Introduction
MicroRNAs (miRNAs) are composed of approximately
20–25 nucleotide-non-coding RNAs that may silence
post-transcriptional protein expression through two axes: first, by
binding to complementary target messenger RNAs to target them for
degradation; and second, inhibiting mRNA translation into proteins
(1). There has been significant
evidence showing that miRNAs regulate as many as 30% of the human
protein coding genes (2).
Moreover, they can function as oncogenes or tumor suppressor genes
by altering regulation of their targets in many cancers (3,4).
Indeed, studies have shown that miRNAs are involved in a variety of
processes including tumor cell proliferation, differentiation, and
apoptosis. Moreover, their differing expressions can lead to
different human cancers (5,6).
Subsequently, miRNA research has become a hot spot in breast cancer
research whereby miRNAs are believed to have broad prospects in
terms of diagnosis and treatment of this disease (7–10).
Recent studies indicate that miR-15a is
downregulated in chronic lymphocytic leukemia (11), prostate cancer (12), osteosarcoma (13), keratocystic odontogenic tumors
(14) and breast cancer (15). Furthermore, through overexpression
of miR-15a, curcumin can reduce the expression of Bcl-2 and
subsequently induce apoptosis in MCF-7 breast cancer cells
(15). However, the mechanism by
which miR-15a contributes to breast cancer tumorigenesis is still
unclear.
Cyclin E1 (CCNE1), one member of the cyclin E
family, can associate with and activate cyclin-dependent kinase 2
(CDK2). CCNE1 is a positive regulator of G1/S phase transition and
is essential for cell cycle re-entry from G0 phase. In many human
tumors, CCNE1 is overexpressed and the level of both protein and
kinase activity is often deregulated relative to the cell cycle, as
demonstrated in human breast epithelial cells (16). One study has indicated that
deregulation of CCNE1 is an early event in the development of
breast cancer (17). Furthermore,
overexpression of CCNE1 in patients with breast cancer is
associated with worse prognosis (18,19).
CCNE1 can be repressed by some miRNAs, for example, a study has
demonstrated the involvement of miR-16 in its regulation in human
endothelial progenitor cells (20).
In this study, we first demonstrated that miR-15a
expression is significantly lower in breast cancer specimens when
compared with that of adjacent normal tissues. Its overexpression
inhibited proliferation of MDA-MB-231 breast cancer cells, in
association with inhibition of migration and disruption of the cell
cycle by targeting CCNE1. These results indicate that miR-15a
functions as a tumor suppressor gene, whose dysregulation may be
involved in the development of human breast cancer.
Materials and methods
Specimens
In this study, 40 paired breast cancer specimens and
adjacent normal breast tissues were collected from the Department
of General Surgery of the Shanghai Tenth People’s Hospital. These
samples were immediately snap-frozen in liquid nitrogen. All
samples were confirmed as invasive, ductal breast cancer by trained
pathologists. No patients received chemotherapy or radiotherapy
prior to surgery.
Cell lines and transfection
The MDA-MB-231 breast cancer cells and HEK293T cells
used in this study were purchased from the ATCC (Manassas, VA,
USA). Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM;
Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Gibco),
penicillin (100 U/ml) and streptomycin (100 μg/ml)
(Enpromise, China). Cells were incubated at 37°C in a humidified
chamber supplemented with 5% CO2.
For transfections, cells (2×105) were
added into each well of a 6-well plate and cultured with DMEM
medium without either serum or antibiotics. When the density of
MDA-MB-231 breast cancer cells reached 30–40%, miR-15a mimics
(GenePharma Co., Ltd., Shanghai, China) and Lipofectamine
transfection reagent (Invitrogen, USA) were each diluted in 500
μl DMEM medium, at a ratio of 1 μg:3 μl and
incubated for 5 min at room temperature (RT). The two mixtures were
then gently combined and incubated for a further 20–30 min at RT.
Subsequently, 1,000 μl of the complexes were added to each
well. After 5–6 h of incubation, DMEM medium was replaced by DMEM
with 10% FBS. Cells were incubated at 37°C in a CO2
incubator for 48 h prior to further testing.
Quantitative reverse-transcription
polymerase chain reaction (qRT-PCR)
MicroRNAs were harvested according to the
instructions of the miRcute miRNA isolation kit (Tiangen, Beijing,
China). For miRNA qPCR, the miR-15a primer, U6 primer and EzOmics
SYBR qPCR kit were purchased from Biomics Biotechnology Inc.
(Jiangsu, China). The amplification procedure was as follows: 94°C
for 10 min, followed by 40 cycles at 94°C for 20 sec, 61°C for 30
sec and 72°C for 30 sec.
For quantification of CCNE1 mRNA expression, total
RNA was isolated using TRIzol (Invitrogen) and cDNA was generated
by reverse transcription using the PrimeScript RT-PCR kit in
accordance with the manufacturer’s instructions (Takara). Real-time
PCR was performed on a 7900HT fast RT-PCR instrument using
SYBR-Green and the following primers: CCNE1:
5′-TTTCAGGGTATCAGTGGTG-3′ (sense), and 5′-ACATGGCTTTCTTTGCTC-3′
(antisense); GAPDH: 5′-AAGGTCGGAGTCAACGGATT-3′ (sense), and
5′-CTGGA AGATGGTGATGGGATT-3′ (antisense). The PCR parameters for
relative quantification were as follows: 5 min at 94°C, followed by
30 cycles of 30 sec at 94°C, 45 sec at 57°C and 45 sec at 72°C.
Each sample was tested in triplicate. The relative expression was
calculated following the relative quantification equation =
2−ΔΔCT(21) .
Cell proliferation assay
Cell proliferation was assessed using an MTT assay
kit (Sigma, Santa Clara, CA, USA) in accordance with the
manufacturer’s instructions. Briefly, ∼4–5 h after transfection of
miR-15a mimics, cells administered either 50 or 100 nmol/l miR-15a
mimics or negative control (NC) were trypsinized and counted. Cells
from each condition were plated (3,000/well) in 96-well plates (BD
Biosciences, USA) and incubated at 37°C in a humidified chamber
supplemented with 5% CO2. Cell proliferation was
assessed at 24, 48, 72 and 96 h. The optical density (OD) of each
well was measured with a microplate spectrophotometer at 490 nm.
All experiments were performed in biological triplicate.
Colony formation assay
After transfection with 100 nmol/l miR-15a or NC,
cells were trypsinized, counted, and seeded for colony formation
assay in 6-well plates at 300/well. During colony growth, the
culture medium was replaced every 3 days. On the 8th day after
seeding, the cells were fixed and then stained with crystal violet,
and the number of colonies was counted. The colony was counted only
if it contained >50 cells. Each treatment was carried out in
triplicate.
