Introduction
Human DNA polymerase (pol) β is the primary
polymerase involved in base excision repair (BER) an essential
repair pathway that removes oxidized and alkylated bases from DNA
(1). Its small size and monomeric
nature make it an attractive candidate for biochemical and kinetic
analysis (2). DNA polymerase β has
also been suggested to be involved in DNA gap-filling reactions
during meiotic synapsis (3), the
repair of double-strand DNA breaks during non-homologous end
joining (4), nucleotide excision
repair of bulky DNA lesions (5,6) and
replication (7). Targeted
disruption of pol β in mice results in neonatal lethality, growth
retardation, and apoptotic cell death in the developing nervous
system suggesting a role for pol β in neurogenesis (8).
In contrast to the other human DNA polymerases, the
availability of a high-resolution crystal structure of pol β in
various liganded states provides a foundation to identify
functionally important residues for mechanistic studies, as well as
to interpret kinetic results with site-directed mutants.
Furthermore, pol β shares many structural and mechanistic features
with other DNA polymerases of known structure. For example, the
mechanism of DNA polymerization follows an ordered binding of
substrates to the enzyme, with the DNA template binding first
(9). These attributes make pol β
an excellent model for biochemical study of DNA synthesis and
fidelity.
DNA polymerase β lacks a proofreading exonuclease
domain, but encompasses two main domains: i) an amino-terminal
8-kDa lyase domain (responsible for the removal of the
5′-deoxyribose phosphate intermediate formed during BER) and ii) a
carboxyl-terminal 31-kDa polymerase domain (10). The polymerase domain (residues
91–335) can be further separated into three functionally distinct
subdomains, which correspond to the palm (C, catalytic), thumb (D,
duplex DNA binding) and fingers (N, dNTP selection or nascent base
pair binding) subdomains, according to the nomenclature that uses
the architectural analogy to a right hand (11).
Comparisons of pol β structures in various liganded
states have shown that upon dNTP binding to the binary (pol β/DNA)
complex, the N-subdomain rotates from an open to closed state
sandwiching the nascent base pair (dNTP-templating nucleotide)
between α-helix N and the primer terminus base pair (12,13).
These structures have also shown that productive binding of pol β
to both gapped and nicked DNA requires a 90° bend in the DNA
template strand at the 5′-phosphodiester linkage of the templating
residue. The bend allows residues of the N-subdomain to interact
with the nascent base-pair in the closed conformation (Fig. 1). Various mutagenesis studies have
shown that the pol β/dNTP contacts produced by this bend are
important for both polymerization efficiency and fidelity (reviewed
in ref. 2).
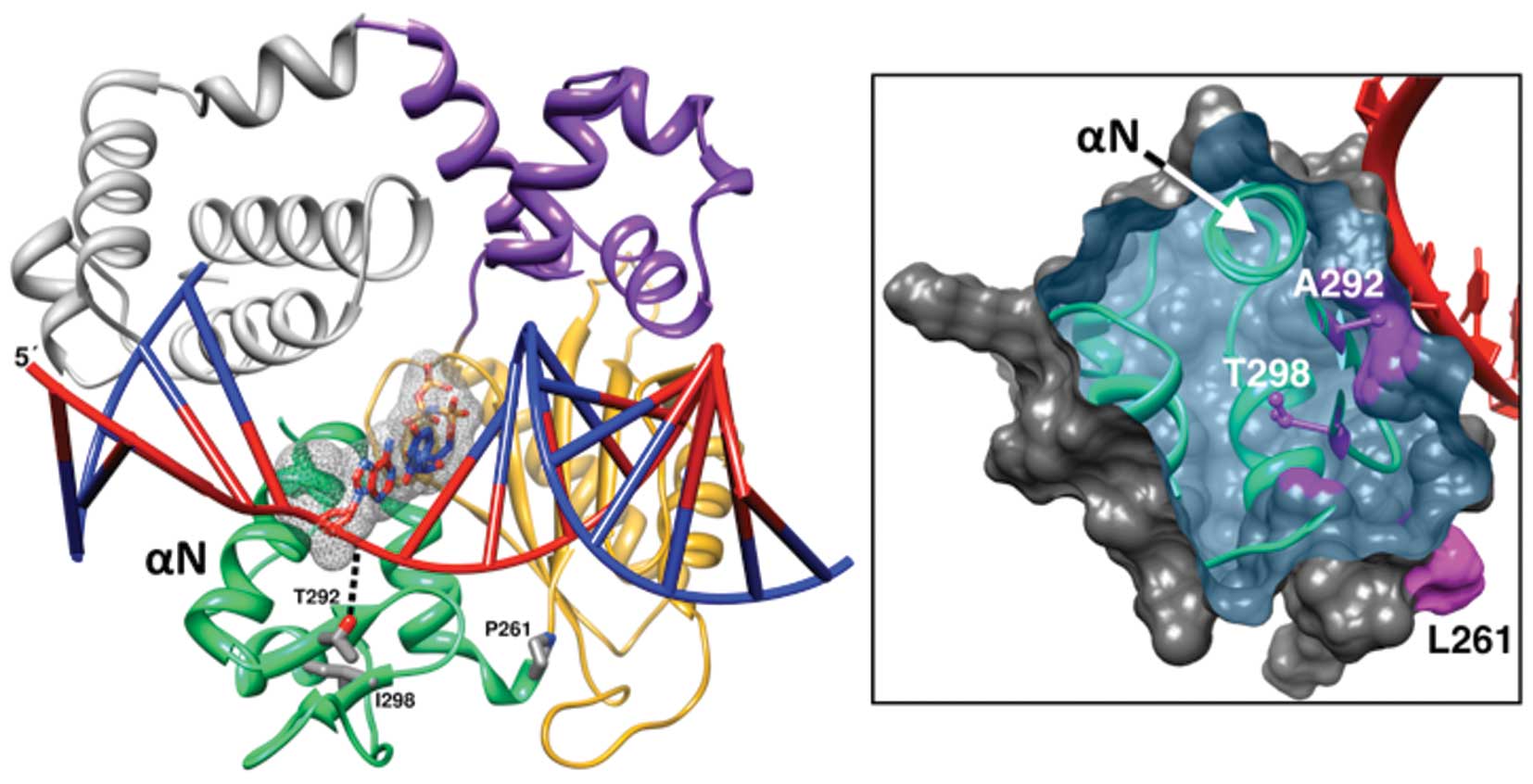 | Figure 1.Position of the variant residues of
the triple mutant in the structure of the DNA polymerase β ternary
substrate complex. Left panel, a ribbon representation of pol β
illustrating the polymerase (colored) and amino-terminal lyase
(grey) domains (PDB code 2FMS). The polymerase domain is composed
of three subdomains: D, purple; C, gold; and N, green. These
correspond to the thumb, palm, and fingers subdomains of DNA
polymerases that utilize an architectural analogy to a right-hand,
respectively. The single-nucleotide gapped DNA is illustrated in a
ladder representation with a red template strand and blue primer
and downstream DNA strands. The 5′-end of the template strand is
indicated. The nascent base pair (templating nucleotide and
incoming nucleoside triphosphate) is shown in a stick
representation with a mesh surface. The three residues (Pro261,
P261; Thr292, T292; and Ile298, I298) of the N-subdomain altered in
the triple mutant and α-helix N are shown. Right panel, the
molecular surface of the N-subdomain is clipped to expose the
internal position of the altered Thr298 (T298) and the surface
exposed Ala292 (A292). The altered residues are shown in magenta as
ball-and-sticks and their contribution to the surface is also
shaded in magenta. Although the residue cannot be seen in this
view, the surface contribution of Leu261 (L261) is indicated. The
altered side chains were modeled based on probable rotamers from
the Dunbrack library (36) and
exhibited no clashes with nearby residues. The red template strand
in the vicinity of the N-subdomain is also shown and α-helix N that
interacts with the nascent base pair is labeled. The molecular
images were produced in Chimera (35). |
Mutations in the pol β gene are commonly found in
tumor tissues (14) and in several
instances, these alterations are associated with diminished
polymerase fidelity (15),
catalytic activity (16), and
increase in cellular transformation (17). By screening the complete coding
region of the pol β gene in 26 prostate cancer tissues, we
identified 20 somatic mutations, nine of them missense (18). Subsequent biochemical analysis of
all missense pol β mutations demonstrated much smaller changes in
enzyme activity for all mutants compared to the triple mutant,
p.P261L/T292A/I298T, which had dramatically decreased activity
(19). The pol β triple mutant was
identified in an early-onset prostate cancer patient (18). No normal allele was present in the
patient’s tumor, unlike the adjacent normal prostate that was
wild-type, suggesting a clonal event during tumor evolution. In
order to appreciate the significance of the triple mutant, it is
essential to understand the molecular mechanism of the effects of
the mutations. In addition, information about the
structure-activity relationships of the mutations will enhance the
understanding of the pol β polymerase activity.
