Contents
Introduction
Complex networks in biology
Cancer-associated networks
Network-based approaches for chemotherapy
Network-based approaches for radiotherapy
Targeting network flexibility
Conclusion
Introduction
Cancer is characterized as a complex and
heterogeneous disease involving an orchestration of distinct
cellular signaling events that can be affected by the aberrant
expression or mutation of genes and chromosomes, tumor
microenvironments and tissue origin of the tumor. Experimental and
literature-based analyses of signaling networks supported the
hypothesis that a common network and its components are the same
for all cells (1–3). Therefore, it has been proposed that
differentially expressed tumor phenotypes might result from
distinct interactions and consequent activation of specific
subnetworks (2). Thus,
single-target therapies using highly specific compounds will likely
fail as a cancer treatment unless the compounds are able to disrupt
an actual network. In order to deal with this problem,
network-based approaches have emerged (4). Most cellular components interact with
each other to carry out biological functions within the same cell
or between cells. These intra- and intercellular interactions form
a complex and flexible network, and are dynamically altered on
temporal and spatial scales. This is responsible for the
determination of tumor phenotypes. Therefore, cancer-associated
molecular networks and their dynamics could be potential targets
for therapeutic intervention. With chemo- and radiotherapies,
dynamic alteration of signaling networks occurs in tumor cells as
protective processes against stress stimuli. These cellular
responses are the main cause of resistance to therapies. To develop
better cancer treatment strategies, it is important to determine
which subnetworks are activated and which factors play a crucial
role in network alteration upon chemo- and radiotherapies. In this
review, we will describe the complex networks associated with
cancers, their properties, and further strategies targeting these
networks for development of efficient anticancer therapies.
Complex networks in biology
Cells can be depicted as complex networks of
macromolecular interactions (5).
Most biological processes are executed through multi-scale dynamic
complex systems formed by interacting macromolecules, metabolites,
cells and tissues (6). At a highly
abstract level, components of a network can be reduced to a series
of nodes that are interconnected by several links with each link
representing the interactions between two components (7). Nodes are basic components of a
network and are connected by links. In biological networks,
proteins, metabolites, DNA and RNA correspond to nodes and their
interactions represent links.
Several model organisms have produced a bulk of
information for understanding biological networks (8). Based on these data, several studies
examining human-specific networks have been recently performed.
Many groups have focused on molecular networks such as
protein-protein interaction networks, metabolic networks, DNA/RNA
networks and gene regulatory networks. In networks of
protein-protein interaction, proteins serve as nodes and are linked
to each other by physical interactions (8–10).
Metabolic networks consist of metabolites as nodes that are linked
if they play a role in the same biochemical pathway (11,12).
In DNA/RNA networks, regulatory RNA molecules (such as microRNA and
small interfering RNA) and DNA are considered nodes and their links
indicate functional interactions that influence the regulation of
gene expression (13–15). In gene regulatory networks, direct
links of each node represent the regulatory relationships between a
transcriptional factor and a gene (13,16).
Each of these networks is fundamentally present in all cells
(3). However, these networks are
differentially modulated in a cell type-specific manner. Due to the
complexity and dynamics of the networks, cells are able to develop
unique phenotypes (2). It means
that each cell may distinctively respond to the same stimulus.
Thus, it is encouraged to understand the common network structures
and their signaling process by downstream effectors that are
associated with specific phenotype, which can be applied to the
implication of cancer cell-specific responses to targeted
therapies.
Network structures are divided into two major
classes based on the distribution of connections, indicating the
probability that one node in the network is linked to other nodes.
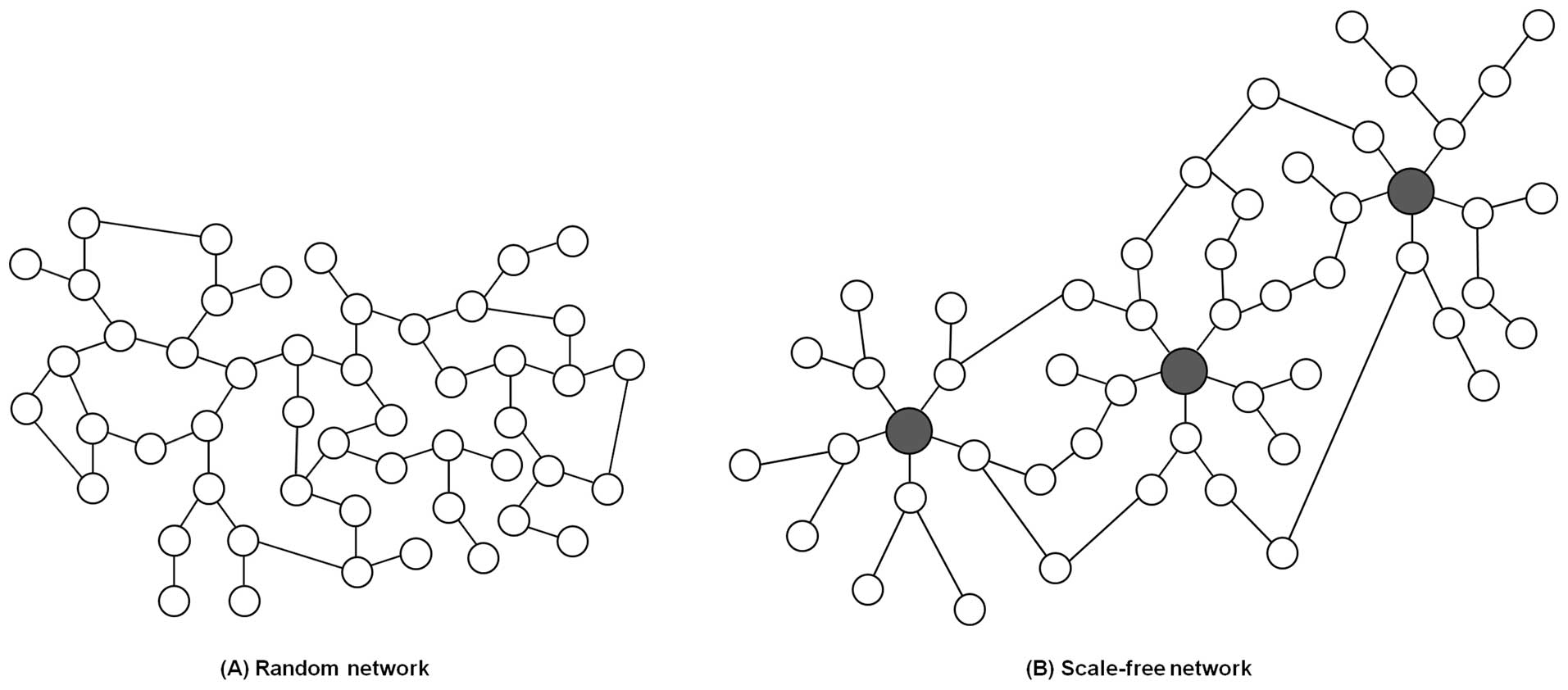
One class of networks is characterized as a random network, and
follows a Poisson distribution (Fig.
