to their ability to compete with 17β-estradiol
antagonizing ER downstream signaling events (21). Several studies have demonstrated
the in vivo and in vitro proapoptotic potential of
SERMs in various ER-negative tissues and cells including bladder,
glioma, melanoma or breast cancer (1,22–24).
The in vitro effect of the SERMs was
concentration-dependent, as growth arrest was induced by nanomolar
concentrations while cell death was achieved in the micromolar
range (1). In addition to the
specific antagonistic effects of SERMs in the breast, other data
suggest antitumorigenic effect of SERMs that are independent of ERα
signaling. For example, raloxifene was shown to modulate
phospholipase D activity (25) and
more recently both raloxifene and tamoxifen were shown to reduce
glutamine uptake (26).
In our study, we observed that raloxifene affects
the expression of EGFR, a protein known to have a prominent role in
the development of TNBC. EGFR expression is found in 45–70 of TNBC
(27) and is a putative biomarker
associated with an unfavorable prognosis (28). Analysis by western blot analysis
showed that EGFR expression in tumors was decreased by a daily dose
of raloxifene. The present study suggests that its effects on cell
proliferation and apoptosis would stem from a decrease in EGFR and
its clustering within endosomes. Furthermore, previous studies have
demonstrated that internalized EGFR can promote caspase-3 mediated
apoptosis (29).
Interestingly, SERMs such as raloxifene and
tamoxifen were shown to bind with high affinity to the microsomal
anti-estrogen binding site (AEBS) (30). AEBS is a protein complex composed
of two enzymes and acts as a cholesterol epoxyde hydrolase, an
enzyme involved in cholesterol metabolism (31). AEBS has no affinity for estrogens
and in the MDA-MB-468 cells, an ERα-negative breast cancer cell
line, it is highly expressed in comparison to the ERα-positive cell
line, MCF-7 (32). Recently, a
study demonstrated that tamoxifen binding to AEBS induces the
formation of small cytoplasmic vesicles or phagosomes in MCF-7
cells and their accumulation promoted cellular apoptosis (30). Few of these phagosomes contain the
early endocytic marker EEA1 (33).
These vesicles can fuse with endosomes leading to the degradation
of their content (34). Moreover,
caveolae vesicles have been shown to mediate the internalization of
proteins at the plasma membrane into cells, such as the EGFR
(35). Internalization of EGFR
could follow two distinct endocytotic routes: the
clathrin-dependent one and the clathrin-independent route mediated
by caveolin (36). The
EGFR-caveolin interaction reduces the activation of EGFR signaling
(37). Furthermore, a recent study
demonstrated that the accumulation of EGFR in the endosomes of
MDA-MB-468 cells induced EGFR-mediated apoptosis (15).
However, the effects of raloxifene on EGFR
expression and localization could be explained by another potential
mechanism routed in the possible ability of ERβ to control EGFR
signaling. Expression of ERβ has been detected in 20% of TNBC
(38) and at lower level in
MDA-MB-468 cells (39). However,
its role, if any, in the development and progression of breast
cancer remains unclear. Raloxifene binds with high affinity to ERα
and ERβ and usually acts as an antagonist in the breast (40). Interestingly, a recent study has
identified a physical interaction between ERβ, EGFR and caveolin-1
which reduces EGFR signaling in lung cancer (41), a similar mechanism involving
raloxifene binding to ERβ and the triggering of EGFR endocytosis
may be responsible for the tumor suppressing effect of raloxifene
treatment in vivo and warrants further analysis.
Several studies have identified a truncated variant
of 36-kDa of ERα (ERα-36), which is frequently expressed in TNBC
(42). ERα-36 has no intrinsic
transcriptional activity (43) but
mediates non-genomic estrogen signaling through the EGFR/Src/ERK
signaling pathway in TNBC (44).
ERα-36 was shown to possibly enhance tamoxifen agonist activity in
endometrial cancer cell lines suggesting the capability of SERMs to
directly bind to ERα-36 (45).