Crystallographic analyses provide insight into the
potential importance of the residues altered in the triple mutant.
Threonine-292 (Thr292) is a surface exposed residue that is not
part of the active site, but hydrogen bonds with the template
backbone immediately upstream of the coding templating nucleotide
(Fig. 1) (20). Alanine substitution would abolish
this hydrogen bonding interaction, and thus may affect template
binding, activity and/or fidelity. Proline-261 (Pro261) is situated
at the boundary between the C- and N-subdomains that reposition
themselves in response to nucleotide binding. Isoleucine-298
(Ile298) is distant from the active site and appears to form
packing interactions in the mobile N-subdomain (Fig. 1). Threonine substitution would
result in a buried side chain with hydrogen bonding capacity
(Fig. 1, right panel). Thus the
p.I298T substitution may alter the folding and/or stability of the
N-subdomain. Given these relatively mild consequences expected by
the single point mutations that comprise the triple mutant, the
complete loss of pol β activity observed previously was
unexpected.
We have constructed, purified and biochemically
analyzed all single and double mutant variants in vitro that
comprise the triple mutant in order to understand this apparent
paradox regarding the activity of pol β. Herein we demonstrate that
the loss of function exhibited by the triple mutant can be
separated into distinct enzyme activity and enzyme stability
changes, mainly afforded by the p.T292A and p.I298T point
mutations, respectively.
Materials and methods
Bacterial strains and growth
conditions
The strain BL21 DE3 was used for protein expression.
E. coli DH5α, BL21 (DE3) and recombinant E. coli
harboring pol β genes were cultured in LB medium containing
kanamycin (50 μg/ml) when appropriate.
Construction of pol β variants
Wild-type (WT) pol β was obtained from J.
Sweasy at Yale University. The mutants were obtained by the
Stratagene Quick-change Site-Directed Mutagenesis kit according to
the protocol of the manufacturer using the pET28a(+)-WT bacterial
expression vector as a template (21). Successful mutagenesis was confirmed
by DNA sequencing with BigDye chemistry on a 3100 ABI sequencer
(Perkin-Elmer).
Expression and purification of mutant
enzymes
E. coli strain BL21 (DE3) carrying
pET-28a(+)/pol β were grown at 37°C in LB medium containing 50
μg/ml kanamycin with 1 mM IPTG. The cells were harvested by
centrifugation, resuspended in 40 mM Tris, pH 8.0, 500 mM NaCl, 10
mM imidazole, and Protease Inhibitor Cocktail (as recommended by
the manufacturer; Sigma). Resuspended cells were lysed by
sonication. Extracts were cleared by centrifugation (15,000 rpm, 15
min at 4°C), and then loaded onto HisTrap FF crude Kit according to
the manufacturer’s instructions (GE Healthcare). Proteins were
eluted with 500 mM imidazole in 0.5 M NaCl. The elutants were
loaded onto a HiTrap SP HP column (GE Healthcare). The column was
washed with 100 mM NaCl and proteins were eluted with 2 M NaCl and
stored at −80°C in 50 mM Tris, pH 8.0, 1 mM EDTA, 2 M NaCl, 10%
glycerol and protease inhibitors as above (22,23).
We then estimated enzyme homogeneity based on Coomassie
Blue-stained SDS-PAGE gels. All proteins were quantified by
Bradford protein assay (Sigma). This quantification together with
the % homogeneity assessed by Coomassie Blue-stained gels (above)
allowed us to quantify the enzyme amounts for each mutant (used in
calculating Kcat below).
Western blot analysis
Expressed His-tagged proteins were identified by
western blot analysis (24).
Proteins were electrophoresed in a 12% SDS-PAGE gel and transferred
to a polyvinylidenedifluoride membrane (Thermo Scientific). Blots
were blocked by 5% non-fat dry milk in Tris-buffered saline
Tween-20 (0.1% Tween-20) and incubated with anti-His Tag antibody
(Sigma) according to the manufacturer’s protocol. For detection, we
used IRDye 800CW goat anti-rabbit IgG (LI-COR Biosciences) and the
Odyssey imaging system (LI-COR Biosciences).
DNA substrate
All oligonucleotides were synthesized and
high-pressure liquid chromatography-purified by Integrated DNA
Technologies. A 20-mer primer (5′-GCA GGA AAG CGA GGG TAT CC-3′)
and 20-mer downstream oligonucleotide (5′-ACA AAG TCC AGC GTA CCA
TA-3′) were annealed to a 46-mer template (5′-TAT GGT ACG CTG GAC
TTT GTG GGA TAC CCT CGC TTT CCT GCT CCT G-3′) to generate a
one-nucleotide gapped DNA substrate with a templating guanine
(25). The 20-mer primer was
5′-labeled with [γ-32P]-ATP (3,000 Ci/mmol;
Perkin-Elmer) using T4 polynucleotide kinase (US Biochemical Corp.)
according to the manufacturer’s protocol. The
5′-32P-labeled primer was then purified from
unincorporated label by a Microspin™ G-50 (GE Healthcare) column.
The downstream oligonucleotide was 5′-phosphorylated by Integrated
DNA Technologies. The oligonucleotides were annealed at a
primer:template:downstream oligonucleotide molar ratio of 1:1.2:1.3
in 50 mM Tris, pH 8.0, 250 mM NaCl, in order to create a single
nucleotide gap. The mixture was incubated at 95°C for 5 min, slow
cooled to 50°C over 30 min, and incubated at 50°C for 20 min and
then transferred to ice. Annealing of primer was confirmed on an
18% polyacrylamide (acrylamide/bis-acrylamide: 29:1) native gel
followed by autoradiography as described (26,27).
Protein stability assay
Protein stability was assessed by incubating pol β
for varying lengths of time (3, 6, 9 and 12 min) at 37°C or room
temperature (RT, 22°C). All reactions (20 μl) were performed
in 50 mM Tris-Cl, pH 8.0, 10 mM MgCl2, 2 mM DTT, 20 mM
NaCl, 0.2 mg/ml BSA, 2.5% glycerol with 40 nM pol β and 50 nM DNA.
All concentrations refer to the final concentration after mixing.
The reaction mixtures were pre-incubated in the absence of dCTP and
reactions were initiated by the addition of 12.8 μM dCTP.
After incubation for 2 min at 37°C and 6 min at RT, the reactions
were quenched by adding 20 μl of formamide loading buffer
(900 μl formamide, 22.2 μl 0.5 M EDTA, pH 8.0 and
77.8 μl water) and boiled for 10 min, and then transferred
to ice. Products were resolved on a 15% polyacrylamide
(acrylamide/bis-acrylamide: 29:1) gel containing 7 M urea. Gels
were dried and the products were quantified with a PhosphorImager
(Molecular Dynamics).
Kinetic characterization
The conditions of all incorporation reactions were
the same as those described above for the protein stability assay.