1A). In a random network, most nodes generally have the same
number of links, resulting in a fairly homogeneous network. In
contrast, many real systems including biological networks belong to
a class of heterogeneous networks called scale-free networks
(Fig. 1B). This type of network
follows a power-law for which a few nodes have a lot of links and
most nodes have a few links (1 or 2 links) (17,18).
The few highly connected nodes are known as hubs. Perturbing these
hubs could be catastrophic since they render the whole network
structure more stable and robust (19). Ironically, existence of hubs could
cause network fragility by offering reasonable targets for attacks.
However, natural events that cause network disruption (such as
mutations) are classified as failures rather than attacks since
these are non-specific events. In the absence of specific attacks,
hubs are relatively free from damage owing to dilution effect of a
number of non-hub nodes when a given network is disrupted in a
non-specific or random manner. Due to the existence of hubs,
networks can display a robustness to random errors (20).
As mentioned above, most biological networks
characterized as heterogeneous scale-free networks have the unique
property of possessing hubs. In a scale-free network, hub
disruption can lead to a major loss of connectivity that induces
network disturbance (21). Due to
this property, it is very likely that the proteins identified as
hubs are critical players in various biological processes. In
particular, studies using model organisms have shown that hub
proteins are more closely associated with essential genes. Due to
their functions and essentiality, the genes encoding hub proteins
also tend to be more conserved and evolve more slowly than ones
encoding non-hub proteins (19,22).
In addition to protein networks, disruption of the networks
containing essential metabolites (hub metabolites) could cause
severe cellular damage that would negatively impact cell survival
and growth. However, when the processes involving non-essential
metabolites are interfered with, cellular functions are not
significantly affected (23).
These data demonstrate that targeting hubs can cause considerable
damage to an overall cellular network compared to targeting non-hub
molecules. Another study was conducted to identify cancer-specific
network signatures using proteomic analysis (24). Several proteins including PTEN,
cyclin B1, p-CREB and vascular endothelial growth factor (VEGF)
were considered to be hubs of cancer-associated networks, which
could provide the implication to cancer diagnostic markers,
prognostic markers and potential therapeutic targets. Thus,
extracting hub elements could serve as an invaluable strategy for
drug discovery. The importance of hubs is also emphasized by their
potential use as biological markers.
Based on spatial and chemical connections for
cellular functionality in a process, a biological network is
subdivided in a modular manner (25,26).
Several functional modules (subnetworks) could determine phenotypic
changes. In disease processes, these modules are called disease
modules, and their development is associated with disease
occurrence (27). Since various
modules composed of a complex web of node interactions are densely
integrated, modular organization for a disease process is not
immediately apparent in a whole network and it is difficult to
specify a disease-associated module correctly. Previous studies
have been conducted to characterize specific networks and their
hubs, including ones associated with cancer, that might be used as
disease markers (28–32). Furthermore, disease incidence,
state and severity could be evaluated based on network signatures.
Altogether, identification of the hub molecules in biological
complex networks is important because these hubs could serve as
specific biomarkers and drug targets.
Cancer-associated networks
Cancer is one of complex diseases caused by
alterations of various signaling networks responsible for major
cellular functions such as proliferation, survival and apoptosis.
These complex networks consisting of several signaling modules are
driven by dynamics of various components, including DNA, RNA, and
proteins, as well as their connections leading to many
intracellular pathways such as crosstalk and feedback loops in
response to internal and external stimuli. Components in each
module function within different temporal and spatial scales,
leading to cancer heterogeneity. The integration of dynamic
signaling modules ultimately influences cell phenotype and may
result in tumorigenesis. Thus, understanding the complex networks
and their modules associated with cancer can be promising for the
development of novel therapeutic strategies.
Much effort has been devoted to understanding
tumor-associated networks involved in tumor initiation,
progression, and metastasis using various high-throughput
techniques (microarray, next-generation sequencing, and
2-dimensional electrophoresis/mass spectrometry) and bioinformatics
(computational algorithms and statistical/analytical tools).
Detailed interactome maps of several tumor types may help identify
network nodes as potential targets for therapeutic strategies that
are more effective than traditional approaches such as gene-focused
therapies which hardly consider the biological context of the
targets (31,33–36).
Out of approximately 25,000 human genes, only hundreds are
considered to represent essential diseases genes. A large
proportion of these genes has been identified as tumor-associated
hubs and includes ones encoding epidermal growth factor receptor
(EGFR), Ras, Akt, PTEN and p53 (37–40).