Therefore, the effect of raloxifene could be mediated through
ERα-36 interaction leading to a decreased EGFR expression and its
downstream signaling pathways.
The majority of cancer-related deaths are caused by
metastasis, a multistep process that depends on alterations of
tumor microenvironment, survival of cancer cells in the circulation
and colonization of a distant organ (46). Previous studies have shown that in
TNBC, a low level of EGFR expression correlates with a reduced
incidence of metastases (47).
Inhibition of invasive potential is important for the prevention of
tumor recurrence. Raloxifene treatment effectively reduced
migration, invasion and the malignancy potential of MDA-MB-231
cells in vitro and reduced microvessel density in
vivo. However, further in vivo studies are needed to
definitively prove the potential of raloxifene alone or in
combination to prevent ERα-negative metastasis.
Collectively these data show that an oral daily dose
of raloxifene suppressed tumor growth in two relevant mouse
xenograft models of TNBC. Moreover, raloxifene treatment acted
independently of ERα and this was mediated by decreased EGFR
protein levels and their altered localizations within endosomes.
Overall, this study shows that raloxifene can be a valuable
treatment for TNBC and significant new targets can be identified
with further studies. This mechanistic information may lead to the
development of new therapies for TNBC.
Acknowledgements
This study was supported by grants from the Otago
Medical Research Foundation (AG302-ST), UORG (ST), Health Research
Council of New Zealand (ST) and Lottery Health (ST). We would like
to thank Ms. Hayley Nehoff and Ms. Céline Bourdon for the editorial
assistance.
References
|
1
|
Sporn MB, Dowsett SA, Mershon J and Bryant
HU: Role of raloxifene in breast cancer prevention in
postmenopausal women: clinical evidence and potential mechanisms of
action. Clin Ther. 26:830–840. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jordan VC and Koerner S: Inhibition of
oestradiol binding to mouse uterine and vaginal oestrogen receptors
by triphenylethylenes. J Endocrinol. 64:193–194. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Croxtall JD, Emmas C, White JO, Choudhary
Q and Flower RJ: Tamoxifen inhibits growth of oestrogen
receptor-negative A549 cells. Biochem Pharmacol. 47:197–202. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Couldwell WT, Weiss MH, DeGiorgio CM, et
al: Clinical and radiographic response in a minority of patients
with recurrent malignant gliomas treated with high-dose tamoxifen.
Neurosurgery. 32:485–490. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Del Prete SA, Maurer LH, O’Donnell J,
Forcier RJ and LeMarbre P: Combination chemotherapy with cisplatin,
carmustine, dacarbazine and tamoxifen in metastatic melanoma.
Cancer Treat Rep. 68:1403–1405. 1984.PubMed/NCBI
|
|
6
|
Murphy LC and Sutherland RL: Differential
effects of tamoxifen and analogs with nonbasic side chains on cell
proliferation in vitro. Endocrinology. 116:1071–1078. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Plowman PN: Tamoxifen as adjuvant therapy
in breast cancer. Current status Drugs. 46:819–833. 1993.PubMed/NCBI
|
|
8
|
Mandlekar S and Kong ANT: Mechanisms of
tamoxifen-induced apoptosis. Apoptosis. 6:469–477. 2001. View Article : Google Scholar
|
|
9
|
Carey LA, Dees EC, Sawyer L, et al: The
triple-negative paradox: primary tumor chemosensitivity of breast
cancer subtypes. Clin Cancer Res. 13:2329–2334. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kaplan HG and Malmgren JA: Impact of
triple-negative phenotype on breast cancer prognosis. Breast J.
14:456–463. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Somers-Edgar TJ, Taurin S, Larsen L,
Chandramouli A, Nelson MA and Rosengren RJ: Mechanisms for the
activity of heterocyclic cyclohexanone curcumin derivatives in
estrogen receptor negative human breast cancer cell lines. Invest
New Drugs. 29:87–97. 2011. View Article : Google Scholar
|
|
12
|
Taurin S, Sandbo N, Qin Y, Browning D and
Dulin NO: Phosphorylation of β-catenin by cyclic AMP-dependent
protein kinase. J Biol Chem. 281:9971–9976. 2006.
|
|
13
|
Reagan-Shaw S, Nihal M and Ahmad N: Dose
translation from animal to human studies revisited. FASEB J.