Kinetic reactions were performed at 37°C for stable variants and at
RT for all variants. To determine Km,dNTP, the
reaction mixtures contained 2.5 nM purified pol β for correct
incorporation and 50 nM pol β for incorrect incorporation with 50
nM annealed DNA substrate. All reactions were performed by first
pre-incubating the DNA substrate with pol β for 3 min without
dNTPs. Reactions were initiated by the addition of a single dNTP
(0.1–2,000 μM) and incubated for 2 min at 37°C (for stable
variants) and for 6 min at RT (for all variants). For
Km,DNA determinations, RT reactions contained 20 nM WT
or T292A pol β and 40 nM triple mutant; at 37°C, 8 nM enzyme was
used. Reactions were initiated by addition of enzyme mixtures to
annealed single-nucleotide gapped DNA substrate (0.01–3.2
μM) and incubated for 4 min at RT or 37°C. The template
nucleotide in the gap was deoxyguanosine and the dCTP concentration
was 100 μM. After incubation, the reactions were quenched as
described above for the protein stability assay and quantified as
above to obtain the percentage of product formed. Time courses were
linear for the chosen enzyme concentration and time interval.
Data analysis
The kinetic data were extracted from Lineweaver-Burk
plots. We determined the values of kcat and
Km,dNTP from trend line equations calculated from
these plots with Microsoft Excel software (Microsoft). Apparent
kcat was calculated from Vmax,
where kcat = Vmax/(apparent
enzyme). The apparent enzyme concentration was estimated from total
protein. Fidelities for misinsertion reactions were calculated
using the following equation: fidelity =
[(kcat/Km,correct) +
(kcat/Km,incorrect)]/(kcat/Km,incorrect).
Results
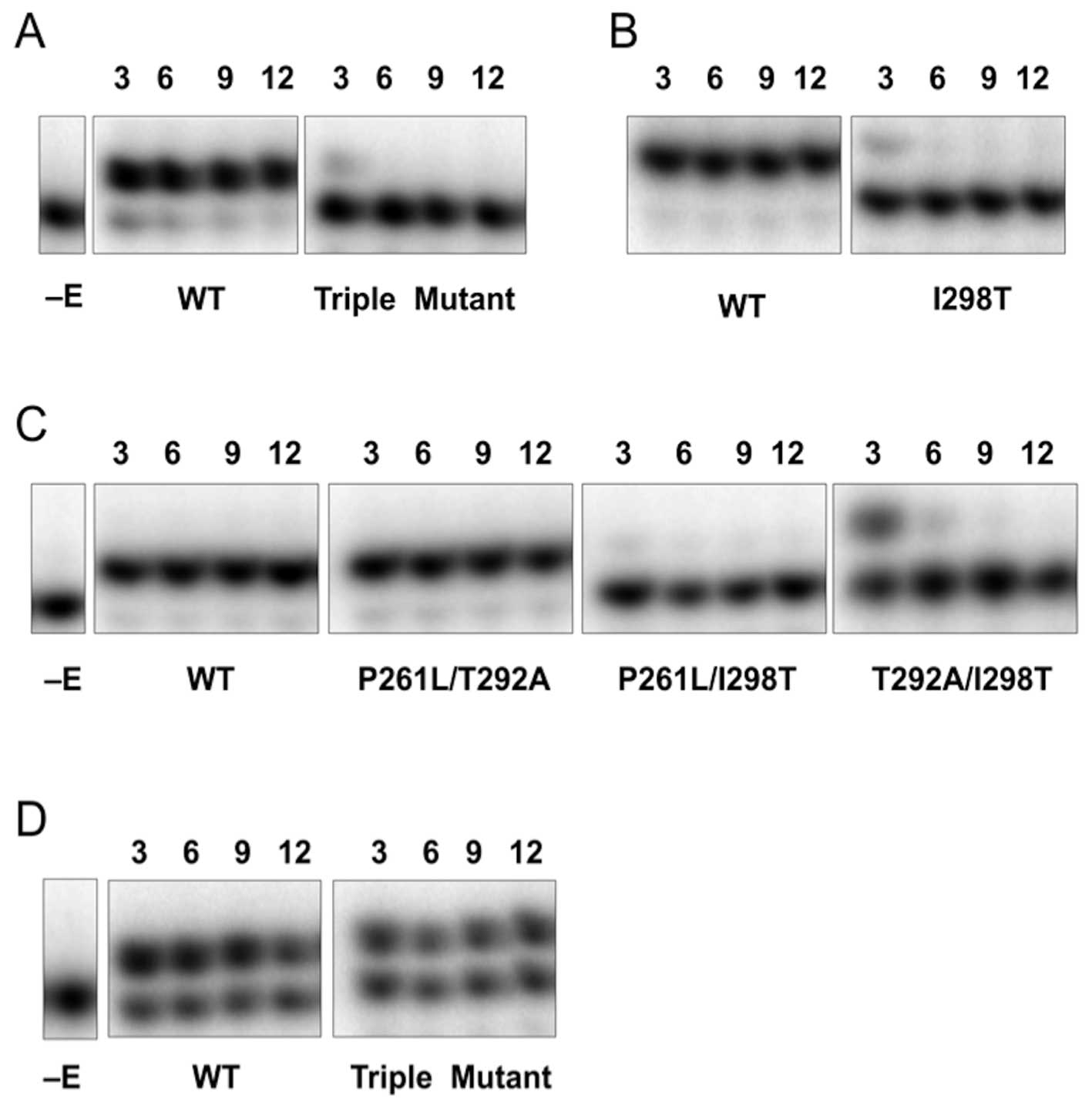
Triple mutant stability
Variants of a triple mutant of human pol β
identified previously in prostate cancer tissues (18) were obtained by site-directed
mutagenesis. The WT, triple mutant (p.P261L/T292A/I298T), and all
single and double mutant variants of pol β that comprise the triple
mutant were expressed in E. coli and purified as described
in Materials and methods. After purification, WT and the variants
of pol β were analyzed by SDS-PAGE and identified by western blot
analysis (data not shown), and proteins were quantified by Bradford
protein assay. Expression of pol β was poor when the p.P261L
mutation was included in the protein. Thus, p.P261L, p.P261L/T292A,
p.P261L/I298T, and triple mutant were partially purified to 49, 60,
50 and 51% homogeneity, respectively. The lack of any polymerase or
exonuclease activity after prolonged incubations at 37°C in the
stability measurements described below (Fig. 2) indicates that E. coli
polymerases do not likely contribute to product formation in these
preparations at room temperature or 37°C and that our purification
protocol removes contaminating activities from our enzyme
preparations. In contrast, the WT, p.T292A, p.I298T and
p.T292A/I298T variants were greater than 90% homogenous (19 and
data not shown).
Following enzyme purification, we performed assays
of pol β activity by using a DNA substrate that was previously used
for pol β fidelity studies (25).
We determined pol β catalytic efficiency based on kinetic analyses
of single-nucleotide addition opposite template dG with
single-nucleotide gapped DNA substrate. Since the catalytic
efficiency (kcat/Km,dCTP) of
the triple mutant on a gapped DNA substrate was dramatically
reduced at 37°C (Fig. 2) and it
was difficult to purify the triple mutant to more than 50%
homogeneity, we hypothesized that this mutant may be unstable at
37°C and was degraded during bacterial expression.
To probe this hypothesis, we tested the stability of
WT and all single, double and triple mutant variants of pol β by
examining the sensitivity of the dCTP incorporation reaction to
incubation time at 37°C. Interestingly, the triple mutant, p.I298T,
p.P261L/I298T and p.T292A/I298T variants are unstable at 37°C:
after 3 min of pre-incubation at 37°C, the activity of those
variants was dramatically decreased (Fig. 2A–C). In contrast, the p.P261L/T292A
variant and its respective single mutant variants are stable at
37°C (Fig. 2C and data not shown).