EGFR network is important for tumor growth, progression and
metastasis in human cancers, since EGFR have been identified to
play a critical role in DNA repair, cell cycle progression,
proliferation, and cell motility (39). It has been reported that EGFR
mutations were accumulated in patients with non-small cell lung
cancer and these mutant EGFR could be indicators for tumor behavior
and poor prognosis (41). In case
of p53, a tumor suppressor gene, DNA damage response can induce the
expression of this gene, consequently leading to cell cycle arrest
and apoptosis. p53 has been identified to have loss-of-function
mutations in various types of cancers (37). Furthermore, it is very likely that
disruption of p53 is highly associated with tumor initiation and
development (42). These hub genes
usually have been revealed to be dysregulated in many cancers,
leading to hyper-activation of proliferative networks, distant
metastatsis, and evasion of apoptotic cell death. Due to the
functional importance of hubs in cellular systems, modulation of
such hub networks is highly responsible for decision of cancer
phenotypes. In addition, these hubs may serve to the clinical
implications in promising design for therapeutic strategy.
Network-based approaches for
chemotherapy
Over the past decades, chemotherapeutic drug
discovery and development have focused on specific inhibitors that
target hub or hub-associated proteins (Table I). Although these strategies
initially increased curative efficacy with great potency of
targeting hub element in a given disease network, these target
therapies have generally been inappropriate for cancer treatment
due to their side-effects such as the induction of drug resistance.
It is very likely that network structures have remarkable
flexibility achieved through the alteration of subnetworks,
including pathway reprogramming and activation of a crosstalk
pathway, in response to external stimuli (43,44).
Indeed, biological components are highly redundant due to gene
duplication and the existence of protein isomers and families,
which have different properties despite the inclusion of very
closely related proteins. This redundancy enables the maintenance
of an entire biological network through the activation of
compensatory or detouring networks in response to stress-induced
damage (45–47). For example, the MEK kinase
inhibitor GSK1120212 inhibits MEK1 but not MEK2 that lacks a
binding site for the inhibitor (48). Therefore, MEK2 could escape
drug-induced inhibition. Undisturbed MEK2 could then reactivate the
pathway blocked by GSK1120212 via the activation of mediator
proteins instead of MEK1. This event is a consequence of network
reprogramming (49). In another
study, herceptin was developed for targeting cases of breast cancer
that express HER2, a member of the EGFR family (50,51).
Herceptin-resistance is sometimes acquired during treatment due to
the activation of other signaling subnetworks, including the
Akt-induced glycolytic pathway or Bcl-2-mediated anti-apoptotic
pathway, as compensatory crosstalk networks (52,53).
Additionally, for treating metastatic renal cell carcinomas,
therapies that target both the VEGF receptor (VEGFR) and the
mammalian target of rapamycin complex 1 (mTORC1) showed better
therapeutic efficiency than previous traditional treatments
(54). Unexpectedly, cancer cells
have acquired resistance to the combination therapies via
inhibition of VEGFR and mTORC1 and patients eventually suffered
from tumor relapse with re-established tumor vasculature. Acquiring
drug-resistant mechanisms was accomplished by the loss of negative
feedback networks involving suppression of mTORC2 and Akt
signaling, which could consequently result in mTORC2-mediated
signaling activation as a crosstalk network for drug-induced mTORC1
inhibition. This event caused the mTORC2-mediated Akt and
hypoxia-inducible factor-1 (HIF-1) activation for angiogenesis,
leading to poor prognosis in metastatic renal cell carcinomas.
 | Table I.Hub elements and their functions in
cancer-related networks. |
Table I.
Hub elements and their functions in
cancer-related networks.
| Hub element | Biological
effect | Targeted drug | Refs. |
|---|
| VEGF | Invasion,
angiogenesis, metastasis | Bevacizumab,
Ranibizumab | (24,54,71–73) |
| EGFR/Her2 | Proliferation,
invasion, metastasis, cell cycle progression, DNA repair,
anti-apoptosis | Lapatinib,
Erlotinib, Cetuximab, Trastuzumab | (39,41,50–53,77,78) |
| NF-κB | Inflammation,
proliferation, survival, radioresistance | Denosumab (RANKL
inhibitor) | (70) |
| PI3K/Akt | Proliferation,
metabolism, survival, anti-apoptosis | GS-1101 (phase II),
PX-866 (phase II), KRX-0401 (phase III) | (53,54) |
| HIF-1 | Hypoxia response,
glycolytic switch, survival, invasion, angiogenesis,
metastasis | EZN-2208 (phase I),
EZN-2968 (phase I), PX-478 (phase I) | (54,71–73) |
| p53 | Tumor suppressor
activity, DNA repair, cell cycle arrest, senescence, apoptosis | | (37,42) |
| PTEN | Senescence,
anti-proliferation, tumor suppressor activity | | (24) |
These findings demonstrate that altered network
states including network reprogramming and the existence of
crosstalk network can complement a hub-associated network disrupted
by a specific drug, and are major causes of drug resistance.
Single-target drugs might not only have beneficial effects on
dysfunctional aspects of disease-associated modules in entire
complex networks of cancer, but they could also turn-on other
components in nearby disease-associated modules showing
side-effects.
Network-based approaches for
radiotherapy
Radiotherapy is one of the major modalities of
cancer management. More than 50% of the patients with cancer have
undergone radiation treatment. Ionizing radiation (IR) generates
intermediate free radicals and reactive oxygen species leading to
DNA double-strand breaks (DSBs) (55). Unless cells repair this type of
injury properly, they directly or indirectly undergo cell death.
Although many tumor cells immediately die via apoptosis after
radiation exposure, some of cells can survive through activation of
DSB repair pathway including homologous recombination and
non-homologous end-joining (56,57).