Transwell chamber migration assay
The transwell migration assay was performed in a
24-well transwell chamber system. The filter was washed with the
serum-free DMEM, and placed between the lower and upper chambers.
The lower chambers contained DMEM with 10% FBS. The miR-15a or NC
transfected MDA-MB-231 cells were trypsinized, resuspended in DMEM
with 0.1% BSA, transferred to the upper chambers, and incu bated at
37°C in 5% CO2. After 20 h, the filter was removed, the
upper surface of the filter containing non-migrating cells was
cleared using a wet cotton swab, and the cells remaining on the
underside were stained with crystal violet. Five fields of each
well were randomly gated and counted. Then, glacial acetic acid was
used to dissolve crystal violet and the OD was measured at 573 nm.
Each treatment was carried out in triplicate.
Cell cycle and apoptosis assay
Thirty-six hours after transfection with the miR-15a
mimics, or NC, cells were trypsinized and centrifuged at 1,000 rpm
for 5 min, followed by two washes in cold PBS. Then, 3.0 ml
ice-cold ethanol was added in a dropwise fashion and cells were
allowed to fix for ≥30 min. A total of 250 μl 0.05 g/l
propidium iodide (PI) staining solution was added into each sample
and incubated for 30 min at RT. Cells were then analyzed on a flow
cytometer (FACSCanto™ II, BD Biosciences).
For Annexin V staining, miR-15a and NC groups of
adherent cells were harvested and incubated with Annexin V
incubation reagent (prepared by combining 10 μl 10X binding
buffer, 10 μl PI, 1 μl Annexin V-FITC and 79
μl deionized, distilled H2O) at a ratio of
105–106 cells/100 μl for 15 min at RT
in the dark. All samples were processed by flow cytom etry
(FACSCanto™ II, BD Biosciences). FACS analyses were performed at
least three times with reproducible results.
Luciferase assay
We used a total PCR reaction volume of 50 μl
to amplify the 3′-UTR of CCNE1 containing the predicted miR-15a
binding site using the Primer star kit (Takara), in accordance with
the manufacturer’s instructions. The primers used were:
5′-ATTCTAGGCGATCGCTCGAGC CACCCCATCCTTCTCCA-3′ (sense);
5′-TTTATTGCGGCC AGCGGCCGCTCAAAAACAGTATTATCTTTATTAAA-3′ (antisense).
Fragments were then subcloned into the XhoI site in the
3′-UTR of firefly luciferase of the psiCHECK-2 reporter vector.
psiCHECK-2/CCNE1 3′-UTR reporter plasmids (100 ng) were
co-transfected with the miR-15a mimics or NC (100 nmol/l) into
HEK293T cells, at 70% confluence, using Lipofectamine 2000
(Invitrogen), according to the manufacturer’s instructions. After
30 h, cells were lysed and reporter activity was assessed using the
Dual-luciferase reporter assay system (Promega, USA) in accordance
with the manufacturer's protocol. Firefly luciferase activity was
normalized to renilla luciferase activity.
Western blot analysis
The protein expression levels were detected by
western blotting. Whole cell protein extracts [lysis buffer: 50 mM
Tris-HCl (pH 7.5), 150 mM NaCl, 1% NP40, 1 mM phenylmethylsulfonyl
fluoride, and 19 mM protease inhibitor cocktail (Sigma-Aldrich,
USA)] were quantified by bicinchoninic acid assay (Pierce, USA).
Protein samples were separated by 10% sodium dodecyl sulfate
polyacrylamide gel electrophoresis and transferred onto
nitrocellulose membranes (Beyotime, China). Immune complexes were
formed by incubation of membranes with primary antibody (Epitomics,
USA) overnight at 4°C. Blots were washed and incubated for 1 h with
horseradish peroxidase-conjugated anti-rabbit secondary antibody.
Immunoreactive protein bands were detected using an Odyssey
Scanning system.
Statistical analysis
Data are presented as the mean ± standard error of
mean from at least three independent experiments. The two-tailed
t-test was used to draw a comparison between groups. The null
hypothesis was rejected at the 0.05 level.
Results
Expression of miR-15a is decreased in
human breast cancer
To investigate the expression level of miR-15a in
breast cancer, we analyzed levels of miR-15a in 40 paired invasive
ductal breast cancer specimens and associated normal adjacent
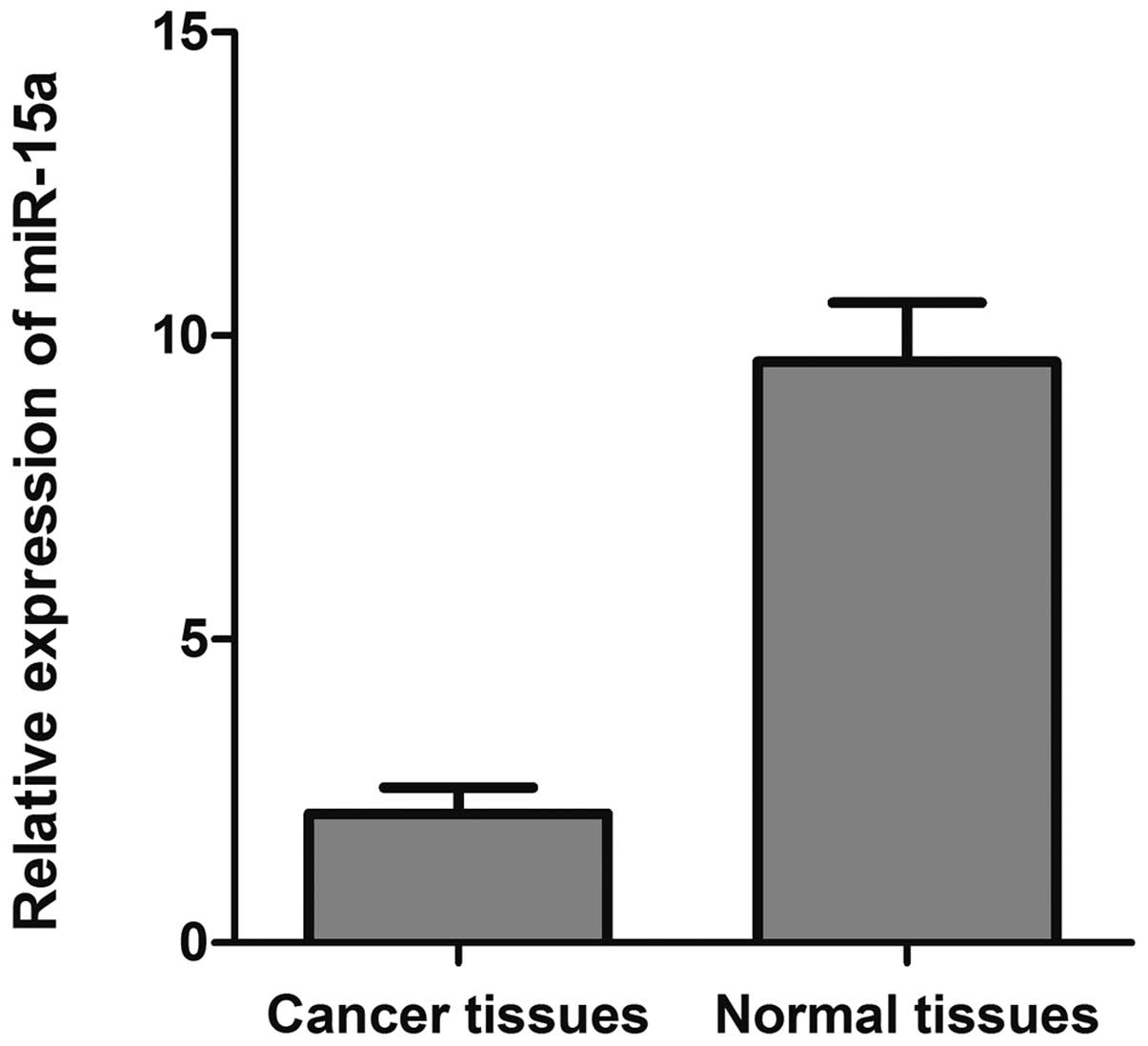
tissues by qRT-PCR. As depicted in Fig. 1, the 2−ΔΔCt value of
miR-15a was significantly decreased in breast cancer tissues
(2.125±0.096) compared with that of normal adjacent tissues
(9.570±0.337) (p<0.05).