These results suggest that the stability of the triple mutant at
37°C was affected specifically by the p.I298T alteration. At room
temperature, the stability of the triple mutant is increased and
comparable to WT (Fig. 2D). Since
several of the mutant forms (e.g., p.I298T, p.P261L/I298T,
p.T292A/I298T, p.P261L/T292A/I298T) do not exhibit activity at long
incubation periods at 37°C, these preparations provide an
opportunity to ascertain whether contaminating activities exist.
The lack of any activity indicates that E. coli polymerases
do not contribute to product formation in these preparations at
room temperature and that our purification protocol removes
contaminating polymerase activities from our enzyme
preparations.
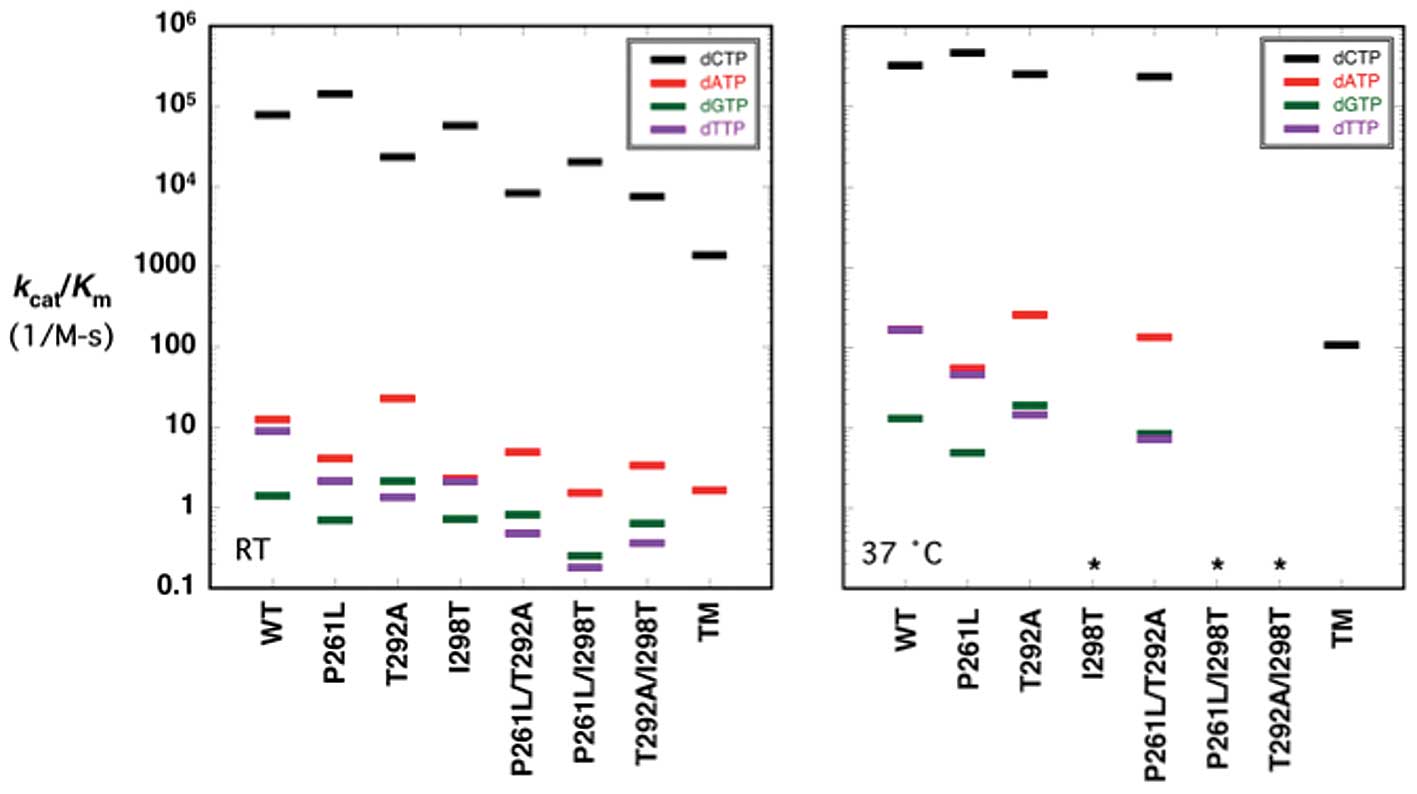
Catalytic efficiencies for correct
incorporation
Following the confirmation of decreased stability
conferred by the triple mutant, we decided to test whether
stability alone explains the dramatic reduction in activity at 37°C
(Fig. 2), by measuring the
steady-state kinetic parameters for all of the variants at room
temperature (Table I; Fig. 3, left panel). As expected, the
apparent kcat values of the pol β variants that
could be measured at 37°C (Table
II) were lower at room temperature.
 | Table I.Kinetic summary for single-nucleotide
gap-filling opposite a templating guanine at room temperature
(22°C) by wild-type pol β and variants. |
Table I.
Kinetic summary for single-nucleotide
gap-filling opposite a templating guanine at room temperature
(22°C) by wild-type pol β and variants.
| Enzyme | dNTP |
kcata 10−2
s−1 |
Km,dNTP μM |
kcat/Km
10−2 μM−1s−1 |
|---|
| WTb | dCTP | 3.24 (0.27) | 0.42 (0.04) | 771 (150) |
| p.P261L | 5.57 (0.33) | 0.39 (0.04) | 1,430 (80) |
| p.T292A | 4.98 (0.13) | 2.16 (0.16) | 231 (11) |
| p.I298T | 1.35 (0.30) | 0.24 (0.03) | 563 (51) |
| p.P261L/T292A | 4.98 (0.52) | 6.06 (0.20) | 82 (8) |
| p.P261L/I298T | 2.50 (0.27) | 1.25 (0.10) | 200 (27) |
| p.T292A/I298T | 3.25 (0.08) | 4.38 (0.35) | 74 (9) |
| TMc | 2.36 (0.34) | 17.08 (4.48) | 14 (1) |
| WT | dATP | 0.164 (0.012) | 131 (22) | 0.125 (0.029) |
| p.P261L | 0.135 (0.005) | 331 (14) | 0.041 (0.001) |
| p.T292A | 0.223 (0.010) | 98 (11) | 0.227 (0.015) |
| p.I298T | 0.091 (0.006) | 397 (64) | 0.023 (0.003) |
| p.P261L/T292A | 0.095 (0.001) | 195 (21) | 0.049 (0.008) |
| p.P261L/I298T | 0.037 (0.005) | 244 (44) | 0.015
(<0.001) |
| p.T292A/I298T | 0.125 (0.018) | 373 (60) | 0.035 (0.002) |
| p.TM | 0.046 (0.002) | 282 (27) | 0.016 (0.002) |
| WT | dGTP | 0.110 (0.007) | 787 (55) | 0.014 (0.001) |
| p.P261L | 0.211 (0.069) | 3,036 (1,397) | 0.007 (0.001) |
| p.T292A | 0.113 (0.009) | 531 (44) | 0.021
(<0.001) |
| p.I298T | 0.064 (0.006) | 880 (262) | 0.007 (0.001) |
| p.P261L/T292A | 0.024 (0.003) | 293 (66) | 0.008 (0.001) |
| p.P261L/I298T | 0.055 (0.010) | 2,205 (589) | 0.003
(<0.001) |
| p.T292A/I298T | 0.019 (0.002) | 293 (80) | 0.006 (0.002) |
| TM | NDd | ND | ND |
| WT | dTTP | 0.244 (0.008) | 275 (52) | 0.089 (0.013) |
| p.P261L | 0.143 (0.017) | 665 (65) | 0.021 (0.001) |
| p.T292A | 0.110 (0.021) | 822 (164) | 0.013 (0.001) |
| p.I298T | 0.089 (0.026) | 424 (188) | 0.021 (0.003) |
| p.P261L/T292A | 0.017 (0.001) | 367 (75) | 0.005 (0.001) |
| p.P261L/I298T | 0.019 (0.002) | 1,068 (77) | 0.002
(<0.001) |
| p.T292A/I298T | 0.017 (0.003) | 483 (102) | 0.004 (0.001) |
| TM | ND | ND | ND |
| Table II.Kinetic summary for single-nucleotide
gap-filling opposite a templating guanine at 37°C by wild-type pol
β and variants. |
Table II.
