Introduction
Statins, the 3-hydroxy-3-methylglutaryl coenzyme A
(HMGCoA) reductase inhibitors, are a class of drugs that inhibit
the rate-limiting steps in the cholesterol biosynthetic pathway
(1). Cholesterol is an important
structural component of the cell membrane and the physiological
requirements derive by endogenous synthesis or exogenous supply.
Increases in lipid levels lead to atherosclerosis and narrowing of
the blood vessels, which in turn may affect the blood supply to the
heart, brain and peripheral circulation, leading to morbidity or
mortality (2).
Statins, by inhibiting cholesterol biosynthesis,
emerged as a principal agent in lowering the incidence of
cardiovascular disease. However, it must be considered that any
compound leading to depletion of cholesterol, which is the main
structural component of cell membranes, affects various cellular
events and impairs homeostasis. Statins, potent inhibitors of
cholesterol synthesis, act by inhibiting 3-hydroxy-3-methylglutaryl
CoA (HMG-CoA) reductase, which catalyzes the conversion of HMG-CoA
to mevalonate (3). In addition to
the cholesterol-lowering property, many biological effects of
statins can be derived from cholesterol-independent pleiotropic
mechanisms, which are likely a consequence of blocking
intracellular signaling (4).
The role of statins extends beyond its
lipid-lowering effects, as they are known to improve endothelial
functions and participate in plaque stabilization, immune
modulation and antioxidant activity and also acts as
anti-inflammatory and anticancer agents. Their pleiotropic or
cholesterol-independent effects at the cellular and molecular
levels are highly related to numerous cellular functions, such as
proliferation and differentiation. Treatment with simvastatin,
mevastatin, atorvastatin, or pravastatin induces morphological
change and decrease cell proliferation. It has been observed that
the use of simvastatin was more effective in cancer cells and
embryonic stem cells (ESCs), in relation to normal cells. In ESC,
the loss of self-renewal by simvastatin was characterized by marked
downregulation of several genes with function of ESC markers as
alkaline phosphatase, Oct4, Nanog, Rex-1 and SSEA-1. Simvastatin
effects were selectively reversed by either mevalonate or its
metabolite, geranylgeranyl pyrophosphate (GGPP), but not by
cholesterol or farnesyl pyrophosphate (5).
Besides their use in the treatment of lipid
disorders, statins have been studied for their anti-carcinogenic
effects in several models, including carcinomas of the colon and
rectum, prostate, breast, lung and skin (6,7).
Many studies have shown the anti-proliferative and
pro-apoptotic effects of statins to a greater degree both in
malignant and in non-malignant cells (8,9).
Statins can also trigger different tumor cells to undergo apoptosis
in vitro and suppress tumor growth (10,11).
The role of cholesterol in cancer progression
remains to be resolved but many tumor cell lines and tissues
exhibit higher levels of cholesterol than their normal counterparts
(12,13). Some reports indicate that
hypocholesterolemia occurs in cancer due to increased use of
cholesterol by tumors (14)
whereas other reports have associated lower tissue cholesterol with
malignancy (15). Epidemiological
studies, the meta-analyses of statins use, and cancer risk in the
general population have provided conflicting results. Some studies
have shown cancer risk reduction associated with statins use
(16–18) while other studies have reported no
effect from its use (19–21) or even an increased risk (22). Unexpectedly, the typical response
to simvastatin was greater in poorly-differentiated cells when
compared to the well-differentiated cells (23).
In addition, substantial experimental and clinical
evidence suggests that statins exhibit anticancer effects mediated
by apoptosis and cell cycle arrest induction (24) through various signaling pathways.
It has been hypothesized that statin-induced apoptosis is mediated
by regulating BCL2 family members involved in mitochondrial
apoptosis pathway of various cells types (1,9,11,25–28).
Interestingly, the same authors (29) found that p54nrb, a novel
RNA binding protein with high homology to the PSF splicing factor
has a high affinity for RNA via its N-terminus and can bind
pre-mRNA and RNA implying a role in RNA processing (30,31).
As a binding protein of single stranded RNA it mediates the
splicing of several RNAs (32).
Furthermore, p54 is known as a transcription factor activating the
expression of several genes (33).
The nuclear p54nrb protein, also called
NonO (non-POU-domain-containing octamer binding protein), is an
RNA-binding molecule of 54 kDa containing two RNA recognition
motifs. p54nrb is able to bind double-stranded DNA,
single-stranded DNA and RNA, allowing the conclusion that
p54nrb has important roles in transcription and splicing
(33). p54nrb can
either form heterodimers with protein-associated splicing factors
or act as a monomer (32–39).
It may be speculated that p54 and NonO are
needed for the proper expression of proteins leading to a
stabilization of the transcription machinery, promoting the
survival of cells (40,41).
The concerted expression of genes involved in
different mechanisms of cell protection, like p54nrb and
NonO may contribute to the survival, promoting response
counteracting the apoptosis in cancer cells.
Malignant melanoma is the most dangerous variant of
skin cancer and prognosis of patients suffering from metastatic
melanoma is poor. One of the key molecules regulating melanoma
progression is the protein melanoma inhibitory activity
(MIA) which is strongly expressed in malignant melanoma but absent
in normal human melanocytes (42).
Several studies suggest an important role for MIA also in the early
tumor formation steps by regulating melanoma-related pathways and
molecules (42). Recent
investigations in mesenchymal stem cells hinted to protein
p54nrb as one of the MIA-regulated proteins (43). On these bases p54nrb or
the murine NonO seem to be a new molecule that function as
regulator of malignant melanoma progression (44).
In this report we have investigated the role of
simvastatin in the cancer cell growth inhibition showing the
capacity of this drug to reduce cancer progression.