Suppression of breast cancer cell
proliferation by miR-15a
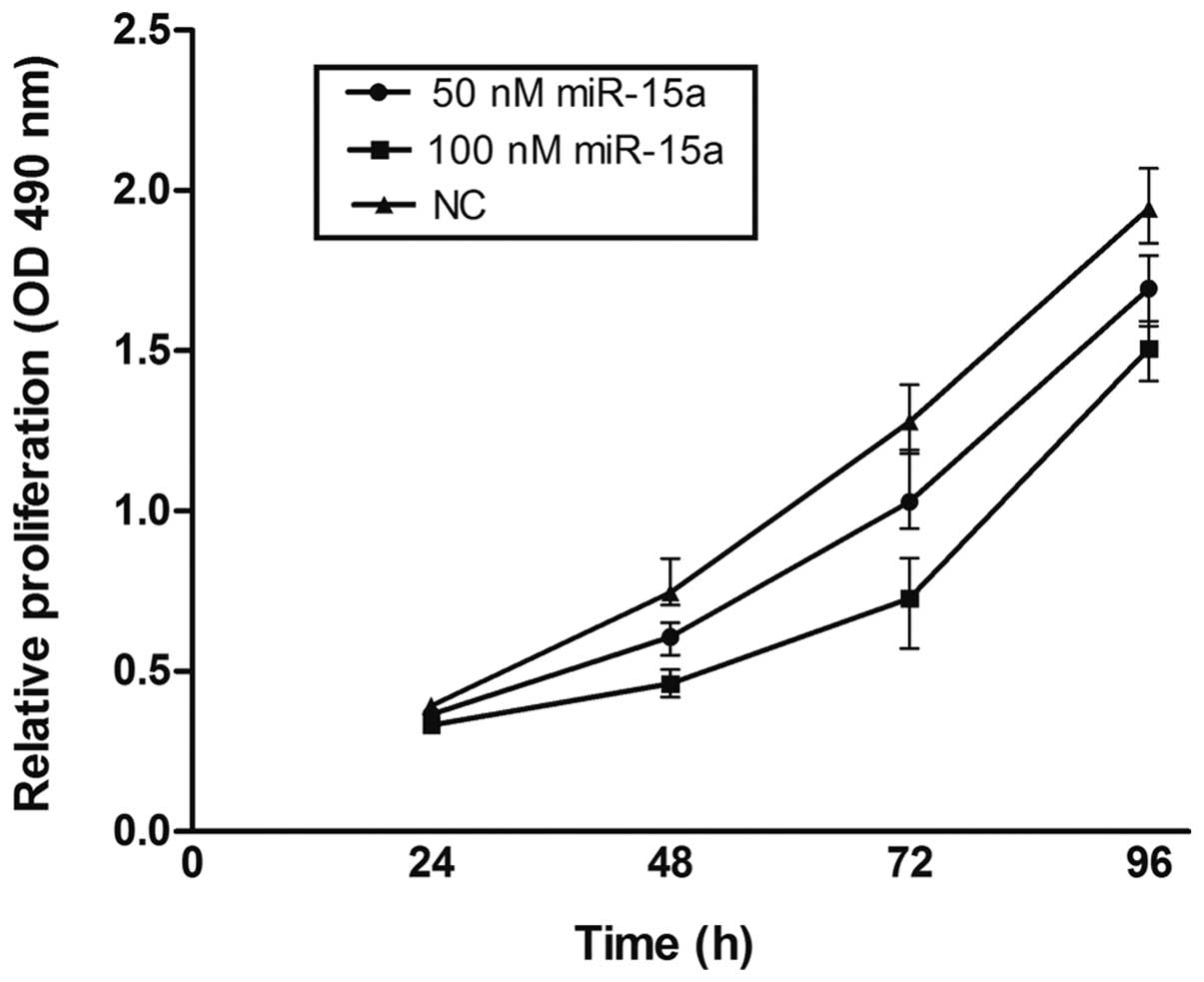
To explore the potential impact of miR-15a on the
proliferation of breast cancer cells, miR-15a mimics were used and
viability was measured by the MTT assay in MDA-MB-231. Compared
with the NC group, miR-15a significantly repressed the growth of
breast cancer cells. Suppression of cell growth by miR-15a was
time- and dose-dependent, whereby miR-15a at a concentration of 100
nmol/l at 72 h showed the greatest inhibitory effect (p<0.05)
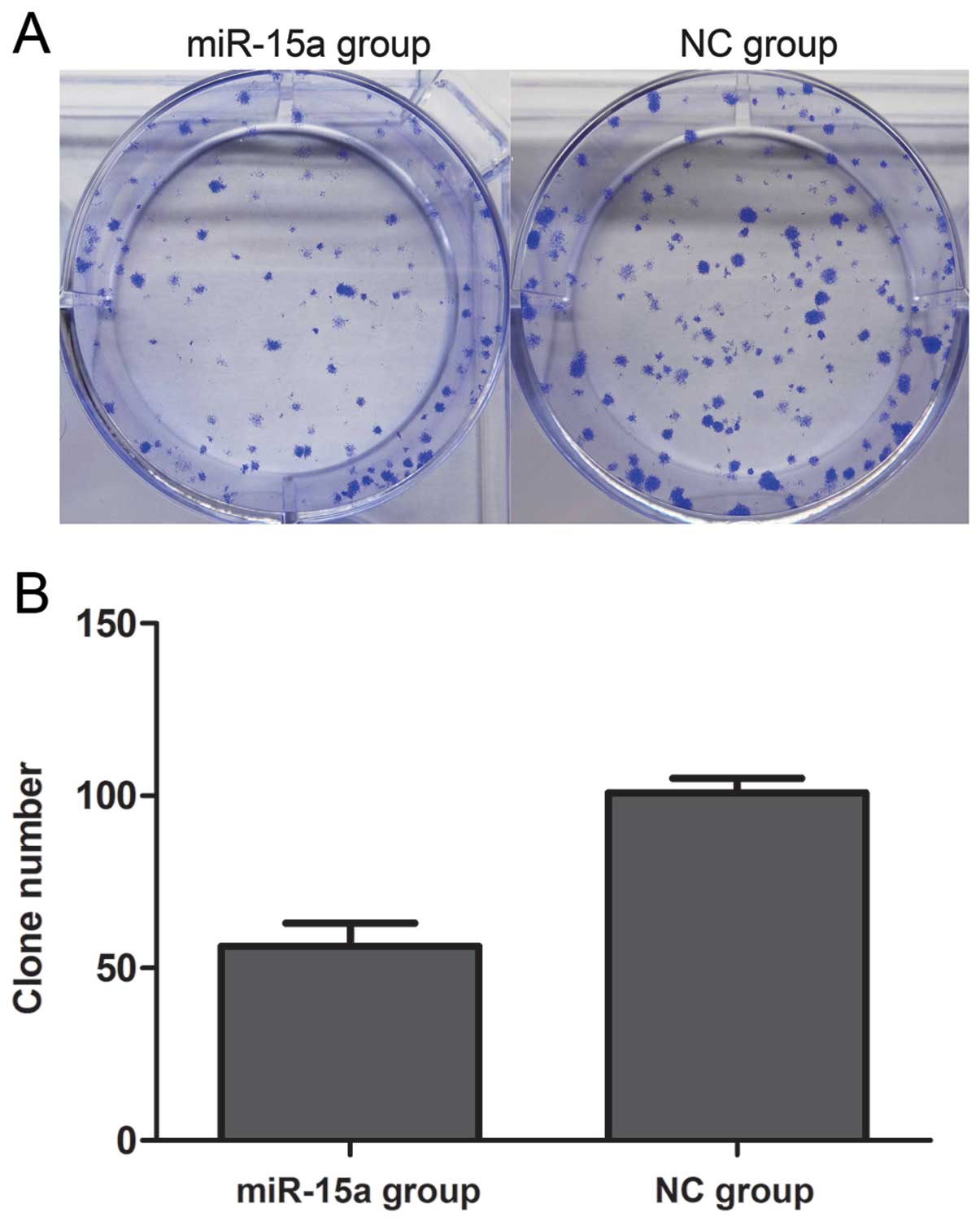
(Fig. 2). Proliferation was also
assessed by colony formation assay (Fig. 3). We found that the number of
colonies of the miR-15a group was 56.25±4.151, which was
significantly less than that of the NC group (100.8±2.175)