Kinetic summary for single-nucleotide
gap-filling opposite a templating guanine at 37°C by wild-type pol
β and variants.
| Enzyme | dNTP |
kcata 10−2
s−1 |
Km,dNTP μM |
kcat/Km
10−2 μM−1s−1 |
|---|
| WTb | dCTP | 8.09 (2.09) | 0.25 (0.10) | 3,240 (920) |
| p.P261L | 13.6 (0.9) | 0.29 (0.07) | 4,690 (1,450) |
| p.T292A | 17.3 (1.2) | 0.68 (0.28) | 2,540 (730) |
| p.I298T | NDc | ND | ND |
| p.P261L/T292A | 25.6 (1.2) | 1.08 (0.15) | 2,370 (300) |
| p.P261L/I298T | ND | ND | ND |
| p.T292A/I298T | ND | ND | ND |
| TMd | 0.040 (0.003) | 3.72 (0.17) | 1.08 (0.08) |
| WT | dATP | 0.786 (0.012) | 47 (2) | 1.680 (0.093) |
| p.P261L | 0.630 (0.024) | 114 (5) | 0.553 (0.026) |
| p.T292A | 0.846 (0.048) | 33 (2) | 2.557 (0.032) |
| p.I298T | ND | ND | ND |
| p.P261L/T292A | 0.792 (0.037) | 59 (4) | 1.348 (0.019) |
| p.P261L/I298T | ND | ND | ND |
| p.T292A/I298T | ND | ND | ND |
| TM | ND | ND | ND |
| WT | dGTP | 0.587 (0.001) | 446 (16) | 0.131 (0.005) |
| p.P261L | 0.527 (0.062) | 1,069 (223) | 0.049 (0.005) |
| p.T292A | 0.709 (0.032) | 376 (42) | 0.189 (0.015) |
| p.I298T | ND | ND | ND |
| p.P261L/T292A | 0.490 (0.017) | 586 (25) | 0.084 (0.001) |
| p.P261L/I298T | ND | ND | ND |
| p.T292A/I298T | ND | ND | ND |
| TM | ND | ND | ND |
| WT | dTTP | 0.870 (0.012) | 52 (2) | 1.667 (0.030) |
| p.P261L | 0.702 (0.028) | 152 (15) | 0.460 (0.030) |
| p.T292A | 0.715 (0.048) | 491 (43) | 0.146 (0.005) |
| p.I298T | ND | ND | ND |
| p.P261L/T292A | 0.410 (0.011) | 569 (34) | 0.072 (0.003) |
| p.P261L/I298T | ND | ND | ND |
| p.T292A/I298T | ND | ND | ND |
| TM | ND | ND | ND |
Since both the triple mutant and all I298T
containing variants are unstable at 37°C, we assayed these variants
at room temperature to ascertain if the modified side chains had
kinetic consequences other than reducing protein stability.
Variants of triple mutant, p.I298T, and p.P261L/I298T displayed a
moderately reduced apparent kcat at RT compared
with WT, whereas the p.T292A/I298T variant showed similar catalytic
activity to that of WT pol β at RT (Table I). Since the active fraction of
enzyme is unknown in these preparations, it is difficult to come to
any definitive conclusions concerning activity alone. However, the
apparent Km values are independent of active
enzyme fraction and thus constitute convenient kinetic parameters
to monitor altered kinetic constants (28). The apparent
Km,dCTP of the triple mutant, p.T292A/I298T, and
p.P261L/I298T variants were increased at room temperature 40-, 10-
and 3-fold, respectively, compared to WT (Table I). At 37°C, the apparent
Km,dCTP of the triple mutant was increased
15-fold compared to WT, similar to the change observed at RT
(Table II). Likewise, the
Km,dCTP of the p.T292A and p.P261L/T292A variants
were significantly increased at both RT and 37°C relative to WT
(Tables I and II).
A comparison of the catalytic efficiencies or
specificity constants for WT and the variants at RT indicates that
the triple mutant exhibits the lowest catalytic efficiency for
correct nucleotide insertion and p.P261L has similar or greater
efficiency than WT (Table I;
Fig. 3, left panel). The catalytic
efficiencies for the other variants were intermediate between these
two extremes (Table I; Fig. 3, left panel). Significantly, all
p.T292A-containing variants reduced catalytic efficiency compared
to WT at RT: the p.T292A variant (by 3.3-fold), the p.P261L/T292A
variant (by 9.4-fold) and the p.T292A/I298T variant (by 10.4-fold)
(Table I). In addition, the
p.P261L/I298T variant decreased catalytic efficiency at RT by
3.9-fold (Table I). These
differences are most easily seen in Fig. 3 where the catalytic efficiencies
for correct insertion (dCTP, black lines) are plotted for WT and
each variant (note the logarithmic ordinate scale). Therefore, the
triple mutant affects catalysis independently of its effect on
stability. Due to the lack of protein stability, we did not measure
kinetic parameters for any of the p.I298T containing mutants at
37°C. The triple mutant displays a catalytic efficiency for correct
insertion 3,000-fold lower than WT at 37°C (19; Table II; Fig. 3, right panel). The catalytic
efficiency of the remaining mutants, p.P261L, p.T292A, and
p.P261L/T292A was similar to that of WT at 37°C (Table II and Fig. 3, right panel).
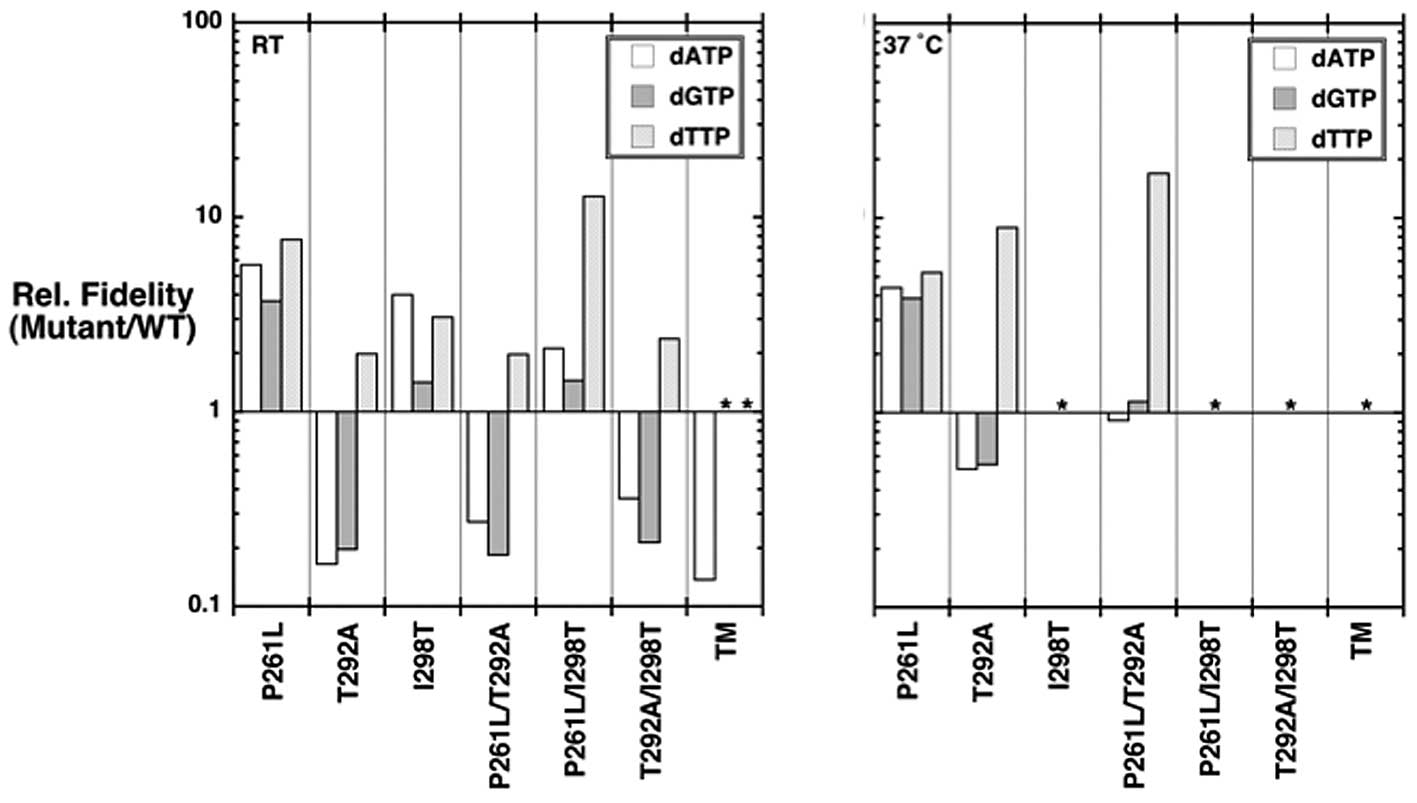
Fidelity of the pol β variants
Misincorporation fidelity studies were performed to
understand the role of the pol β variants on DNA synthesis
fidelity. The apparent kcat,
Km,dNTP, and catalytic efficiency
(kcat/Km,dNTP) values for
misincorporation at room temperature and 37°C are tabulated in
Tables I and II, respectively. The instability of the
p.I298T variants at 37°C precluded fidelity assays for many of the
variants at the elevated temperature. The catalytic efficiencies
for misinsertion are also illustrated in discrimination plots
(Fig. 3) and their relative
fidelities (mutant/WT) shown in Fig.