In the in vivo experiments, in which the
melanoma was induced in C57Bl6 mice with the singenic B16-F10
melanoma cells, we observed an interesting reduction in the tumor
volume and survival increase in the animals treated with
simvastatin compared to the control group.
In addition, simvastatin induced a strong
downregulation of NonO gene-expression, the murine homolog of
p54nrb. On these bases the inhibited expression of NonO,
in simvastatin treated cells, might suggest a possible action
mechanism to explain the reduction of melanoma progression in mice
administered with this drug.
Materials and methods
Ethics statement
All procedures were carried out in strict accordance
with the recommendations in the Guide for the Care and Use of
Laboratory Animals of the National Institutes of Health and mice
were sacrificed using carbon dioxide (CO2) in accordance
to the Guidelines for Humane Endpoints for Animals Used in
Biomedical Research. Human endpoints were employed for the entire
experimentation time and the mice showing signs of excessive
distress or suffering were euthanized and eliminated from the
experiments. We humanely sacrificed and eliminated from the
experimentation any animals showing tumor with a diameter major
>18 mm, that is the maximum tumor size recommended from the
Guidelines of the Animal Experimentation Committee of IGB CNR,
Naples, Italy. The study was approved by the Animal Experimentation
Committee of IGB CNR, Naples, Italy.
Cells culture
The B16-F10 mouse melanoma cells, NIH-3T3 mouse
fibroblast cells, human melanoma SK-Mel-3 and the human melanoma
A375 cells were purchased from American Type Culture Collection.
All cells were grown at subconfluent culture in Dulbecco’s modified
Eagle’s medium or RPMI supplemented with L-glutamine, 100 U/ml
penicillin, 10 μg/ml streptomycin and 10% fetal bovine
serum, in 5% CO2 incubator at 37°C.
Treatment with simvastatin
The simvastatin carboxylate form (Calbiochem-Merck
Co., Darmstadt, Germany) is soluble in di-methyl-sulfoxide (DMSO)
and at a minor rate in ethanol.
In our experiments, simvastatin was dissolved in
DMSO prepared in a 20-mM stock solution stored frozen at −20°C. For
the experiments, the cells were plated and treated with 20
μM simvastatin for 48 or 24 h in normal culture conditions.
At the end of treatment, cells were washed with PBS and used for
TUNEL apoptosis analysis or harvested by scraping in TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) for mRNA expression analysis.
TUNEL analysis
Apoptotic cells were detected with TUNEL using the
Roche in situ cell death fluorescein detection kit (Roche
Diagnostics Mannheim, Germany) following the manufacturer’s
protocol. In brief, the plated cells were maintained in the
presence of 20 μM simvastatin for 48 h, after the treatment
cells were washed with PBS, fixed with 2% PFA, permeabilized with
0.1% Triton X-100 in 0.1% sodium citrate solution, TUNEL reaction
mix was added to the cells and incubated in a humidified chamber at
37°C for 60 min in the dark.
RNA purification and cDNA synthesis
Total mRNA was extracted from control NIH-3T3 mouse
fibroblast cells and simvastatin treated B16-F10 mouse melanoma
cells, using TRIzol reagent (Invitrogen Co. Carlsbad, CA, USA) and
the integrity of purified RNA was verified by agarose gel
electrophoresis.
For cDNA synthesis 2 μg of total RNA in a
final volume of 25 μl was reverse-transcribed with Avian
myeloblastosis virus (AMV) reverse transcriptase (Gibco-BRL,
Invitrogen), in the presence of random examer primers (Promega) at
37°C per 60 min, according to the manufacturer’s instructions. The
cDNA was controlled by PCR with housekeeping GAPDH or actin
primers.
PCR analysis
Three different samples of the B16 melanoma cells
and 3T3 control fibroblast were cultured for 24 h in DMEM
containing 10% FCS and 20 μM simvastatin. Control cells were
cultured in identical culture conditions without simvastatin. Total
RNA and cDNA synthesis was performed according the procedure
described. PCR analysis of NonO gene expression was performed by
using a GeneAmp PCR system 9700 (Applied Biosystem) and hot start
Taq Gold (Applera). Mouse GAPDH or actin was used as a housekeeping
control gene. The sequences of primers used were: mouse NonO Fw,
TTA ACT TGG AGA AGC AGA ATC; mouse NonO Rv, CAG GCA AAG CGC ACT CGC
AGC; actin Fw, GAC TAC CTC ATG AAG ATC CT; and actin Rv, GCT TGC
TGA TCC ACA TCT GC; mouse GAPDH Fw, TCC CTC AAG ATT GTC AGC AA;
mouse GAPDH Rv, AGA TCC ACA ACG GAT ACA TT. PCR conditions for
mouse GAPDH and actin were: initial denaturation at 95°C for 10 min
followed by 35 cycles: 95°C for 45 sec, 60°C for 45 sec and 72°C
for 45 sec with a final extension at 72° for 10 min. For mouse NonO
amplification the annealing temperature was 54°C and 42 PCR cycles.
The amplification products were analyzed on agarose gel to control
the amplicons length.
B16-F10 cell wound healing assay
In order to understand the effect of simvastatin on
progression and invasion of B16 cancer cells, we performed a
wound-healing assay. In brief, cells were seeded onto 60-mm dishes
at 5×105 cells per plate. When the confluence reached
90%, a single scratch wound was created on the plate with a pipette
yellow tip. After 24 and 48 h, in untreated cells or in presence of
20 μM simvastatin, the cell capacity to grown through the
scratch was verified. A quantitative analysis of cells migration
was performed in the Boyden chamber.
Boyden chamber cell migration assay
This analysis was carried out in a Boyden chamber
under serum-free conditions. Polycarbonate filters, 10-μm
pore size, were coated with 5 μg/ml fibronectin. After
treatment with 20 μM simvastatin for 24 and 48 h,
2×105 B16-F10 cells were trypsinized and placed in the
upper compartment of Boyden chambers in serum-free medium, while in
the lower compartment, FBS was introduced as the chemoattractant.