(p<0.05). Thus, these data suggest that miR-15a significantly
suppresses the proliferation of MDA-MB-231 breast cancer cells.
miR-15a inhibits migration of MDA-MB-231
cells
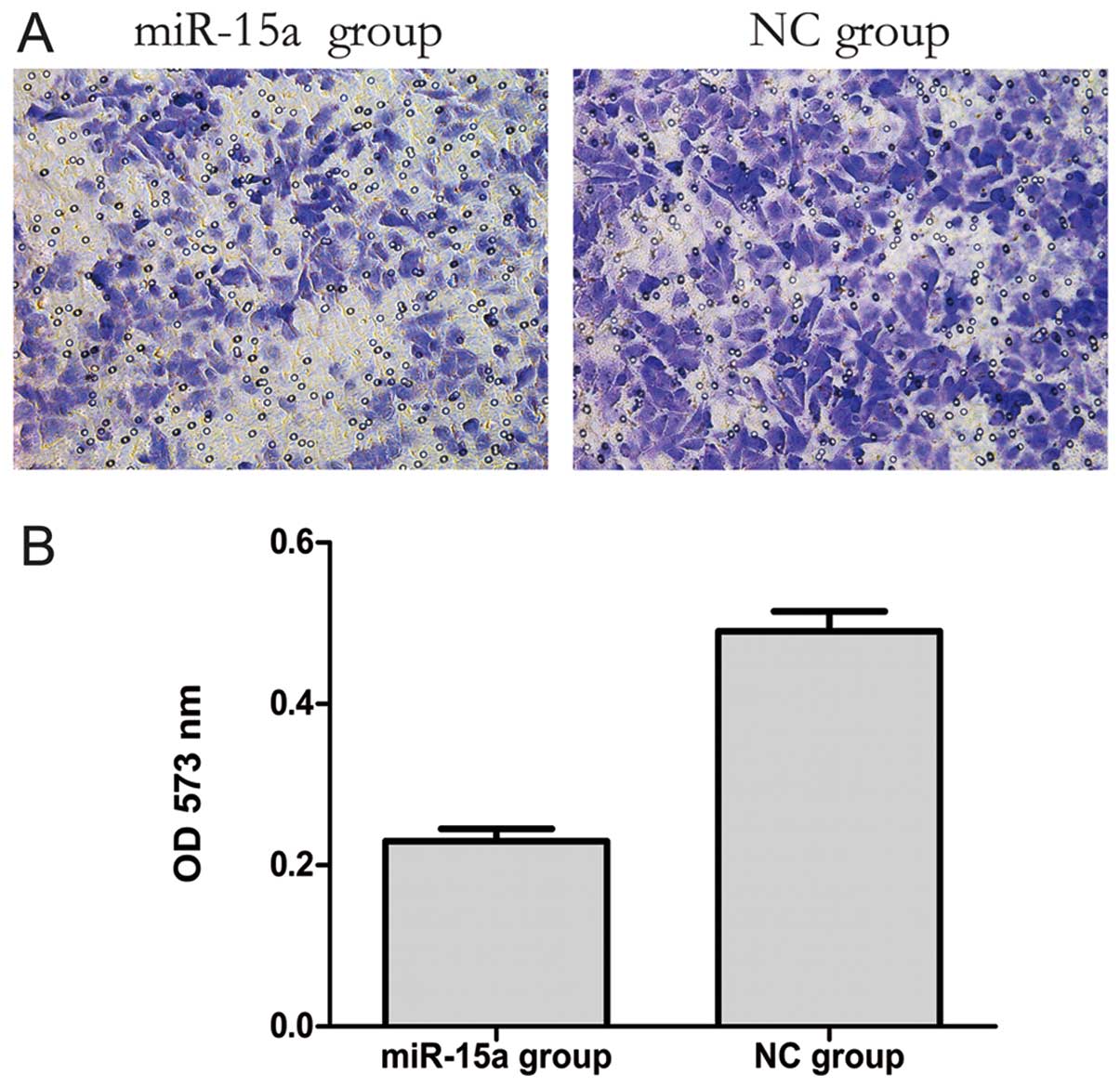
The transwell migration assay is a useful method to
investigate migratory ability. Our results showed that 20 h after
transfection the number of migrating cells in the miR-15a group was
less than that in the NC group. Furthermore, the OD 573 nm values,
derived by solubilization of crystal violet staining, revealed a
significant decreased from 0.497±0.009 to 0.229±0.010 (p<0.05)
in the NC and miR-15a groups, respectively. These data indicate
that the migratory ability of MDA-MB-231 cells might be inhibited
by miR-15a (Fig. 4).