4. In discrimination plots, the distance between correct
insertion (black lines) to that of a misinsertion (colored lines)
is directly proportional to fidelity or misinsertion frequency
(29). Accordingly, the shorter
the distance between these points (correct insertion and
misinsertion), the lower the fidelity. For example, focusing on the
kinetic constants shown in Fig. 3
for the p.P261L mutant indicates that fidelity for all three
misinsertions is greater than that for WT (e.g., efficiency for
dCTP insertion is increased relative to WT and misinsertions are
reduced compared to WT).
We were unable to measure the catalytic activity for
dTTP and dGTP misincorporation for the triple mutant due to its
very low misinsertion efficiency (even at very high enzyme
concentrations). With regards to catalytic efficiency for
misincorporation
(kcat/Km,dNTP), the
kcat/Km,dNTP for the p.T292A
variant was increased for both dATP and dGTP misincorporations and
decreased for dTTP. The remaining variants showed decreased
catalytic efficiencies for all misincorporations (Table I, Fig.
3). The results of misincorporation at 37°C are tabulated in
Table II and illustrated in
Fig. 3 (right panel). For the
variants that could be examined, the results parallel those
observed at room temperature (Fig. 3,
left panel).
Fidelity is defined as the ratio of the sum of
catalytic efficiencies of correct and incorrect nucleotide
incorporation over the catalytic efficiency for misinsertion. Since
the efficiency for misinsertion is much lower than that for correct
insertion (Tables I and II), a simplified view is that fidelity is
simply the ratio of catalytic efficiencies (correct/incorrect). As
explained above, the distance between the plotted catalytic
efficiencies for correct and incorrect insertions is directly
proportional to fidelity (Fig. 3).
The relative fidelities (mutant/WT) can easily be illustrated
(Fig. 4). The triple mutant has a
7-fold lower fidelity than WT for dATP transversions at room
temperature. In contrast, the p.P261L variant had a significantly
higher fidelity than WT at both room temperature and 37°C. The
p.I298T and p.P261L/I298T variants also exhibited a higher fidelity
at room temperature for all three misinsertions that were
assayed.
The p.T292A variant had decreased relative
misinsertion fidelity for dATP and dGTP at room temperature and
37°C (Fig. 4), and an increased
relative fidelity for dTTP at both temperatures. Thus, the p.T292A
variant displays higher relative fidelity for transitions and lower
relative fidelity for transversions. The p.P261L/T292A and
p.T292A/I298T variants (both containing p.T292A) behave like the
p.T292A variant (Fig. 4). Indeed,
although the p.P261L and p.I298T variants have higher relative
fidelity for dATP and dGTP misincorporation compared to the p.T292A
variant, the p.T292A appears to be dominant over the other mutation
in the double mutants p.P261L/T292A and p.T292A/I298T.
DNA substrate binding
Given the dramatic effect of the triple and T292A
mutations on the apparent Km,dNTP (Tables I and II) and the structural observation that
Thr292 forms a hydrogen bond with template strand (Fig. 1), we investigated its effect on the
affinity for the gapped DNA substrate. A steady-state kinetic
analysis indicates that DNA binding is weaker for these mutants
(Table III) and that the elevated
Km,dNTP is in part due to weaker DNA binding
(28). Accordingly, the poor
catalytic efficiency of the triple mutant at room temperature is
due to its poor DNA binding (Table
III).
| Table III.Dependence of DNA synthesis on
single-nucleotide gapped DNA concentration at room temperature and
37°C by wild-type pol β and variants. |
Table III.
Dependence of DNA synthesis on
single-nucleotide gapped DNA concentration at room temperature and
37°C by wild-type pol β and variants.
| Enzyme | Temperature |
kcata 10−2
s−1 |
Km,DNA μM |
kcat/Km
10−2 μM−1s−1 |
|---|
| WTb | RT | 7.8 (0.2) | 0.16 (0.01) | 4,875 (75) |
| 37°C | 9.8 (0.5) | 0.07
(<0.01) | 14,000 (240) |
| TMc | RT | 40.4 (2.0) | 1.5 (0.1) | 2,700 (20) |
| 37°C | NDd | ND | ND |
| T292A | RT | 3.6 (0.1) | 0.25 (0.02) | 1,440 (40) |
| 37°C | 38.6 (7.3) | 1.48 (0.32) | 2,610 (60) |
Discussion
Biochemical analyses of all the single and double
mutant variants that comprise the triple pol β mutant demonstrate
that the loss of function shown by the triple mutant can be
separated into distinct enzyme activity and stability changes. More
specifically, the p.I298T pol β variant is responsible for the
instability shown by the triple mutant at 37°C (Fig. 2), while the p.T292A variant is
responsible for the loss of DNA synthesis fidelity (Fig. 4), and most of the reduction in
catalytic efficiency seen at RT (Fig.
3, left panel). However, the triple mutant exhibits an
efficiency significantly lower than that of p.T292A alone (Table I). Thus, the lower catalytic
efficiency of the triple mutant represents a synergistic effect of
all three mutations. The third variant, p.P261L, exhibits a modest
increase in fidelity relative to WT enzyme. Interestingly, all
three alterations of the triple mutant are in the same pol β
subdomain (N-subdomain; Fig. 1).
The N-subdomain (residues 261–335) undergoes a large conformational
change upon dNTP binding to interact with the nascent base pair
(2). Accordingly, alterations to
this subdomain that interfere with conformational
changes/adjustments or interactions with the nascent base pair
would be expected to impact enzyme activity and/or fidelity.
The dramatic effect of the p.I298T pol β variant on
enzyme stability observed at 37°C was surprising, but can be
rationalized considering the pol β structure (Fig. 1). The hydrophobic isoleucine
residue found in the wild-type enzyme would be expected to provide
good packing for the interior of the N-subdomain. The threonine
substitution buries a hydroxyl group that would be expected to
lower the stability of this subdomain. Also noteworthy, residues
292 and 298 are adjacent to one another in anti-parallel β-strands
(Fig. 1). Thus, the loss of
activity at 37°C is most easily explained by the decreased
stability of the N-subdomain with the p.I298T substitution. Since
there is activity, albeit low, at room temperature, the N-subdomain
must be at least partially folded at the lower temperature to
permit interactions with the nascent base pair. Previous work had
demonstrated that loss of the Arg283 interaction with the template
strand by site-directed mutagenesis results in a >30,000-fold
loss in catalytic efficiency (30,31).