Cells were allowed to migrate for 4 h at 37°C in 5% CO2,
fixed in ethanol and stained with haematoxylin. Ten random
fields/filter were counted at ×200 magnification (45). In parallel, control cells were
assessed for viability and counted using the trypan blue exclusion
technique. Number of cells that had migrated was normalized to
analyse the effects on cell viability.
In vivo experiments
Animal experiments were performed in C57BL/6 mice of
20–25 g body weight, housed at 22–24°C under a 12-h light/dark
cycle and with free access to water and food. The melanoma was
induced in all C57BL6 mice by subcutaneus injection in the flank of
syngenic B16 melanoma cells at a concentration of 1×105
cells in 100 μl PBS. All the animals were injected precisely
with the same quantity of cells to avoid any difference in the
tumor development derived from difference in cells inoculation.
Drug-treated mice were administered, at alternate days, with
simvastatin dissolved in DMSO at a concentration of 25
μg/100 μl DMSO intraperitoneally (simvastatin dose
was 1 μg/g body weight) while control animals were
administered i.p. at alternate days with 100 μl DMSO. The
intraperitoneal administration was chosen to have a strong
absorption and avoid differences in the treatment. The simvastatin
administration started at the same time as the B16 cell injection.
The tumor volume measures were made every 3 days starting after 10
days from cell injection (when the mass was measurable).
The experiment was conducted for 20 days before the
animals died and the tumor became too large inducing animal
sufferings. According to the Guidelines of the Animal
Experimentation Committee of IGB CNR, we humanely sacrificed and
eliminated from the experimentation any animals showing tumor with
a diameter >18 mm.
The tumors diameters were measured using a caliper
and tumor volume was calculate with the ellipsoid volume formula V
= 4/3 πRxRyRz, that considering
the small radius on the y-axis equal to the small radius on z-axis,
were simplified in V = 1/6π (small diameter)2 (large diameter). In
the experiment 22 mice were administered with simvastatin while 20
control mice received only DMSO.
Survival rate
The survival analysis was conducted in accordance to
the Guidelines for Humane Endpoints for Animals Used in Biomedical
Research within an interval time of 18 days. To avoid animal
suffering, we sacrificed all the mice when the tumor was >20 mm,
that occur after 18 days from tumor onset. The mice were sacrificed
using carbon dioxide (CO2). The number of dead mice was
recorded every day and plotted on the Kaplan-Meier diagram.
Statistical analysis
Tumor volumes during the growth were analyzed by
Student’s t-test, and significance was set at p<0.05. For all
the animal groups the mean and the standard error are reported
(SEM).
Results
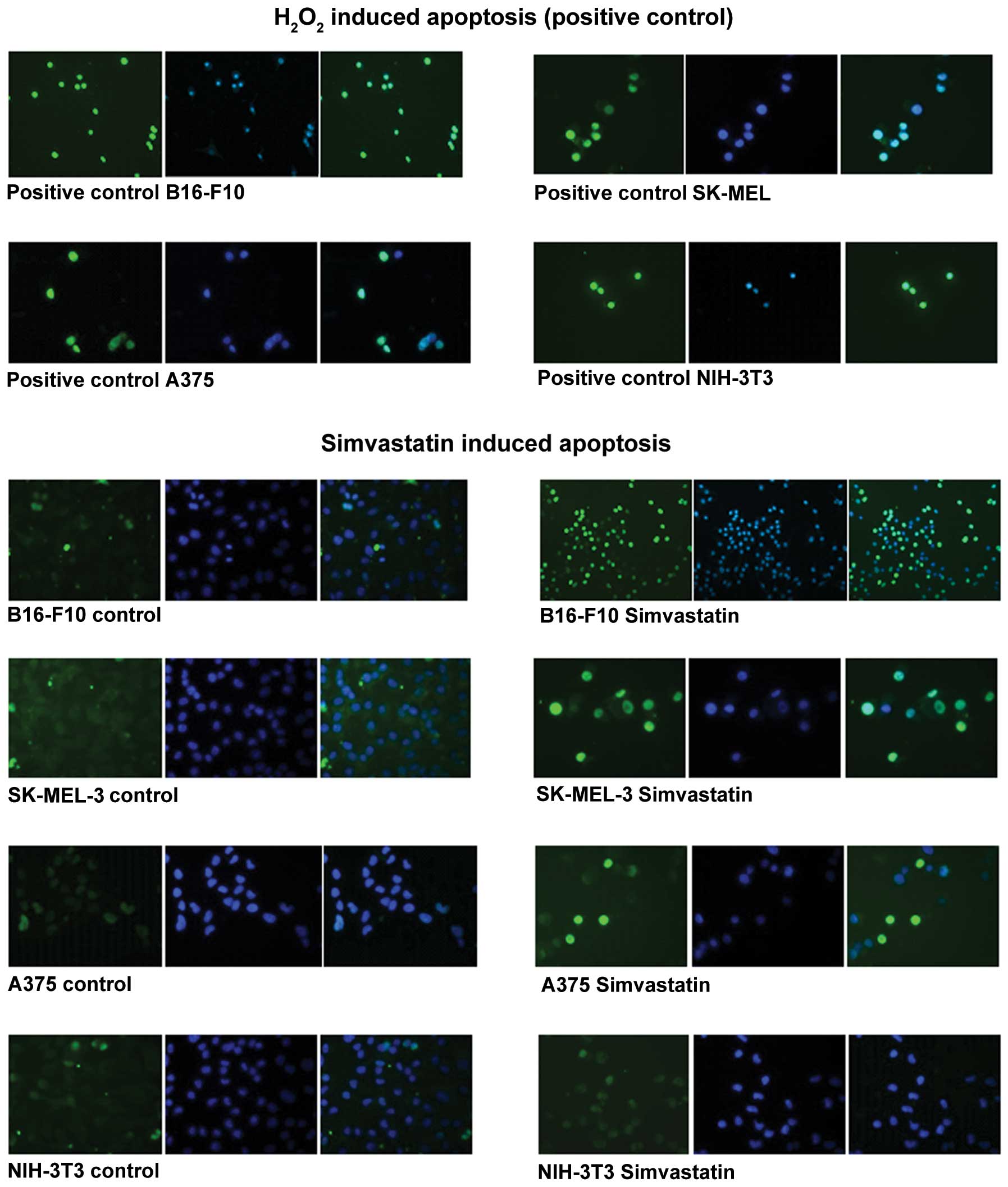
TUNEL analysis
The TUNEL analysis clearly showed that 48-h
treatment with 20 μM simvastatin induced apoptosis in three
different types of melanoma cancer cells, the mouse B16-F10
melanoma cells and the two human SK-Mel-3 and A375 melanoma cells.