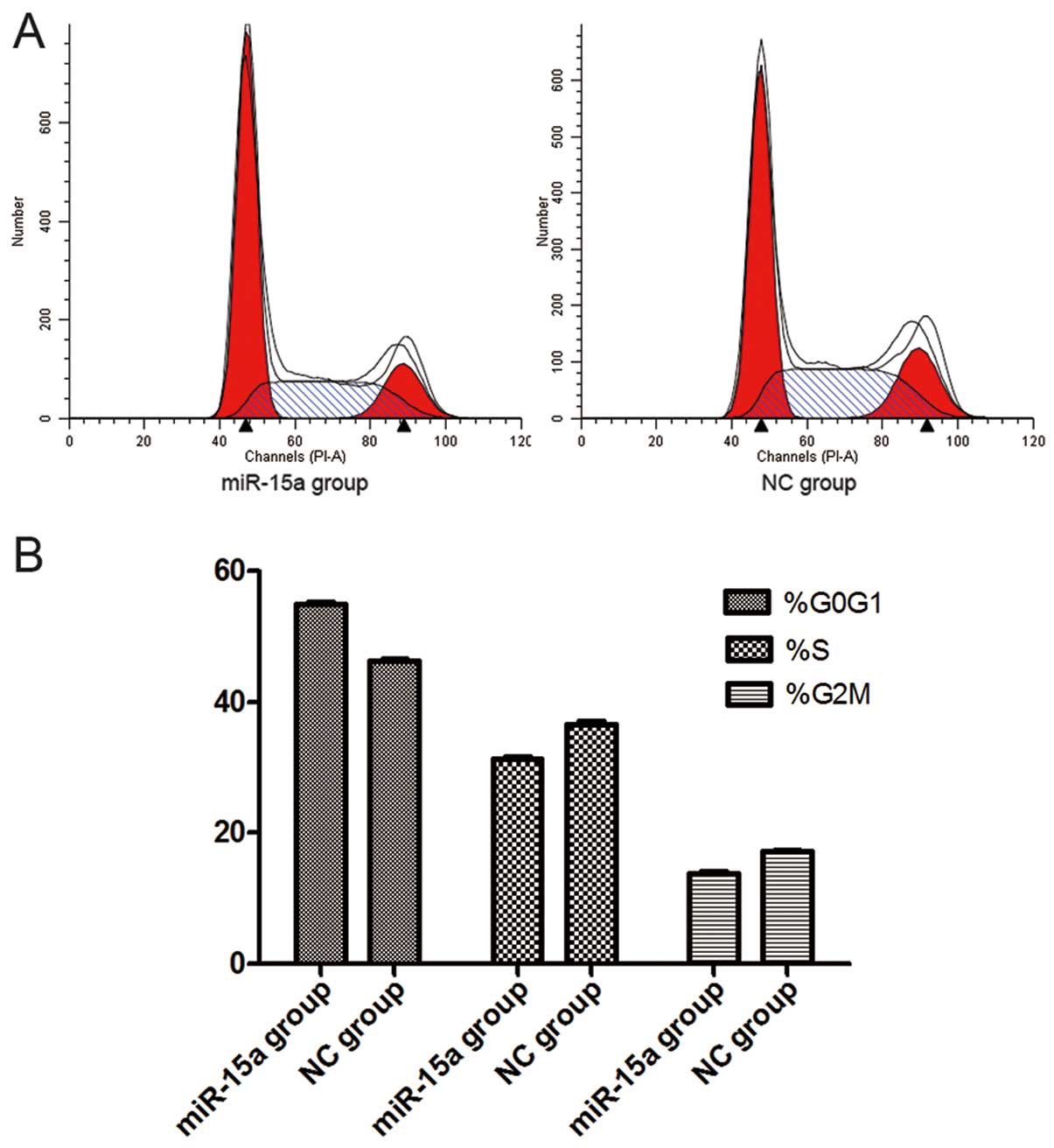
miR-15a disrupts the cell cycle of
MDA-MB-231 cells
Thirty-six hours after the transfection of miR-15a
mimics (100 nmol/l), flow cytometry analysis revealed that the
percentage of G0/G1 phase cells (54.88±0.175%) dramatically
increased in the miR-15a group, when compared with that of the NC
group (46.16±0.182%) (p<0.05), while the proportion of S-phase
cells decreased in the miR-15a group (31.30±0.116%) compared with
that of the NC group (36.62±0.205%) (p<0.05). The percentage of
G2/M phase cells also decreased in the miR-15a group (13.71±0.229%)
compared with that of the NC group (17.23±0.076%) (p<0.05).