In light of such a dramatic effect, the decrease in catalytic
efficiency afforded by the triple mutant is in reasonable
context.
Although the p.I298T variants are unstable at 37°C,
they exhibit significant activity at room temperature permitting
kinetic characterization (Table
I). While the activity of the pol β variants assayed at RT was
similar to wild-type enzyme, catalytic efficiency was significantly
decreased for the triple mutant and all p.T292A-containing variants
(Table I; Fig. 3, left panel), mostly due to a
significant increase in the apparent Km,dNTP
(Table I). The
Km,dNTP can increase due to an increase in the
dissociation constant Kd,dNTP, decreased rate of
dNTP insertion, or decreased DNA binding affinity (i.e., increased
dissociation rate constant, koff) (28). The p.T292A substitution is
predicted to eliminate an important hydrogen bond with the DNA
backbone immediately upstream of the templating (coding) nucleotide
(Fig. 1). The observed increase in
Km,dNTP for the p.T292A variants may thus be
partially due to decreased DNA binding affinity and is consistent
with the observed increased catalytic activity of these variants
(Table I) since increasing the DNA
dissociation rate constant can enhance catalytic cycling (32). Consistent with this idea, the
Km,DNA for the T292A variant and triple mutant
are increased relative to wild-type enzyme (Table III). However, this interpretation
must be tempered since the active enzyme fraction is not known. An
alternate explanation is that the loss of the hydrogen bond
destabilizes the coding templating base making it more difficult
for the polymerase to identify its correct base pairing partner.
This could result in both an increase in the binding affinity for
the incoming nucleotide and/or a decrease in the rate of
insertion.
Since DNA synthesis fidelity approximates the ratio
of catalytic efficiencies for right and wrong nucleotides, these
efficiencies must be differentially altered for a variant
polymerase to exhibit an altered fidelity. There are several
examples where mutations distant from the pol β active site have
impacted DNA synthesis fidelity (2). The only single nucleotide alteration
of the triple mutant that directly contacts the DNA substrate,
p.T292A, exhibits a significantly lower fidelity for transversions
(dATP and dGTP misinsertions opposite dG; Figs. 3 and 4). Importantly, the double mutants
containing p.T292A (p.P261L/T292A and p.T292A/I298T) also exhibit
the transversion over transition bias. The triple mutant likewise
exhibits a lower fidelity than WT for dATP misinsertion opposite G.
Interestingly, the p.P261L single mutant has a significantly higher
fidelity than WT at room temperature and 37°C, but the
p.P261L/T292A double mutant does not. These data suggest that the
p.T292A mutation is dominant compared to both p.P261L and p.I298T
with regards to fidelity. Several previously characterized pol β
mutants, including some found in tumors, exhibit a base
substitution bias. For example, the p.D246V pol β mutant, present
in the flexible loop in the catalytic C-subdomain (Fig. 1), shows a preferential misinsertion
of dTTP opposite guanine relative to WT (33). This misincorporation bias makes the
p.D246V enzyme a mutator mainly for C to T transitions. In
contrast, all of the remaining pol β mutants we identified in
prostate tumors do not exhibit this bias (19). However, fidelity is dependent on
DNA sequence and dNTP pool imbalances, so a strict correlation
between in vitro polymerase fidelity measurements with
altered polymerases and their cellular impact on mutagenesis is
qualitative.
Crystallographic analyses of pol β have shown that
productive binding of pol β to both gapped and nicked DNA template
requires a 90° bend in the DNA template (12,13).
This 90° bend allows α-helix N residues of the N-subdomain of pol β
to interact with the nascent base-pair in the closed conformation
(Fig. 1). Various mutagenesis
studies have shown that the pol β/dNTP contacts produced by this
90° bend are important for both polymerization efficiency and
fidelity (10,30,32).
Thus, residues that influence the equilibrium between the open and
closed forms could potentially modulate enzyme activity or fidelity
if they were modified. Thr292 is far from the template strand in
the open binary DNA complex, but as discussed above, the hydrogen
bond between Thr292 and the template strand would be expected to
stabilize the closed pol β form as well as assist template base
positioning. Consistent with this prediction, removal of this
hydrogen bond through alanine substitution (p.T292A) results in
increases of both the apparent Km for the DNA and
the incoming dNTP. Curiously, Pro261 is situated at the boundary
between the C- and N-subdomains, at a position critical for the
transition from open to closed form (2,13).
The observation that leucine substitution of Pro261 increases
fidelity of the mutant due to a decrease in catalytic efficiency
for misinsertions (Fig. 3)
suggests that the closed conformation is destabilized in the
p.P261L mutant during incorrect nucleotide incorporation.
In summary, the in vitro kinetic analyses
presented here demonstrate that the loss of function shown by the
pol β early onset prostate tumor-associated triple mutant can be
separated into distinct changes in catalytic efficiency and
fidelity (primarily the p.T292A point mutation) as well as an
enzyme stability defect (p.I298T mutation). The results explain the
mechanistic underpinning of the dramatic loss in enzymatic activity
of this tumor-associated mutant form of pol β. The fact that the
wild-type allele of pol β was not found in the tumor indicates that
this tumor was pol β null. This genotype would be expected to
result in a base excision repair defect and thus genomic
accumulation of single base lesions. Alternatively, a less faithful
DNA polymerase may fulfill gap-filling DNA synthesis during repair.
Previously it was shown that the loss of pol β function in mouse
embryonic fibroblasts results in an increase in spontaneous and
alkylation-induced mutation frequency (34). The therapeutic implications of
these data are interesting to consider.
Acknowledgements
This research was supported by
Research Project Numbers Z01-ES050158 and Z01-ES050161 in the
Intramural Research Program of the National Institutes of Health,
National Institute of Environmental Health Sciences and was in
association with the National Institutes of Health Grant
1U19CA105010. NMM is supported by grant number P20RR020152-06 from
the National Institutes of Health, and PC094628 from the Department
of Defense. Molecular graphics images were produced using the
Chimera package (35) from the
Resource for Biocomputing, Visualization, and Informatics at the
University of California, San Francisco (supported by NIH P41
RR-01081).
References
|
1.
|
Friedberg EC: DNA damage and repair.
Nature. 421:436–440. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Beard WA and Wilson SH: Structure and
mechanism of DNA polymerase β. Chem Rev. 106:361–382. 2006.
|
|
3.
|
Kidane D, Jonason AS, Gorton TS, Mihaylov
I, Pan J, Keeney S, de Rooij DG, Ashley T, Keh A, Liu Y, Banerjee
U, Zelterman D and Sweasy JB: DNA polymerase β is critical for
mouse meiotic synapsis. EMBO J. 29:410–423. 2010.
|
|
4.
|
Wilson TE and Lieber MR: Efficient
processing of DNA ends during yeast nonhomologous end joining:
evidence for a DNA polymerase β (POL4)-dependent pathway. J Biol
Chem. 274:23599–23609. 1999.PubMed/NCBI
|
|
5.
|
Horton JK, Srivastava DK, Zmudzka BZ and
Wilson SH: Strategic down-regulation of DNA polymerase β by
antisense RNA sensitizes mammalian cells to specific DNA damaging
agents. Nucleic Acids Res. 23:3810–3815. 1995.
|
|
6.
|
Oda N, Saxena JK, Jenkins TM, Prasad R,
Wilson SH and Ackerman EJ: DNA polymerases α and β are required for
DNA repair in an efficient nuclear extract from Xenopus
oocytes. J Biol Chem. 271:13816–13820. 1996.
|
|
7.
|
Jenkins TM, Saxena JK, Kumar A, Wilson SH
and Ackerman EJ: DNA polymerase β and DNA synthesis in
Xenopus oocytes and in a nuclear extract. Science.