On the contrary, the non-cancer NIH-3T3 fibroblasts did not show
apoptosis signs even when the drug treatment was prolonged >48
h. As positive control of TUNEL assay, apoptosis was induced by
30-min treatment with hydrogen peroxide. For all cell lines treated
with simvastatin, the nuclei DAPI staining (blue) and the apoptotic
nuclei (green) and the merge image were observed (Fig. 1).
The merge image clearly demonstrate that apoptotic
signal (green) was localized only in the nuclei, showing that 48-h
treatment with 20 μM simvastatin induced strong apoptosis in
cancer cells but not in non-transformed fibroblasts. In NIH-3T3
control cells, after 2 days of treatment, there was no apoptotic
stain in the nuclei. On the contrary, apoptosis induction, with a
strong green nuclear staining, was evident in B16 murine melanoma
cells. Similar results were observed in the human melanoma SK-MEL
and A375 cells. In all cancer cell lines, but principally in the
human melanoma cells, there was a high number of non-viable cells
after a 48-h drug treatment, and only some cells remained attached
on the culture plate. In these residual cells, a very strong nuclei
green coloration was observed due to apoptosis induction (Fig. 1). This experiment was performed
three times confirming apoptosis induced by simvastatin in cancer
cells, but not in normal fibroblasts.
NonO expression in simvastatin treated
cells
In our experiments 24-h treatment with simvastatin
completely inhibits the expression of NonO in melanoma cells
(Fig. 2). After 24 h of
simvastatin treatment there were no apparent signs of apoptosis and
the downregulation in NonO expression may not be a consequence of
the simvastatin induced cells damage or death. NonO is involved in
mRNA transcription, in splicing and consequently in protein
synthesis and it may play an important role in cell proliferation
and probably also in B16 melanoma progression. As shown in Fig. 2, simvastatin completely switched
off the NonO expression in B16-F10 melanoma cells, while the
expression of this gene was present in untreated B16 cells. NonO
expression is not visible in simvastatin treated cells also after
42 PCR cycles, while it was clearly expressed in control untreated
cells. In normal non-cancer fibroblast, the simvastatin did not
inhibit the NonO gene expression, in fact, its expression did not
changed in simvastatin-treated NIH-3T3 cells (Fig. 2). These data, strongly suggest a
possible action mechanism of this drug in the cancer cell growth