The surviving tumor cells accompany unavoidable gene mutations due
to the properties of DSB repair modules, leading to additional
effects including hyper-activation of crosstalk signaling to
compensate the loss of a certain gene that account for
radioresistance. Indeed, other modules are also activated and
integrated for helping tumor cells overcome IR-induced stress. It
is supported by several interactome and gene profiling analyses
using various types of cancer cells treated with radiation
(58–63). The results indicated that numerous
cellular networks, including modules for DNA repair, survival,
apoptosis, cell cycle, cell migration, protein localization, RNA
processing, antioxidant defense, inflammation and cell
proliferation, are altered by radiation exposure and help determine
tumor cell fate. For example, p53-related genes and DNA-damage
response genes are generally activated by irradiation in
susceptible lung cancer cells while the networks associated with
these genes are disrupted in radioresistant lung cancer cells
(60,62,63).
In addition to drug-induced network alterations,
radiotherapy could also contribute to the activation of new
subnetworks, resulting in network flexibility (64–66).
Unlike drugs which act on specific target molecules, ionizing
radiation exerts effects on whole cell components (67). Thus, the proportion of hub elements
that are functionally disrupted by radiotherapy is relatively low.
This is due to the dilution effect of non-hub elements since these
elements are more abundant and consequently subjected to greater
damage (18). Hubs that remain
undamaged could eventually activate other radiation-responsive
signaling networks, reintegrate network topologies and establish
networks more resistant to radiotherapy, thus leading to
radioresistance (68,69). For example, NF-κB-mediated
inflammatory signaling cascade in cancer cells could be activated
in response to reactive oxygen species generated by irradiation
(70). Activated inflammatory
network with various chemokines and cytokines is associated with
cancer cell survival activity for overcoming radiotherapy-induced
inflammatory stress. This event might result in acquired
radio-resistance in cancer cells and consequently reduced efficacy
of following radiotherapy in tumor. During tumor development,
cancer cells partially undergo hypoxic condition due to
insufficient vasculature systems. Thus, these cells show
hyper-activation of HIF-1 network for adaptation to hypoxic stress.
It has been well-studied that cancer cells show more resistance to
irradiation under hypoxia than under normoxia (71). Several studies have revealed that
radiation induced HIF-1 stabilization and activate its signaling
module in a solid tumor via oxidative stress leading to the
increase in VEGF expression, which is well-known to play a
protective role in endothelial cells from the radiation-induced
cytotoxic effects (72,73). Consequently, tumor cells can be
supplied oxygen and nutrients from the protected tumor vasculature,
leading to tumor radioresistance and tumor growth progression.
Since exposure of tumor cells to radiation could
impact a large number of proteins simultaneously, it might take
much time and effort to identify hubs as specific drug targets
among the proteins affected by IR. Instead, understanding the
altered activation patterns of various IR-responsive modules could
allow us to hypothesize which proteins are responsible for critical
functions in each module. This will help identify hubs and
consequently promote the development of novel radiotherapy
strategies. Moreover, therapeutic efficacy could be improved by
administrating radiotherapy in conjunction with chemotherapeutic
agents such as radiosensitizers or inhibitors targeting hub
molecules associated with radioresistance.
Targeting network flexibility
As mentioned above, a large number of traditional
strategies that do not account for the dynamics of complex
systematic networks are not satisfactory cancer treatments.
Biological networks in tumors gradually adapt to chemo- and
radiotherapy. Tumor cells are thus able to maintain their
tumorigenic properties through compensatory mechanisms such as
crosstalk circuits (74,75). To improve cancer therapeutic
efficacy, targeting the hub itself might be insufficient.
Additionally, hub-associated network flexibility developed in
response to therapeutic challenges could be targeted. Consequently,
the field of network medicine has recently emerged (4,27,43).
Based on numerous system biological studies, several investigations
have been recently conducted to identify and discover ways to
regulate back-up networks activated during chemotherapy, and
develop network medicine to overcome chemoresistance (28,76,77).
For the patients with Her2-positive breast cancer, lapatinib was
approved as the first dual inhibitor of EGFR/Her2. However, the
efficiency of this drug was not prolonged due to acquired
resistance. A network-based computational analysis showed that,
while lapatinib initially induced inhibition of glucose uptake and
energetic stress leading to apoptosis in Her2-positive cancer
cells, the glucose deprivation response network is gradually
activated as a compensatory mechanism in response to the inhibition
of the Her2-mediated oncogenic network by lapatinib, which
eventually result in drug resistance (Fig. 2) (77). It was suggested that novel
combinations should be administered to simultaneously target Her2
networks and metabolic networks to treat cases of Her2-positive
breast cancer that had acquired drug resistance. For the patients
with hormone receptor-positive metastatic breast cancer, letrozole,
an inhibitor for aromatase (estrogen producing enzyme), could be
treated for endocrine therapies (78). However, this endocrine therapy
showed unsatisfactory effect due to the activation of cross-talk
pathways involving EGFR/Her2 and estrogen receptor (ER), leading to
resistance to therapies. It was presented that the combined
therapies with lapatinib (or herceptin) and letrozole could be a
promising strategy for endocrine resistance by blocking ER-mediated
hormone signaling as well as compensatory EGFR/Her2 networks for
survival signaling.