22:659–661. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nielsen TO, Hsu FD, Jensen K, et al:
Immunohistochemical and clinical characterization of the basal-like
subtype of invasive breast carcinoma. Clin Cancer Res.
10:5367–5374. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rush JS, Quinalty LM, Engelman L, Sherry
DM and Ceresa BP: Endosomal accumulation of the activated epidermal
growth factor receptor (EGFR) induces apoptosis. J Biol Chem.
287:712–722. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ueno NT and Zhang D: Targeting EGFR in
triple-negative breast cancer. J Cancer. 2:324–328. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nickerson NK, Mohammad KS, Gilmore JL, et
al: Decreased autocrine EGFR signaling in metastatic breast cancer
cells inhibits tumor growth in bone and mammary fat pad. PLoS One.
7:e302552012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zoncu R, Perera RM, Balkin DM, Pirruccello
M, Toomre D and De Camilli P: A phosphoinositide switch controls
the maturation and signaling properties of APPL endosomes. Cell.
136:1110–1121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pol A, Lu A, Pons M, Peiró S and Enrich C:
Epidermal growth factor-mediated caveolin recruitment to early
endosomes and MAPK activation. J Biol Chem. 275:30566–30572. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Freedman VH and Shin SI: Cellular
tumorigenicity in nude mice: correlation with cell growth in
semi-solid medium. Cell. 3:355–359. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Riggs BL and Hartmann LC: Selective
estrogen-receptor modulators - mechanisms of action and application
to clinical practice. N Engl J Med. 348:618–629. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Perry RR, Kang Y and Greaves B: Effects of
tamoxifen on growth and apoptosis of estrogen-dependent and
-independent human breast cancer cells. Ann Surg Oncol. 2:238–245.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gelmann EP: Tamoxifen induction of
apoptosis in estrogen receptor-negative cancers: new tricks for an
old dog? J Natl Cancer Inst. 88:224–226. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim HT, Kim BC, Kim IY, et al: Raloxifene,
a mixed estrogen agonist/antagonist, induces apoptosis through
cleavage of BAD in TSU-PR1 human cancer cells. J Biol Chem.
277:32510–32515. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Eisen SF and Brown HA: Selective estrogen
receptor (ER) modulators differentially regulate phospholipase D
catalytic activity in ER-negative breast cancer cells. Mol
Pharmacol. 62:911–920. 2002. View Article : Google Scholar
|
|
26
|
Todorova VK, Kaufmann Y, Luo S and
Klimberg VS: Tamoxifen and raloxifene suppress the proliferation of
estrogen receptor-negative cells through inhibition of glutamine
uptake. Cancer Chemother Pharmacol. 67:285–291. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bosch A, Eroles P, Zaragoza R, Vina JR and
Lluch A: Triple-negative breast cancer: molecular features,
pathogenesis, treatment and current lines of research. Cancer Treat
Rev. 36:206–215. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cho EY, Chang MH, Choi YL, et al:
Potential candidate biomarkers for heterogeneity in triple-negative
breast cancer (TNBC). Cancer Chemother Pharmacol. 68:753–761. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hyatt DC and Ceresa BP: Cellular
localization of the activated EGFR determines its effect on cell
growth in MDA-MB-468 cells. Exp Cell Res. 314:3415–3425. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
de Medina P, Payre B, Boubekeur N, et al:
Ligands of the antiestrogen-binding site induce active cell death
and autophagy in human breast cancer cells through the modulation
of cholesterol metabolism. Cell Death Differ. 16:1372–1384.