inhibition, but further experimentation are required to confirm
NonO as a simvastatin target.
B16-F10 cell wound healing analysis
Simvastatin treatment clearly inhibited B16 melanoma
cell growth after 24 and 48 h of culture in presence of the drug.
Fig. 3 clearly shows that in the
untreated cells the scratch wound is repopulated after 48 h,
whereas in the plate administered with 20 μM simvastatin,
the cell growth is inhibited and the scratch remains empty after 24
and 48 h, demonstrating that simvastatin might inhibit cancer cells
growth and migration. This experiment has been replicated three
times confirming the results obtained.
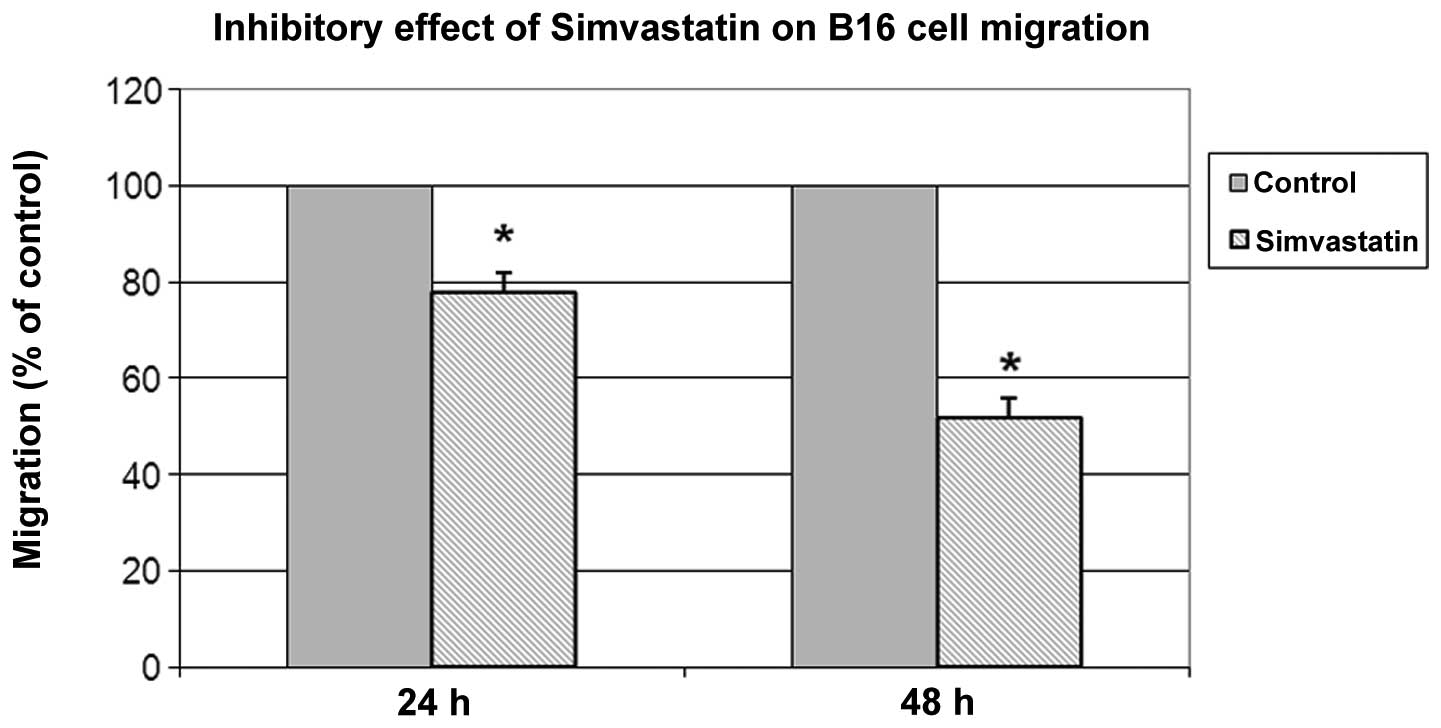
Boyden chamber cell migration assay
To investigate if simvastatin was also associated
with reduced cell invasion ability, a cell migration assay was
performed. As shown in Fig. 4
simvastatin affected B16-F10 cell migration. After 24 and 48 h of
treatment, cell migration inhibition, in comparison with untreated
cells, was ∼22 and 48%, respectively.
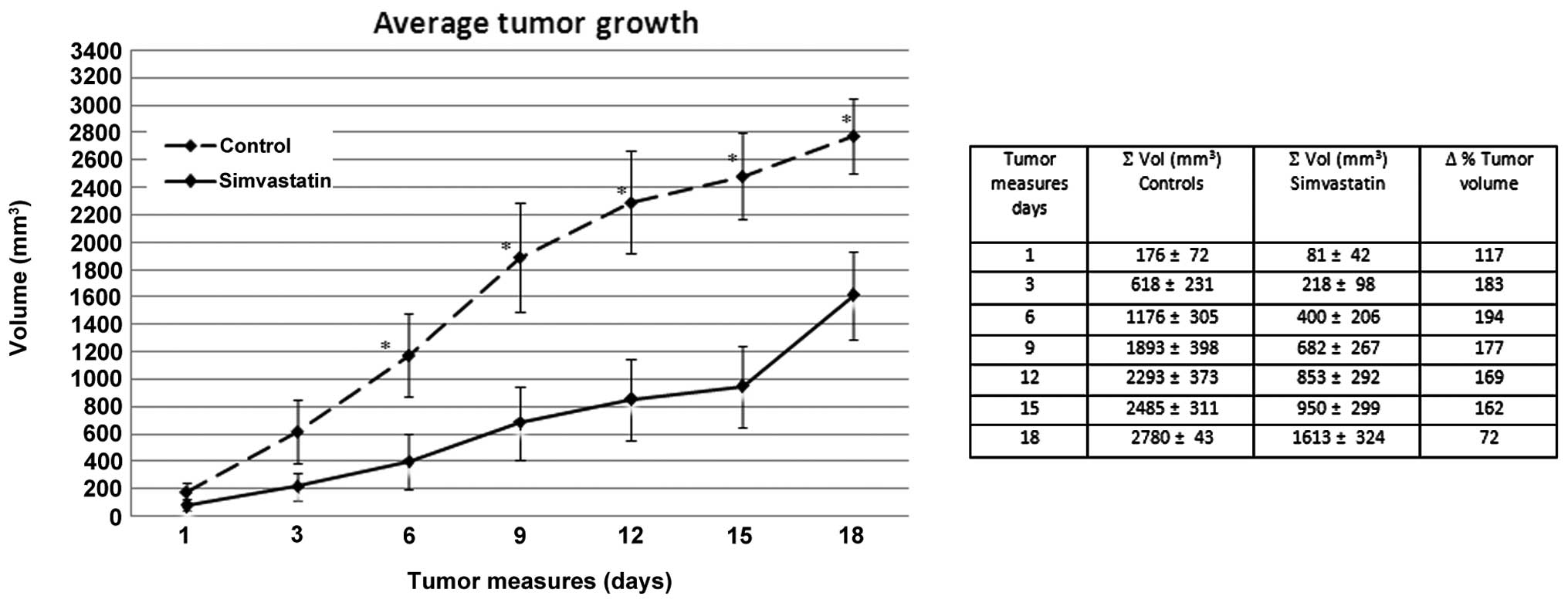
Tumor growth inhibition in simvastatin
treated mice
To verify the hypothesis that simvastatin could
limit melanoma growth in vivo, we administered the drug on
B16 melanoma-bearing mice. Simvastatin at a dose of 1 μg/g
body weight dissolved in DMSO, was given intraperitoneally at
alternate days for 20 days, starting the treatment contemporary
with the tumor cell inoculation. The effect of this drug on tumor
growth is shown in Fig. 5, in
which the tumor volume reduction compared to the controls, is
evident. Fig. 5 shows the average
tumor size of simvastatin treated mice, compared to the average
size in the control group during the treatment follow-up. We
observed, already at the first tumor measure, 10 days after cells
injection, a clear delay in tumor development in the simvastatin
treated group. In these mice we observed a volume average of 81
mm3 whereas it was 176 mm3 in the control
group. Furthermore, 6 days after the begin of tumor measures, the
average tumor volume in the control group was ∼1200 mm3
compared to 950 mm3, the average volume reached in the
group treated with simvastatin after >15 days of cancer
development. These data, clearly demonstrate that in a murine in
vivo model, simvastatin inhibits melanoma growth. In our
experiment the tumor development, as a volume measure, is on
average <150% in simvastatin treated animals compared to the
control group for all the experimental time. The differences were
significant with p-value ranging from 0.05 to <0.01.