These findings suggest that miR-15a can initiate G0/G1 phase arrest
and that upregulation of miR-15a expression could lead to the
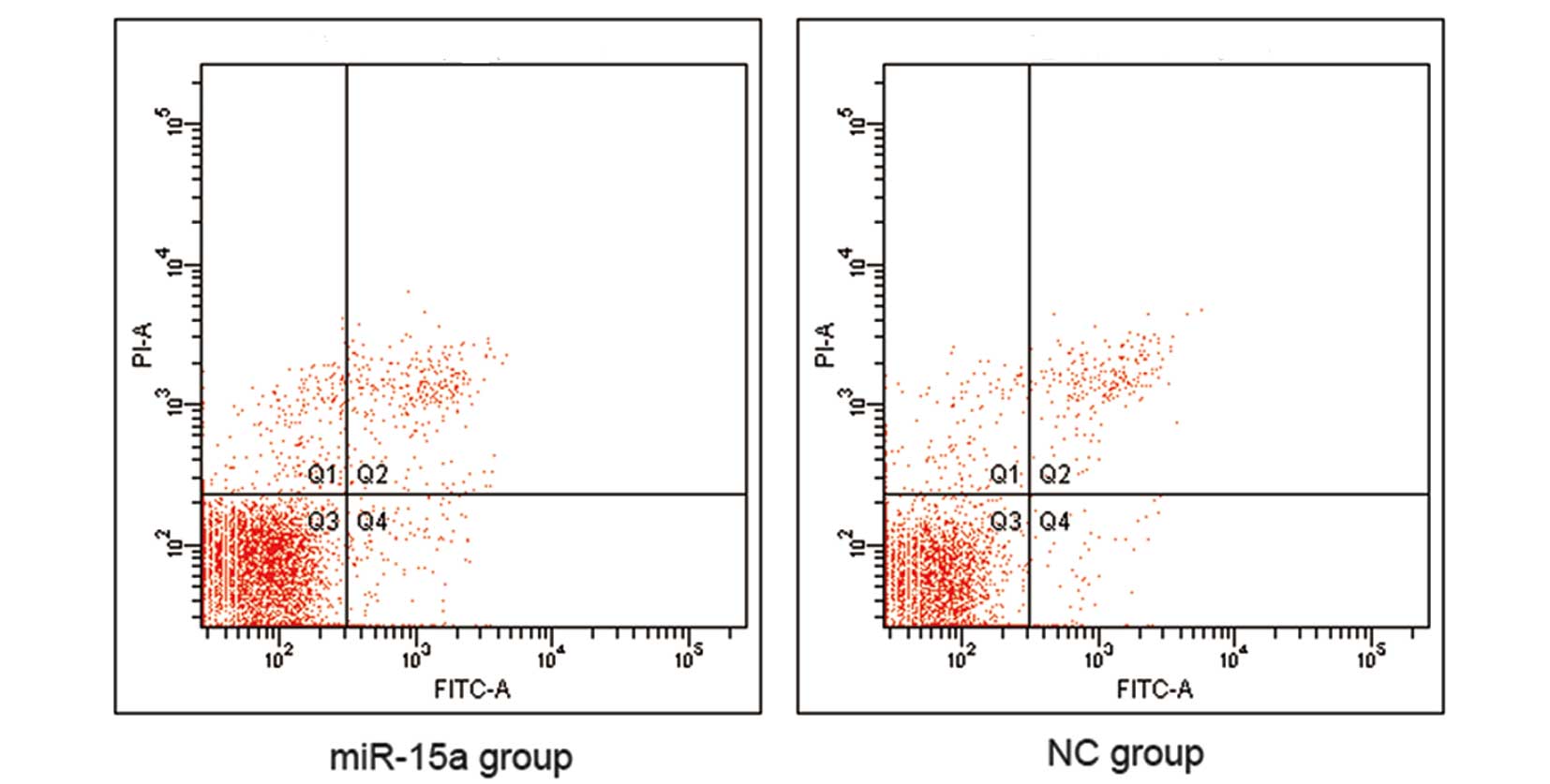
reduction of S-phase and G2/M phase cells (Fig. 5). However, our data indicate that
there was no difference in apoptosis between the miR-15a and NC
groups (Fig. 6).
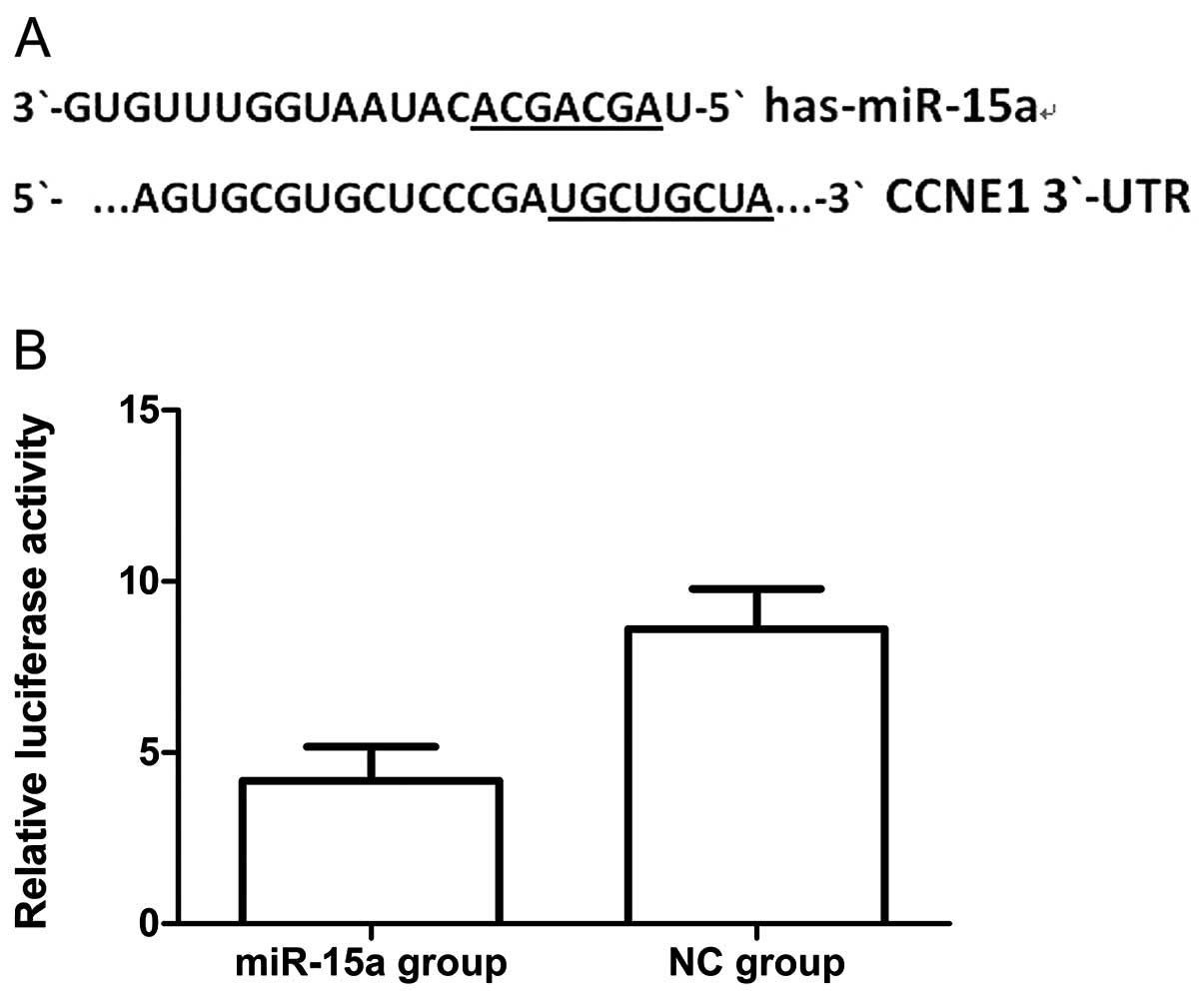
MiR-15a regulates CCNE1 expression by
targeting CCNE1 in MDA-MB-231 cells
To validate the possibility that miR-15a may target
CCNE1 in breast cancer cells we first searched for putative targets
using the miRanda, targetscan and miRBase databases. We found a
potential binding site for miR-15a in the 3′-UTR of CCNE1 mRNA,
which was located 247–254 bp downstream from the 5′-end of the
CCNE1 3′-UTR. We then cloned the putative binding site into a
luciferase reporter construct and used it to measure the effects of
miR-15a mimics in MDA-MB-231 cells. We found that luciferase
activity was significantly lower in cells co-transfected with
psiCHECK-2/CCNE1 3′-UTR and miR-15a, when compared with that of
co-transfection with NC (Fig. 7)
(p<0.05). Thus the results of this experiment show that miR-15a
could directly interact with the CCNE1 3′-UTR fragments of the
psiCHECK-2 reporter plasmid, which could lead to the degradation of
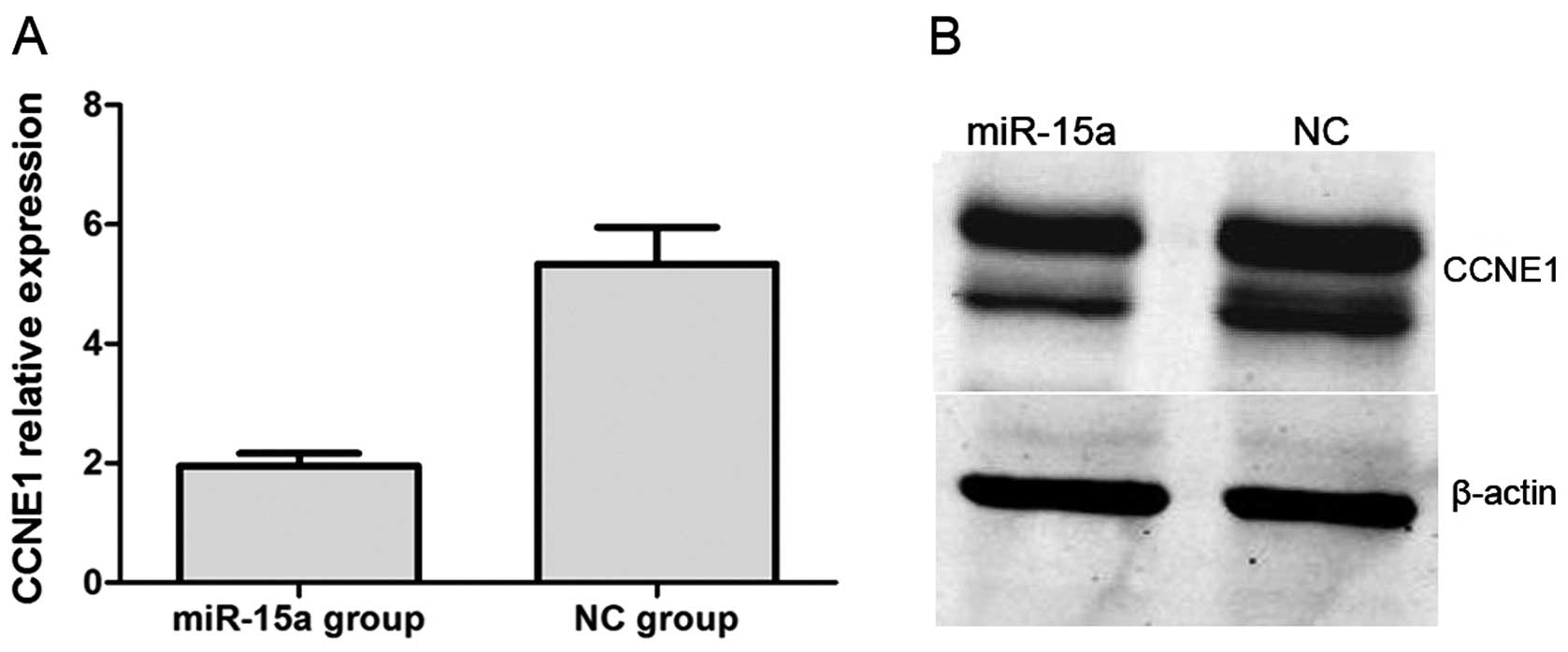
renal luciferase mRNA. Moreover, we performed qPCR and western blot
analysis. QPCR indicated that CCNE1 expression was significantly