2009.PubMed/NCBI
|
|
31
|
de Medina P, Paillasse MR, Segala G,
Poirot M and Silvente-Poirot S: Identification and pharmacological
characterization of cholesterol-5,6-epoxide hydrolase as a target
for tamoxifen and AEBS ligands. Proc Natl Acad Sci USA.
107:13520–13525. 2010.PubMed/NCBI
|
|
32
|
Payre B, de Medina P, Boubekeur N, et al:
Microsomal antiestrogen-binding site ligands induce growth control
and differentiation of human breast cancer cells through the
modulation of cholesterol metabolism. Mol Cancer Ther. 7:3707–3718.
2008. View Article : Google Scholar
|
|
33
|
Simonsen A and Tooze SA: Coordination of
membrane events during autophagy by multiple class III PI3-kinase
complexes. J Cell Biol. 186:773–782. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Razi M, Chan EY and Tooze SA: Early
endosomes and endosomal coatomer are required for autophagy. J Cell
Biol. 185:305–321. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sigismund S, Woelk T, Puri C, et al:
Clathrin-independent endocytosis of ubiquitinated cargos. Proc Natl
Acad Sci USA. 102:2760–2765. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Aguilar RC and Wendland B: Endocytosis of
membrane receptors: two pathways are better than one. Proc Natl
Acad Sci USA. 102:2679–2680. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Couet J, Sargiacomo M and Lisanti MP:
Interaction of a receptor tyrosine kinase, EGF-R, with caveolins.
Caveolin binding negatively regulates tyrosine and serine/threonine
kinase activities. J Biol Chem. 272:30429–30438. 1997. View Article : Google Scholar
|
|
38
|
Litwiniuk MM, Roznowski K, Filas V, et al:
Expression of estrogen receptor beta in the breast carcinoma of
BRCA1 mutation carriers. BMC Cancer. 8:1002008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vladusic EA, Hornby AE, Guerra-Vladusic
FK, Lakins J and Lupu R: Expression and regulation of estrogen
receptor beta in human breast tumors and cell lines. Oncol Rep.
7:157–167. 2000.PubMed/NCBI
|
|
40
|
Kuiper GGJM, Lemmen JG, Carlsson B, et al:
Interaction of estrogenic chemicals and phytoestrogens with
estrogen receptor β. Endocrinology. 139:4252–4263. 1998.
|
|
41
|
Pinton G, Thomas W, Bellini P, et al:
Estrogen receptor β exerts tumor repressive functions in human
malignant pleural mesothelioma via EGFR inactivation and affects
response to gefitinib. PLoS One. 5:e141102010.
|
|
42
|
Lee W-L, Chao H-T, Cheng M-H and Wang P-H:
Rationale for using raloxifene to prevent both osteoporosis and
breast cancer in postmenopausal women. Maturitas. 60:92–107. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang Z, Zhang X, Shen P, Loggie BW, Chang
Y and Deuel TF: A variant of estrogen receptor-α, hER-α36:
transduction of estrogen- and antiestrogen-dependent
membrane-initiated mitogenic signaling. Proc Natl Acad Sci USA.
103:9063–9068. 2006.
|
|
44
|
Zhang XT, Kang LG, Ding L, Vranic S,
Gatalica Z and Wang ZY: A positive feedback loop of ER-alpha36/EGFR
promotes malignant growth of ER-negative breast cancer cells.
Oncogene. 30:770–780. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lin SL, Yan LY, Zhang XT, et al:
ER-alpha36, a variant of ER-alpha, promotes tamoxifen agonist
action in endometrial cancer cells via the MAPK/ERK and PI3K/Akt
pathways. PLoS One. 5:e90132010. View Article : Google Scholar
|
|
46
|
Nguyen DX, Bos PD and Massague J:
Metastasis: from dissemination to organ-specific colonization. Nat
Rev Cancer. 9:274–284. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Viale G, Rotmensz N, Maisonneuve P, et al:
Invasive ductal carcinoma of the breast with the ‘triple-negative’
phenotype: prognostic implications of EGFR immunoreactivity. Breast
Cancer Res Treat. 116:317–328. 2009.
|