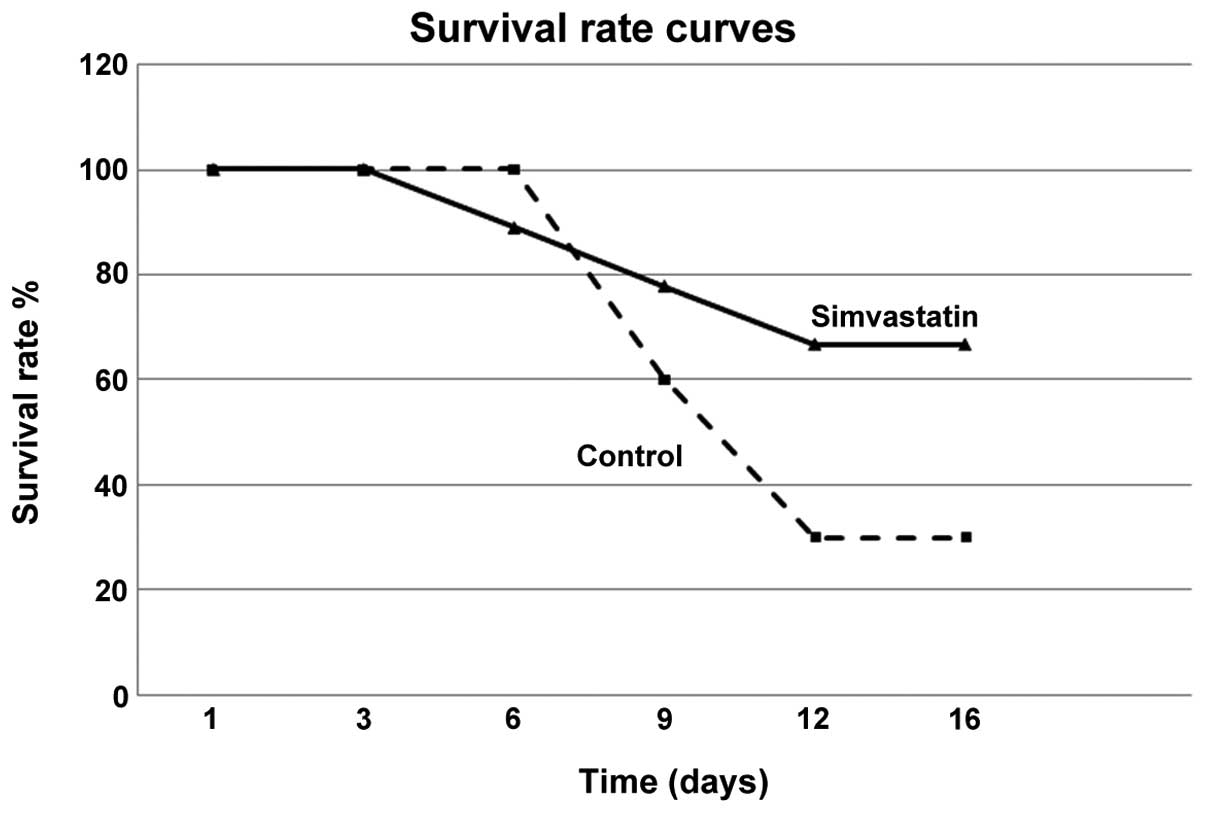
Survival rate curves
To avoid animal suffering all the mice were observed
in an interval of 18 days starting from tumor onset. The
Kaplan-Meier curve, demonstrated that in the control group, after 9
days from tumor onset, the survival rate was 60%, and after 12 only
30%. On the contrary, in simvastatin treated animals, the survival
rate remained ∼70% from day 9 to 16 (Fig. 6). These data confirmed that
simvastatin treatment inhibited melanoma growth and progression in
a well known murine model.
Discussion
Many studies have shown that the role of statins
extends beyond its lipid-lowering effects, as they are known to
improve endothelial functions and antioxidant activity and also
acts as anti-inflammatory and anticancer agents (46).
Their pleiotropic or cholesterol-independent effects
at the cellular and molecular levels are highly related to numerous
cellular functions, such as proliferation and differentiation.
Treatment with simvastatin induced morphological change and
decreased cell proliferation. It has been demonstrated that the use
of simvastatin was more effective in cancer cells and embryonic
stem cells (ESCs), in relation to normal cells (47).
Besides their use in the treatment of lipid
disorders, statins have been studied for their anti-carcinogenic
effects in several models, including carcinomas of the colon,
rectum, prostate, breast, lung and skin (6,7).
Many studies have shown the anti-proliferative and
proapoptotic effects of statins to a greater degree in malignant
than in non-malignant cells (8,9).
Statins also can promote different tumor cells to undergo apoptosis
in vitro and to suppress tumor growth (10,11).
The role of cholesterol in cancer progression remains to be cleared
but various tumor cell lines and tissues exhibit higher levels of
cholesterol than their normal counterparts (12,13).
The splicing mechanism is an important point in the proliferation
processes and all the factors involved in this regulation may be
responsible for growth inhibition.