lower in the miR-15a mimics group compared with that of the NC
group, with relative expression levels of 1.957±0.050 and
5.335±0.169 (p<0.05), respectively (Fig. 8A). In western blot analysis, CCNE1
protein expression was also significantly decreased by the
overexpression of miR-15a (Fig.
8B).
Discussion
Breast cancer is one of the most common malignant
tumors in women. Besides surgery, chemotherapy is the major
therapeutic method. However, there are still patients that exhibit
resistance to chemotherapy, as demonstrated through early
recurrence and metastasis, leading to poor prognosis. This is
especially true for triple-negative breast cancers (22), which do not express estrogen
receptor, progesterone receptor or the human epidermal growth
factor receptor-2. In this case, it is particularly important to
explore new treatments. Currently, the search for novel therapeutic
agents for breast cancer is one of the hot topics of breast cancer
research (23–25).
In this study, we examined the expression level of
miR-15a in human breast cancer and its potential role in disease
pathogenesis. First, we detected the expression level of miR-15a in
human breast cancer specimens by qRT-PCR. The results showed that
miR-15a was significantly lower in breast cancer tissues than in
normal breast tissues. Similar findings have been reported in other
cancer types (11,12), which indicates that downregulation
of miR-15a is common in human cancer specimens and cell lines.