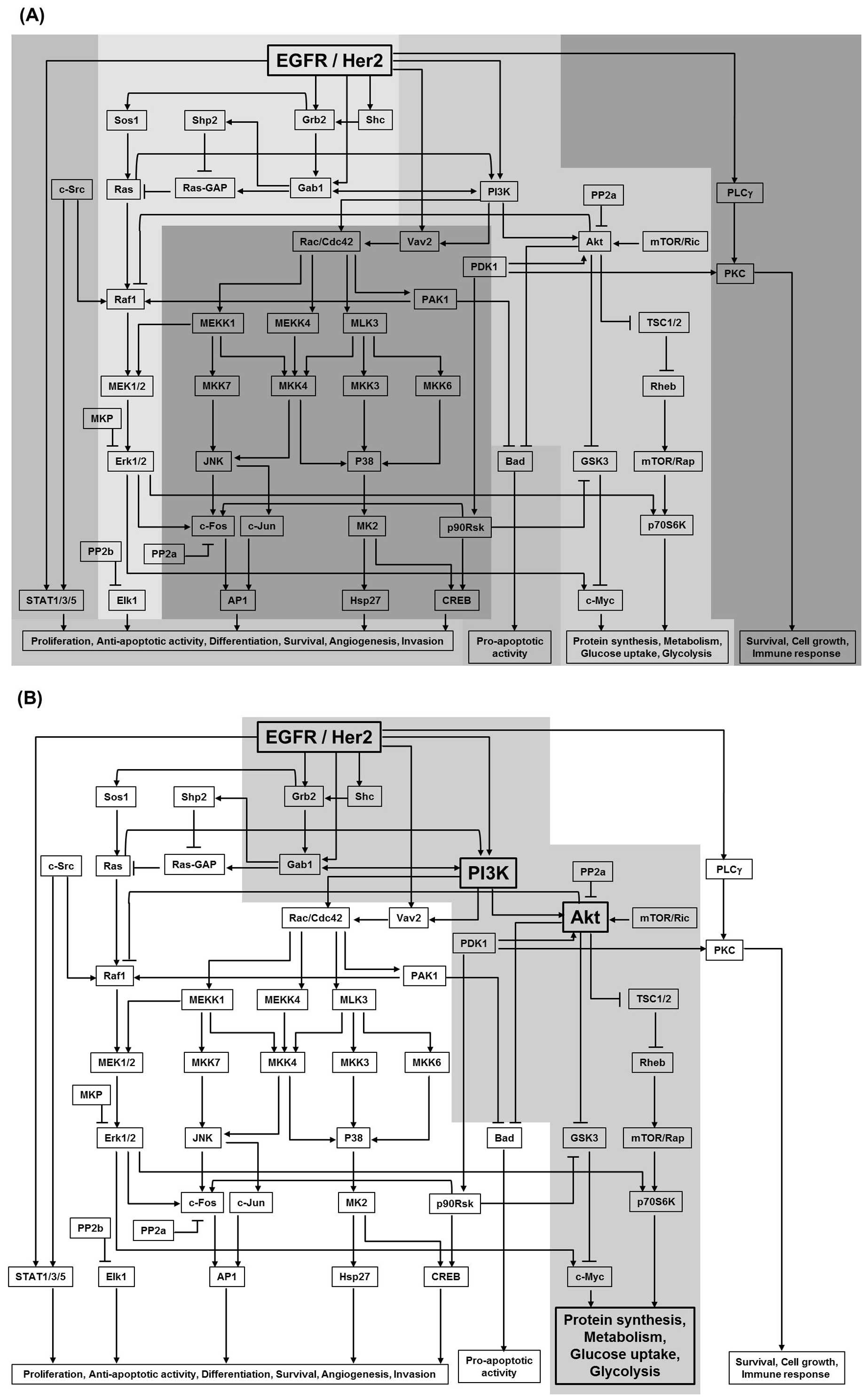 | Figure 2.An EGFR network structure. (A) An
EGFR network including several functional modules in cells is
presented. EGFR and Her2 as well-known receptor tyrosine kinases
can activate various downstream effectors in response to external
stimuli, including their ligands, drugs and radiation.
Consequently, many functional signaling modules are influenced,
which are usually associated with tumor initiation and development,
including cell survival, metabolism activation, cell cycle
progression, proliferation and differentiation. EGFR/Her2-targeted
drugs might interrupt some of these modules, leading to tumor cell
death and therapeutic effects. (B) A compensatory module in
response to lapatinib in drug-resistance cancer cells is presented
as a shaded area. Lapatinib is one of EGFR/Her2 inhibitors.
Although the tumor shrinkage efficiency was shown in Her2-positive
breast cancer patients, it could not be repeatedly administered in
cancer therapy due to acquired drug-resistance. The reason is that
lapatinib-induced glucose deprivation, which led to tumor
cytotoxicity, might activate a cross-talk module for the increase
of glucose uptake and metabolism to adapt to stress condition,
leading to drug-resistance and poor tumor prognosis. |
When it comes to radiotherapy, complex networks that
are critically responsible for network flexibility resulting in
acquired radioresistance are not fully understood. Nevertheless,
some investigations have demonstrated the involvement of several
genes, including HDAC1, MDM2, c-Jun, PKC-β, c-Abl and CDK1, in
cellular responses to radiation (79–81).
Some of these genes have been studied as potential targets for
radiosensitizer development. For example, c-Abl, a non-receptor
tyrosine kinase, plays a critical role in cell survival,
proliferation, and anti-apoptotic activity, leading to
tumorigenesis. In addition to oncogenic properties of c-Abl, a
study using glioma cells showed that c-Abl elevated the expression
of Rad51 in response to radiation, which is a crucial component of
the DNA repair pathway, especially DSBs (82–84).
It means that c-Abl could modulate radio-response through
activating DNA repair module leading to radioresistance. In this
case, STI571, a pharmacological drug of c-Abl kinase, could be used
to block c-Abl-Rad51 signaling for a DNA repair module to render
radiosensitizing effect in glioma cells, but this drug had no
effect in normal cells (82). It
could be concluded that several compensatory modules such as the
cell cycle module, proliferative module, and DNA repair module are
activated after radiotherapy to protect tumor cells against
IR-induced injuries. Activation of these modules would lead to
acquired radioresistance, cancer cell survival, and tumor
re-growth. Thus, it is necessary to use network-specific drugs as
radiotherapeutic adjuvants that suppress the activity of
survival-associated modules and prevent unexpected side-effects. In
order to prevent tumor cells from acquiring resistance to chemo-
and radiotherapy, it is important to understand the rewiring states
of networks and their essential nodes in response to cancer
therapies. In this manner, a network-based combination therapy
targeting the hubs associated with network flexibility can be
formulated to overcome adverse effects induced by current
therapies.