It may be speculated that either p54 or NonO are
needed for the proper expression of proteins leading to a
stabilization of the transcription machinery, protein production
and function, promoting the survival of cells. The concerted
expression of genes involved in different mechanisms of cell
protection, like the human p54nrb and the murine NonO
may contribute to survival, promoting response counteracting the
apoptosis in cancer cells. Our data suggest that either
p54nrb or NonO seem to be new molecules that function as
regulator of progression of malignant melanoma cells (44).
In the present study we investigated the
anti-proliferative effects of simvastatin on several types of
melanoma cells and the inhibition of melanoma cell progression
in vivo. In order to evaluate the growth inhibition induced
by simvastatin treatment, we injected this drug at alternate days
intraperitoneally. According to our results, the cancer cell growth
inhibition is due to the deregulation of apoptosis induction and to
the inhibition in the expression of the gene NonO. Our data suggest
that simvastatin treatment might play an important role in
inhibition of tumor progression, although more detailed studies are
necessary to validate this hypothesis. Further experimentation is
required to demonstrate if NonO is a common target of statin
treatment and if the downregulation is common to other cancer cells
apart from B16 melanoma cells and whether this effect is dose- and
time-dependent.
In in vivo experiments, we demonstrated that
simvastatin reduces tumor growth. In particular, we observed a
delay in the tumor development in almost 50% of the animals treated
with simvastatin respect to the control group. In these animals,
for the first 10 days the tumor appeared very small, and in one of
the simvastatin treated mouse, the tumor appeared after >20 days
from cell inoculation whereas in the control mice and the majority
of tumor-injected mice, tumors were evident after 10–12 days from
the initial B16 cell injection.
The other very clear effect of simvastatin is the
strong reduction in the tumor dimension as shown in Fig. 5.
Also the survival rate, as shown in Fig. 6, is significantly higher in mice
that received simvastatin. In these animals after 12–16 days we
recorded increased survival, that reached a difference of ∼150%
respect to the controls.
The tumor growth reduction in mice is supported by
the results of in vitro cell migration, as both by wound
healing test, and by migration assay, simvastatin was shown to
reduce cell migration.
The simvastatin ability to influence cell migration
probably occurs by affecting integrin or downregulating the
metalloproteinase production, two molecular effects that have been
implicated in many steps of tumor dissemination. Also considering
that metalloproteinase can be localized in a proteolytically active
form on the surface of invasive melanoma cells (48). This may support the hypothesis of a
possible function of the simvastatin in the metastasis
inhibition.
Simvastatin induces a strong downregulation in the
expression of NonO, the murine homolog of p54nrb. On
these bases the inhibition of NonO, in cells treated with
simvastatin, could explain the decrease of melanoma progression in
mice administered with this drug, suggesting a possible action
mechanism involving the NonO promoter, that may contain sequences
with an high rate of mutation in cancer cells respect to the normal
cells and that could function as a target for simvastatin. Further
studies are necessary to verify whether NonO has an active role in
cancer cell proliferation.
The results obtained may sustain the many reports on
the anticancer inhibition property of statins and encourage the
experimentation on this drug for the possible use in therapy
probably in combination with the conventional chemotherapy.
Acknowledgements
The authors are grateful to Dr D. Di
Napoli for the important collaboration in the mouse physiological
and pathological condition control during the experiments, Mr. A.
Cucciardi for the competent assistance in animals experiments, Mr.
F. Moscatiello for the skilful technical assistance and Mr. S.
Arbucci for the collaboration in image preparation.
References
|
1.
|
Spampanato C, De Maria S, Sarnataro M,
Giordano E, Zanfardino M, Baiano S, Cartenì M and Morelli F:
Simvastatin inhibit cancer cell growth inducing apoptosis
correlated to activation of Bax and downregulation of BCL2 gene
expression. Int J Oncol. 40:935–941. 2012.PubMed/NCBI
|
|
2.
|
Gauthaman K, Fong CY and Bongso A:
Statins, stem cells, and cancer. J Cell Biochem. 106:975–983. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Goldstein JL and Brown MS: Regulation of
the mevalonate pathway. Nature. 343:425–430. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Liao JK and Laufs U: Pleiotropic effects
of statins. Annu Rev Pharmacol Toxicol. 45:89–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Lee MH, Yee Cho S and Han YM: Simvastatin
suppresses self-renewal of mouse embryonic stem cells by inhibiting
RhoA geranylgeranylation. Stem Cell. 25:1654–1663. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Chan KK, Oza AM and Siu LL: The statins as
anticancer agents. Clin Cancer Res. 9:10–19. 2003.
|
|
7.
|
Demierre MF, Higgins PD, Gruber SB, Hawk E
and Lippman SM: Statins and cancer prevention. Nat Rev Cancer.
5:930–942. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Mantha AJ, Hanson JE, Goss G, Lagarde AE,
Lorimer IA and Dimitroulakos J: Targeting the mevalonate pathway
inhibits the function of the epidermal growth factor receptor. Clin
Cancer Res. 11:2398–2407. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Wong WW, Dimitroulakos J, Minden MD and
Penn LZ: HMBCoA reductase inhibitors and the malignant cell: the
statin family of drugs as triggers of tumor-specific apoptosis.
Leukemia. 16:508–519. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Wu J, Wong WW, Khosravi F, Minden MD and
Penn LZ: Blocking the Raf-MEK-ERK pathway sensitizes acute
myelogenous leukemia cells to lovastatin-induced apoptosis. Cancer
Res. 64:6461–6468. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Cafforio P, Dammacco F, Gernone A and
Silvestris F: Statins activate the mitochondrial pathway of
apoptosis in human lymphoblasts and myeloma cells. Carcinogenesis.
26:883–891. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Mady EA: Association between estradiol,
estrogen receptors, total lipids, triglycerides, and cholesterol in
patients with benign and malignant breast tumors. J Steroid Biochem
Mol Biol. 75:323–328. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Nygren C, von Holst H, Mansson JE and
Fredman P: Increased levels of cholesterol esters in glioma tissue
and surrounding areas of human brain. Br J Neurosurg. 11:216–220.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Peterson C, Vitols S, Rudling M, Blomgren
H, Edsmyr F and Skoog L: Hypocholesterolemia in cancer patients may
be caused by elevated LDL receptor activities in malignant cells.