Next, we transfected miR-15a mimics into MDA-MB-231 cells to
generate its overexpression. This led to significant inhibition of
cellular proliferation as measured by MTT, as well as a reduction
in the colony number as determined by clone formation assay. These
two experiments indicate that miR-15a represses the growth of
breast cancer cells. Using the transwell migration assay, we found
that the overexpression of miR-15a in breast cancer cells could
suppress their migratory ability. We found that miR-15a distinctly
arrests cancer cells at the G1 phase when compared with the cell
cycle of NC groups. However, our study found no significant
difference in apoptosis between the miR-15a and NC groups. The
vitality of cancer cells is very strong, thus we speculate that
this is one possible reason why miRNA-15a could not promote
apoptosis.
To investigate the downstream targets of miR-15a
that may play a role in mediating its cell function, we searched
for putative targets using the miRanda, targetscan and miRBase
databases. Through luciferase assays, we predicated CCNE1 as a
direct target of miR-15a in MDA-MB-231 cells. Additionally, we
found that both the mRNA and protein levels of CCNE1 were
significantly lower in miR-15a than those in NC groups. These
findings support the prediction that CCNE1 is a downstream target
of miR-15a.
Collectively, our findings suggest that miR15a can
disrupt the cell cycle by targeting CCNE1 in MDA-MB-231 cells. We
show that its overexpression can reduce cell proliferation and
inhibit the migratory ability of cancer cells. Thus, it may be
concluded that miR-15a acts as a tumor suppressor gene in breast
cancer. Moreover, the luciferase, qPCR and western blot assays
illustrate CCNE1 as a downstream target of miR-15a. The artificial
upregulation of miR-15a using CCNE1 as a therapeutic agent could
offer a promising new direction for future breast cancer
treatment.
Acknowledgements
This study was made possible with
financial support from the National Natural Sciences Foundation of
China, for the project 81272240, and the Shanghai Science Committee
Foundation (to Lin Fang) (no. STCSM 10411964700). We sincerely
thank all the teachers at the Central Laboratory of the Shanghai
Tenth People’s Hospital for their help and support.
References
|
1.
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Zhang B, Pan X, Cobb GP and Anderson TA:
microRNAs as oncogenes and tumor suppressors. Dev Biol. 302:1–12.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Chen C-Z: MicroRNAs as oncogenes and tumor
suppressors. N Engl J Med. 353:1768–1771. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Noguchi S, Mori T, Hoshino Y, et al:
MicroRNA-143 functions as a tumor suppressor in human bladder
cancer T24 cells. Cancer Lett. 307:211–220. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Pallante P, Visone R, Ferracin M, et al:
MicroRNA deregulation in human thyroid papillary carcinomas. Endocr
Relat Cancer. 13:497–508. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Iorio MV, Ferracin M, Liu CG, et al:
MicroRNA gene expression deregulation in human breast cancer.
Cancer Res. 65:7065–7070. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Ma L, Teruya-Feldstein J and Weinberg RA:
Tumour invasion and metastasis initiated by microRNA-10b in breast
cancer. Nature. 449:682–688. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Shi W, Gerster K, Alajez NM, et al:
MicroRNA-301 mediates proliferation and invasion in human breast
cancer. Cancer Res. 71:2926–2937. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Tang D, Zhang Q, Zhao S, et al: The
expression and clinical significance of microRNA-1258 and
heparanase in human breast cancer. Clin Biochem. 46:926–932. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Sampath D, Liu C, Vasan K, et al: Histone
deacetylases mediate the silencing of miR-15a, miR-16, and miR-29b
in chronic lymphocytic leukemia. Blood. 119:1162–1172. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Musumeci M, Coppola V, Addario A, et al:
Control of tumor and microenvironment cross-talk by miR-15a and
miR-16 in prostate cancer. Oncogene. 30:4231–4242. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Cai C-K, Zhao G-Y, Tian L-Y, et al:
miR-15a and miR-16-1 downregulate CCND1 and induce apoptosis and
cell cycle arrest in osteosarcoma. Oncol Rep. 28:1764–1770.
2012.PubMed/NCBI
|
|
14.
|
Diniz MG, Gomes CC, de Castro WH, et al:
miR-15a/16-1 influences BCL2 expression in keratocystic odontogenic
tumors. Cell Oncol. 35:285–291. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Yang J, Cao Y, Sun J and Zhang Y: Curcumin
reduces the expression of Bcl-2 by upregulating miR-15a and miR-16
in MCF-7 cells. Med Oncol. 27:1114–1118. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Spruck CH, Won KA and Reed SI: Deregulated
cyclin E induces chromosome instability. Nature. 401:297–300. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Shaye A, Sahin A, Hao Q, Hunt K, Keyomarsi
K and Bedrosian I: Cyclin E deregulation is an early event in the
development of breast cancer. Breast Cancer Res Treat. 115:651–659.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Keyomarsi K, Tucker SL, Buchholz TA, et
al: Cyclin E and survival in patients with breast cancer. N Engl J
Med. 347:1566–1575. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Sgambato A, Camerini A, Collecchi P, et
al: Cyclin E correlates with manganese superoxide dismutase
expression and predicts survival in early breast cancer patients
receiving adjuvant epirubicin-based chemotherapy. Cancer Sci.
100:1026–1033. 2009. View Article : Google Scholar
|
|
20.
|
Goretti E, Rolland-Turner M, Leonard F,
Zhang L, Wagner DR and Devaux Y: MicroRNA-16 affects key functions
of human endothelial progenitor cells. J Leukoc Biol. 93:645–655.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
|
|
22.
|
Bryan BB, Schnitt SJ and Collins LC:
Ductal carcinoma in situ with basal-like phenotype: a possible
precursor to invasive basal-like breast cancer. Mod Pathol.
19:617–621. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ljungberg BJ, Jacobsen J, Rudolfsson SH,
Lindh G, Grankvist K and Rasmuson T: Different vascular endothelial
growth factor (VEGF), VEGF-receptor 1 and -2 mRNA expression
profiles between clear cell and papillary renal cell carcinoma. BJU
Int. 98:661–667. 2006. View Article : Google Scholar
|
|
24.
|
Tryfonopoulos D, Walsh S, Collins DM, et
al: Src: a potential target for the treatment of triple-negative
breast cancer. Ann Oncol. 22:2234–2240. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Oliveras-Ferraros C, Vazquez-Martin A,
Lopez-Bonet E, et al: Growth and molecular interactions of the
anti-EGFR antibody cetuximab and the DNA cross-linking agent
cisplatin in gefitinib-resistant MDA-MB-468 cells: new prospects in
the treatment of triple-negative/basal-like breast cancer. Int J
Oncol. 33:1165–1176. 2008.
|