258:475–478. 1992.
|
|
8.
|
Sugo N, Aratani Y, Nagashima Y, Kubota Y
and Koyama H: Neonatal lethality with abnormal neurogenesis in mice
deficient in DNA polymerase β. EMBO J. 19:1397–1404.
2000.PubMed/NCBI
|
|
9.
|
Tanabe K, Bohn EW and Wilson SH:
Steady-state kinetics of mouse DNA polymerase β. Biochemistry.
18:3401–3406. 1979.
|
|
10.
|
Beard WA, Shock DD, Yang XP, DeLauder SF
and Wilson SH: Loss of DNA polymerase β stacking interactions with
templating purines, but not pyrimidines, alters catalytic
efficiency and fidelity. J Biol Chem. 277:8235–8242. 2002.
|
|
11.
|
Ollis DL, Brick P, Hamlin R, Xuong NG and
Steitz TA: Structure of large fragment of Escherichia coli
DNA polymerase I complexed with dTMP. Nature. 313:762–766.
1985.PubMed/NCBI
|
|
12.
|
Pelletier H, Sawaya MR, Wolfle W, Wilson
SH and Kraut J: Crystal structures of human DNA polymerase β
complexed with DNA: implications for catalytic mechanism,
processivity, and fidelity. Biochemistry. 35:12742–12761. 1996.
|
|
13.
|
Sawaya MR, Prasad R, Wilson SH, Kraut J
and Pelletier H: Crystal structures of human DNA polymerase β
complexed with gapped and nicked DNA: evidence for an induced fit
mechanism. Biochemistry. 36:11205–11215. 1997.
|
|
14.
|
Starcevic D, Dalal S and Sweasy JB: Is
there a link between DNA polymerase β and cancer? Cell Cycle.
3:998–1001. 2004.
|
|
15.
|
Lang T, Maitra M, Starcevic D, Li SX and
Sweasy JB: A DNA polymerase β mutant from colon cancer cells
induces mutations. Proc Natl Acad Sci USA. 101:6074–6079. 2004.
|
|
16.
|
Lang T, Dalal S, Chikova A, DiMaio D and
Sweasy JB: The E295K DNA polymerase beta gastric cancer-associated
variant interferes with base excision repair and induces cellular
transformation. Mol Cell Biol. 27:5587–5596. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Sweasy JB, Lang T, Starcevic D, Sun KW,
Lai CC, Dimaio D and Dalal S: Expression of DNA polymerase β
cancer-associated variants in mouse cells results in cellular
transformation. Proc Natl Acad Sci USA. 102:14350–14355. 2005.
|
|
18.
|
Makridakis NM, Caldas Ferraz LF and
Reichardt JK: Genomic analysis of cancer tissue reveals that
somatic mutations commonly occur in a specific motif. Hum Mutat.
30:39–48. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
An CL, Chen D and Makridakis NM:
Systematic biochemical analysis of somatic missense mutations in
DNA polymerase β found in prostate cancer reveal alteration of
enzymatic function. Hum Mutat. 32:415–423. 2011.PubMed/NCBI
|
|
20.
|
Batra VK, Beard WA, Shock DD, Krahn JM,
Pedersen LC and Wilson SH: Magnesium induced assembly of a complete
DNA polymerase catalytic complex. Structure (Camb). 14:757–766.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Dalal S, Hile S, Eckert KA, Sun KW,
Starcevic D and Sweasy JB: Prostate-cancer-associated I260M variant
of DNA polymerase β is a sequence-specific mutator. Biochemistry.
44:15664–15673. 2005.PubMed/NCBI
|
|
22.
|
An CL, Lim WJ, Hong SY, Kim EJ, Shin EC,
Kim MK, Lee JR, Park SR, Woo JG, Lim YP and Yun HD: Analysis of bgl
operon structure and characterization of β-glucosidase from
Pectobacterium carotovorum subsp carotovorum LY34.
Biosci Biotechnol Biochem. 68:2270–2278. 2004.
|
|
23.
|
Kosa JL and Sweasy JB:
3′-Azido-3′-deoxythymidine-resistant mutants of DNA polymerase β
identified by in vivo selection. J Biol Chem. 274:3851–3858.
1999.
|
|
24.
|
Servant L, Cazaux C, Bieth A, Iwai S,
Hanaoka F and Hoffmann JS: A role for DNA polymerase β in mutagenic
UV lesion bypass. J Biol Chem. 277:50046–50053. 2002.
|
|
25.
|
Chagovetz AM, Sweasy JB and Preston BD:
Increased activity and fidelity of DNA polymerase β on
single-nucleotide gapped DNA. J Biol Chem. 272:27501–27504.
1997.
|
|
26.
|
Li SX, Vaccaro J and Sweasy JB:
Involvement of phenylalanine 272 of DNA polymerase beta in
discriminating between correct and incorrect deoxynucleoside
triphosphates. Biochemistry. 38:4800–4808. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Maitra M, Gudzelak A Jr, Li SX, Matsumoto
Y, Eckert KA, Jager J and Sweasy JB: Threonine 79 is a hinge
residue that governs the fidelity of DNA polymerase β by helping to
position the DNA within the active site. J Biol Chem.
277:35550–35560. 2002.PubMed/NCBI
|
|
28.
|
Beard WA, Bebenek K, Darden TA, Li L,
Prasad R, Kunkel TA and Wilson SH: Vertical-scanning mutagenesis of
a critical tryptophan in the minor groove binding track of HIV-1
reverse transcriptase. Molecular nature of polymerase-nucleic acid
interactions. J Biol Chem. 273:30435–30442. 1998. View Article : Google Scholar
|
|
29.
|
Beard WA, Batra VK and Wilson SH: DNA
polymerase structure-based insight on the mutagenic properties of
8-oxoguanine. Mutat Res. 703:18–23. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Beard WA, Osheroff WP, Prasad R, Sawaya
MR, Jaju M, Wood TG, Kraut J, Kunkel TA and Wilson SH: Enzyme-DNA
interactions required for efficient nucleotide incorporation and
discrimination in human DNA polymerase β. J Biol Chem.
271:12141–12144. 1996.PubMed/NCBI
|
|
31.
|
Beard WA, Shock DD, Vande Berg BJ and
Wilson SH: Efficiency of correct nucleotide insertion governs DNA
polymerase fidelity. J Biol Chem. 277:47393–47398. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Vande Berg BJ, Beard WA and Wilson SH: DNA
structure and aspartate 276 influence nucleotide binding to human
DNA polymerase β: implication for the identity of the rate-limiting
conformational change. J Biol Chem. 276:3408–3416. 2001.PubMed/NCBI
|
|
33.
|
Dalal S, Kosa JL and Sweasy JB: The D246V
mutant of DNA polymerase β misincorporates nucleotides: evidence
for a role for the flexible loop in DNA positioning within the
active site. J Biol Chem. 279:577–584. 2004.PubMed/NCBI
|
|
34.
|
Sobol RW, Watson DE, Nakamura J, Yakes FM,
Hou E, Horton JK, Ladapo J, Van Houten B, Swenberg JA, Tindall KR,
Samson LD and Wilson SH: Mutations associated with base excision
repair deficiency and methylation-induced genotoxic stress. Proc
Natl Acad Sci USA. 99:6860–6865. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Pettersen EF, Goddard TD, Huang CC, Couch
GS, Greenblatt DM, Meng EC and Ferrin TE: UCSF Chimera - a
visualization system for exploratory research and analysis. J
Comput Chem. 25:1605–1612. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Dunbrack RL Jr: Rotamer libraries in the
21st century. Curr Opin Struct Biol. 12:431–440. 2002. View Article : Google Scholar : PubMed/NCBI
|