Conclusion
Network-based therapies for treating human cancers
may have various promising biological and clinical applications. In
particular, hub elements in a disease network could function as
biological markers because these hubs are highly connected to
biological scale-free networks and their roles in each network are
essential. It is probable that the fate of tumor cells is
controlled by the regulation of hub elements. Analysis of
biological networks and the discovery of hub elements will help
identify novel drug-targets as well as diagnostic markers for
detecting early stage cancer. Single-target therapies might take
advantage of some aspects of the disease modules associated with
cancer, but these modalities are not generally effective since
complex biological networks consisting of various disease modules
exist in tumor cells and tissues. In addition, simple multi-target
therapies are still not optimal cancer treatments because
biological networks are dynamically altered in a stimulus-dependent
manner to maintain homeostasis (in this case, cancer cell
homeostasis for survival and proliferation). Biological systems are
highly heterogeneous and network structures in the context of
tumors are flexible enough for adaptation to various external
stimuli. Network-based combinational approaches could be the most
promising strategies for silencing specific mediators (i.e., novel
hubs) responsible for the alteration of network states. These
techniques could maximize the effect of more traditional therapies
by simultaneous administration of pharmacological agents that
specifically target hubs and disrupt major disease modules. A large
quantity of integrated bioinformatics data has been gradually
collected over time. Using this information, we can closely examine
entire network structures and their states. In addition, it will be
possible to predict how a network state will be modified in
response to chemo- and radiotherapy. This will facilitate the
development of ideal network-based drug combinations and
personalized therapeutic strategies.
Acknowledgements
This study was supported by the
Bio-Scientific Research Grant funded by the Pusan National
University (PNU-2010-101-249).
References
|
1.
|
Janes KA and Lauffenburger DA: A
biological approach to computational models of proteomic networks.
Curr Opin Chem Biol. 10:73–80. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Miller-Jensen K, Janes KA, Brugge JS and
Lauffenburger DA: Common effector processing mediates cell-specific
responses to stimuli. Nature. 448:604–608. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Jordan JD, Landau EM and Iyengar R:
Signaling networks: the origins of cellular multitasking. Cell.
103:193–200. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Pawson T and Linding R: Network medicine.
FEBS Lett. 582:1266–1270. 2008. View Article : Google Scholar
|
|
5.
|
Vidal M, Cusick ME and Barabasi AL:
Interactome networks and human disease. Cell. 144:986–998. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Vidal M: A unifying view of 21st century
systems biology. FEBS Lett. 583:3891–3894. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Barabasi AL and Oltvai ZN: Network
biology: understanding the cell’s functional organization. Nature
Rev Genet. 5:101–113. 2004.
|
|
8.
|
Ideker T and Sharan R: Protein networks in
disease. Genome Res. 18:644–652. 2008. View Article : Google Scholar
|
|
9.
|
Rual JF, Venkatesan K, Hao T, et al:
Towards a proteome-scale map of the human protein-protein
interaction network. Nature. 437:1173–1178. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Stelzl U, Worm U, Lalowski M, et al: A
human protein-protein interaction network: a resource for
annotating the proteome. Cell. 122:957–968. 2005.PubMed/NCBI
|
|
11.
|
Jeong H, Tombor B, Albert R, Oltvai ZN and
Barabasi AL: The large-scale organization of metabolic networks.
Nature. 407:651–654. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Chang RL, Xie L, Bourne PE and Palsson BO:
Drug off-target effects predicted using structural analysis in the
context of a metabolic network model. PLoS Comput Biol.
6:e10009382010. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Schlitt T and Brazma A: Current approaches
to gene regulatory network modelling. BMC Bioinformatics. 8(Suppl
6): S92007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Zhao Y, He S, Liu C, et al: MicroRNA
regulation of messenger-like noncoding RNAs: a network of mutual
microRNA control. Trends Genet. 24:323–327. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
He X, He L and Hannon GJ: The guardian’s
little helper: microRNAs in the p53 tumor suppressor network.
Cancer Res. 67:11099–11101. 2007.
|
|
16.
|
Zhou Q, Chipperfield H, Melton DA and Wong
WH: A gene regulatory network in mouse embryonic stem cells. Proc
Natl Acad Sci USA. 104:16438–16443. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Barabasi AL and Albert R: Emergence of
scaling in random networks. Science. 286:509–512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Albert R, Jeong H and Barabasi AL: Error
and attack tolerance of complex networks. Nature. 406:378–382.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Jeong H, Mason SP, Barabasi AL and Oltvai
ZN: Lethality and centrality in protein networks. Nature.
411:41–42. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Park J and Newman ME: Statistical
mechanics of networks. Phys Rev E Stat Nonlin Soft Matter Phys.
70:0661172004. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Albert R: Scale-free networks in cell
biology. J Cell Sci. 118:4947–4957. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Fraser HB, Hirsh AE, Steinmetz LM, Scharfe
C and Feldman MW: Evolutionary rate in the protein interaction
network. Science. 296:750–752. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Kim PJ, Lee DY, Kim TY, et al: Metabolite
essentiality elucidates robustness of Escherichia coli
metabolism. Proc Natl Acad Sci USA. 104:13638–13642. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Zhang DY, Ye F, Gao L, et al: Proteomics,
pathway array and signaling network-based medicine in cancer. Cell
Div. 4:202009. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Ravasz E, Somera AL, Mongru DA, Oltvai ZN
and Barabasi AL: Hierarchical organization of modularity in
metabolic networks. Science. 297:1551–1555. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Han JD, Bertin N, Hao T, et al: Evidence
for dynamically organized modularity in the yeast protein-protein
interaction network. Nature. 430:88–93. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Barabasi AL, Gulbahce N and Loscalzo J:
Network medicine: a network-based approach to human disease. Nature
Rev Genet. 12:56–68. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Creixell P, Schoof EM, Erler JT and
Linding R: Navigating cancer network attractors for tumor-specific
therapy. Nat Biotechnol. 30:842–848. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Hwang S, Son SW, Kim SC, Kim YJ, Jeong H
and Lee D: A protein interaction network associated with asthma. J
Theor Biol. 252:722–731. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Jonsson PF and Bates PA: Global
topological features of cancer proteins in the human interactome.
Bioinformatics. 22:2291–2297. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Chuang HY, Lee E, Liu YT, Lee D and Ideker
T: Network-based classification of breast cancer metastasis. Mol
Syst Biol. 3:1402007. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Garcia M, Millat-Carus R, Bertucci F,
Finetti P, Birnbaum D and Bidaut G: Interactome-transcriptome
integration for predicting distant metastasis in breast cancer.
Bioinformatics. 28:672–678. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Pache RA, Zanzoni A, Naval J, Mas JM and
Aloy P: Towards a molecular characterisation of pathological
pathways. FEBS Lett. 582:1259–1265. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Chand Y and Alam MA: Network biology
approach for identifying key regulatory genes by expression based
study of breast cancer. Bioinformation. 8:1132–1138. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Sonachalam M, Shen J, Huang H and Wu X:
Systems biology approach to identify gene network signatures for
colorectal cancer. Front Genet. 3:802012. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Breitkreutz D, Hlatky L, Rietman E and
Tuszynski JA: Molecular signaling network complexity is correlated
with cancer patient survivability. Proc Natl Acad Sci USA.
109:9209–9212. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Vogelstein B, Lane D and Levine AJ:
Surfing the p53 network. Nature. 408:307–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Goh KI, Cusick ME, Valle D, Childs B,
Vidal M and Barabasi AL: The human disease network. Proc Natl Acad
Sci USA. 104:8685–8690. 2007. View Article : Google Scholar
|
|
39.
|
Han W and Lo HW: Landscape of EGFR
signaling network in human cancers: biology and therapeutic
response in relation to receptor subcellular locations. Cancer
Lett. 318:124–134. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Laubenbacher R, Hower V, Jarrah A, et al:
A systems biology view of cancer. Biochim Biophys Acta.
1796:129–139. 2009.PubMed/NCBI
|
|
41.
|
Sharma SV and Settleman J: ErbBs in lung
cancer. Exp Cell Res. 315:557–571. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Chiang YJ, Difilippantonio MJ, Tessarollo
L, Morse HC and Hodes RJ: Exon 1 disruption alters tissue-specific
expression of mouse p53 and results in selective development of B
cell lymphomas. PLoS One. 7:e493052012. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Erler JT and Linding R: Network medicine
strikes a blow against breast cancer. Cell. 149:731–733. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Lee MJ, Ye AS, Gardino AK, et al:
Sequential application of anticancer drugs enhances cell death by
rewiring apoptotic signaling networks. Cell. 149:780–794. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Pilpel Y, Sudarsanam P and Church GM:
Identifying regulatory networks by combinatorial analysis of
promoter elements. Nat Genet. 29:153–159. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Bhan A, Galas DJ and Dewey TG: A
duplication growth model of gene expression networks.
Bioinformatics. 18:1486–1493. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Pastor-Satorras R, Smith E and Sole RV:
Evolving protein interaction networks through gene duplication. J
Theor Biol. 222:199–210. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Gilmartin AG, Bleam MR, Groy A, et al:
GSK1120212 (JTP-74057) is an inhibitor of MEK activity and
activation with favorable pharmacokinetic properties for sustained
in vivo pathway inhibition. Clin Cancer Res. 17:989–1000. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Duncan JS, Whittle MC, Nakamura K, et al:
Dynamic reprogramming of the kinome in response to targeted MEK
inhibition in triple-negative breast cancer. Cell. 149:307–321.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Slamon DJ, Clark GM, Wong SG, Levin WJ,
Ullrich A and McGuire WL: Human breast cancer: correlation of
relapse and survival with amplification of the HER-2/neu oncogene.
Science. 235:177–182. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Hudis CA: Trastuzumab--mechanism of action
and use in clinical practice. N Engl J Med. 357:39–51. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Crawford A and Nahta R: Targeting Bcl-2 in
herceptin-resistant breast cancer cell lines. Curr Pharmacogenomics
Person Med. 9:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Chan CH, Li CF, Yang WL, et al: The
Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis,
herceptin sensitivity, and tumorigenesis. Cell. 149:1098–1111.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Figlin RA, Kaufmann I and Brechbiel J:
Targeting PI3K and mTORC2 in metastatic renal cell carcinoma: new
strategies for overcoming resistance to VEGFR and mTORC1
inhibitors. Int J Cancer. 133:788–796. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55.
|
Li L, Story M and Legerski RJ: Cellular
responses to ionizing radiation damage. Int J Radiat Oncol Biol
Phys. 49:1157–1162. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
Li YH, Wang X, Pan Y, Lee DH, Chowdhury D
and Kimmelman AC: Inhibition of non-homologous end joining repair
impairs pancreatic cancer growth and enhances radiation response.
PLoS One. 7:e395882012. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Sonoda E, Hochegger H, Saberi A, Taniguchi
Y and Takeda S: Differential usage of non-homologous end-joining
and homologous recombination in double strand break repair. DNA
Repair. 5:1021–1029. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Lhakhang TW and Chaudhry MA: Interactome
of radiation-induced microRNA-predicted target genes. Comp Funct
Genomics. 2012:5697312012. View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Ma L, Nie L, Liu J, et al: An
RNA-seq-based gene expression profiling of radiation-induced
tumorigenic mammary epithelial cells. Genomics Proteomics
Bioinformatics. 10:326–335. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Lee YS, Oh JH, Yoon S, et al: Differential
gene expression profiles of radioresistant non-small-cell lung
cancer cell lines established by fractionated irradiation: tumor
protein p53-inducible protein 3 confers sensitivity to ionizing
radiation. Int J Radiat Oncol Biol Phys. 77:858–866. 2010.
View Article : Google Scholar
|
|
61.
|
Kalanxhi E and Dahle J: Genome-wide
microarray analysis of human fibroblasts in response to gamma
radiation and the radiation-induced bystander effect. Radiat Res.
177:35–43. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Kim KH, Yoo HY, Joo KM, et al: Time-course
analysis of DNA damage response-related genes after in vitro
radiation in H460 and H1229 lung cancer cell lines. Exp Mol Med.
43:419–426. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63.
|
Xu QY, Gao Y, Liu Y, Yang WZ and Xu XY:
Identification of differential gene expression profiles of
radioresistant lung cancer cell line established by fractionated
ionizing radiation in vitro. Chin Med J. 121:1830–1837.
2008.PubMed/NCBI
|
|
64.
|
Ding LH, Shingyoji M, Chen F, et al: Gene
expression profiles of normal human fibroblasts after exposure to
ionizing radiation: a comparative study of low and high doses.
Radiat Res. 164:17–26. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Rashi-Elkeles S, Elkon R, Shavit S, et al:
Transcriptional modulation induced by ionizing radiation: p53
remains a central player. Mol Oncol. 5:336–348. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66.
|
Tusher VG, Tibshirani R and Chu G:
Significance analysis of microarrays applied to the ionizing
radiation response. Proc Natl Acad Sci USA. 98:5116–5121. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
67.
|
Somosy Z: Radiation response of cell
organelles. Micron. 31:165–181. 2000. View Article : Google Scholar
|
|
68.
|
Cao N, Li S, Wang Z, et al:
NF-kappaB-mediated HER2 over-expression in radiation-adaptive
resistance. Radiat Res. 171:9–21. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
69.
|
Lee SY, Park HR, Cho NH, et al:
Identifying genes related to radiation resistance in oral squamous
cell carcinoma cell lines. Int J Oral Max Surg. 42:169–176. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
70.
|
Multhoff G and Radons J: Radiation,
inflammation, and immune responses in cancer. Front Oncol.
2:582012. View Article : Google Scholar : PubMed/NCBI
|
|
71.
|
Brown JM and Wilson WR: Exploiting tumour
hypoxia in cancer treatment. Nat Rev Cancer. 4:437–447. 2004.
View Article : Google Scholar
|
|
72.
|
Moeller BJ, Cao Y, Li CY and Dewhirst MW:
Radiation activates HIF-1 to regulate vascular radiosensitivity in
tumors: role of reoxygenation, free radicals, and stress granules.
Cancer cell. 5:429–441. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
73.
|
Moeller BJ and Dewhirst MW: HIF-1 and
tumour radiosensitivity. Br J Cancer. 95:1–5. 2006. View Article : Google Scholar
|
|
74.
|
Kitano H: Biological robustness. Nature
Rev Genet. 5:826–837. 2004. View Article : Google Scholar
|
|
75.
|
Russell RB and Aloy P: Targeting and
tinkering with interaction networks. Nat Chem Biol. 4:666–673.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
76.
|
Shao L, Wang L, Wei Z, et al: Dynamic
network of transcription and pathway crosstalk to reveal molecular
mechanism of MGd-treated human lung cancer cells. PLoS One.
7:e319842012. View Article : Google Scholar : PubMed/NCBI
|
|
77.
|
Komurov K, Tseng JT, Muller M, et al: The
glucose-deprivation network counteracts lapatinib-induced toxicity
in resistant ErbB2-positive breast cancer cells. Mol Syst Biol.
8:5962012. View Article : Google Scholar : PubMed/NCBI
|
|
78.
|
Johnston S, Pippen J Jr, Pivot X, et al:
Lapatinib combined with letrozole versus letrozole and placebo as
first-line therapy for postmenopausal hormone receptor-positive
metastatic breast cancer. J Clin Oncol. 27:5538–5546. 2009.
View Article : Google Scholar
|
|
79.
|
Eschrich S, Zhang H, Zhao H, et al:
Systems biology modeling of the radiation sensitivity network: a
biomarker discovery platform. Int J Radiat Oncol Biol Phys.
75:497–505. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
80.
|
Chiba M: Radiation-responsive
transcriptome analysis in human lymphoid cells. Radiat Prot
Dosimetry. 152:164–167. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81.
|
Stiubea-Cohen R, David R, Neumann Y, et
al: Effect of irradiation on cell transcriptome and proteome of rat
submandibular salivary glands. PLoS One. 7:e406362012. View Article : Google Scholar : PubMed/NCBI
|
|
82.
|
Russell JS, Brady K, Burgan WE, et al:
Gleevec-mediated inhibition of Rad51 expression and enhancement of
tumor cell radiosensitivity. Cancer Res. 63:7377–7383.
2003.PubMed/NCBI
|
|
83.
|
Raderschall E, Stout K, Freier S, Suckow
V, Schweiger S and Haaf T: Elevated levels of Rad51 recombination
protein in tumor cells. Cancer Res. 62:219–225. 2002.PubMed/NCBI
|
|
84.
|
Slupianek A, Hoser G, Majsterek I, et al:
Fusion tyrosine kinases induce drug resistance by stimulation of
homology-dependent recombination repair, prolongation of G(2)/M
phase, and protection from apoptosis. Mol Cell Biol. 22:4189–4201.
2002. View Article : Google Scholar
|