Med Oncol Tumor Pharmacother. 2:143–147. 1985.PubMed/NCBI
|
|
15.
|
Kokoglu E, Gorseval A, Sonmez H and Ozyurt
E: Tissue lipid composition of human gliomas and meningiomas.
Cancer Lett. 65:169–171. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Poynter JN, Gruber SB, Higgins PD, Almog
R, Bonner JD, et al: Statins and the risk of colorectal cancer. N
Engl J Med. 352:2184–2192. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Shannon J, Tewoderos S, Garzotto M, Beer
TM, et al: Statins and prostate cancer risk: a case-control study.
Am J Epidemiol. 162:318–325. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Fowke JH, Motley SS, Barocas DA, Cookson
MS, Concepcion R, Byerly S, et al: The associations between statin
use and prostate cancer screening, prostate size, high-grade
prostatic intraepithelial neoplasia (PIN), and prostate cancer.
Cancer Causes Control. 22:417–426. 2011. View Article : Google Scholar
|
|
19.
|
Kim K: Statin and cancer risks: from
tasseomancy of epidemiologic studies to meta-analyses. J Clin
Oncol. 24:4796–4797. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Dale KM, Coleman CI, Henyan NN, Kluger J
and White CM: Statins and cancer risk: a meta-analysis. JAMA.
295:74–80. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Jacobs EJ, Rodriguez C, Brady KA, Connell
CJ, Thun MJ, et al: Cholesterol-lowering drugs and colorectal
cancer incidence in a large United States cohort. J Natl Cancer
Inst. 98:69–72. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Chang CC, Ho SC, Chiu HF and Yang CY:
Statins increase the risk of prostate cancer: A population-based
case-control study. Prostate. 71:1818–1824. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Menter DG, Ramsauer VP, Harirforoosh S,
Chakraborty K, Yang P, et al: Differential effects of pravastatin
and simvastatin on the growth of tumor cells from different organ
sites. PLoS One. 6:e288132011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Lee SK, Kim YC, Song SB, Young BS and Kim
S: Stabilization and translocation of p53 to mitochondria is linked
to Bax translocation to mitochondria in simvastatin-induced
apoptosis. Biochem Biophys Res Commun. 391:1592–1597. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Blanco-Colio LM, Villa A, Ortego M,
Hernandez-Presa MA, Pascual A, et al: 3-Hydroxy-3-methyl-glutaryl
coenzyme A reductase inhibitors, atorvastatin and simvastatin,
induce apoptosis of vascular smooth muscle cells by down-regulation
of Bcl-2 expression and Rho A prenylation. Atherosclerosis.
161:17–26. 2002. View Article : Google Scholar
|
|
26.
|
Herrero-Martin G and Lopez-Rivas A:
Statins activate a mitochondrial-operated pathway of apoptosis in
breast tumor cells by a mechanism regulated by Erb B2 and dependent
on the prenylation of proteins. FEBS Lett. 582:2589–2594. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Vousden KH and Lu X: Live or let die: the
cell’s response to p53. Nat Rev Cancer. 2:594–604. 2002.
|
|
28.
|
Chipuk JE and Green DR: Dissecting
p53-dependent apoptosis. Cell Death Differ. 13:994–1002. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Stier S, Totzk G, Grünewald E, et al:
Identification of p54nrband the 14-3-3 protein HS1 as
TNF-α-inducible genes related to cell cycle control and apoptosis
in human arterial endothelial cells. J Biochem Mol Biol.
38:447–456. 2005.
|
|
30.
|
Dong B, Horowitz DS, Kobayashi R and
Krainer AR: Purification and cDNA cloning of HeLa cell
p54nrb, a nuclear protein with two RNA recognition
motifs and extensive homology to human splicing factor PSF and
Drosophila NONA/BJ6. Nucleic Acids Res. 21:4085–4092.
2003.PubMed/NCBI
|
|
31.
|
Shav-Tal Y and Zipori D: PSF and
p54(nrb)/NonO - multi-functional nuclear proteins. FEBS Lett.
531:109–114. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Zhang Z and Carmichael GG: The fate of
dsRNA in the nucleus: a p54(nrb)-containing complex mediates the
nuclear retention of promiscuously A-to-I edited RNAs. Cell.
106:465–475. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Basu A, Dong B, Krainer AR and Howe CC:
The intracisternal A-particle proximal enhancer-binding protein
activates transcription and is identical to the RNA- and
DNA-binding protein p54nrb/NonO. Mol Cell Biol.
17:677–686. 1997.PubMed/NCBI
|
|
34.
|
Kameoka S, Duque P and Konarska M:
P54nrbassociates with the 50 splice site within large
transcription/splicing complexes. EMBO J. 23:1782–1791. 2004.
|
|
35.
|
Urban RJ, Bodenburg Y, Kurosky A, Wood TG
and Gasic S: Polypyrimidine tract-binding protein-associated
splicing factor is a negative regulator of transcriptional activity
of the porcine p450scc insulin-like growth factor response element.
Mol Endocrinol. 14:774–782. 2000. View Article : Google Scholar
|
|
36.
|
Mathur M, Tucker PW and Samuels HH: PSF is
a novel corepressor that mediates its effect through Sin3A and the
DNA binding domain of nuclear hormone receptors. Mol Cell Biol.
21:2298–2311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Patton JG, Porro EB, Galceran J, Tempst P
and Nadal-Ginard B: Cloning and characterization of PSF, a novel
pre-mRNA splicing factor. Genes Dev. 7:393–406. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Peng R, Dye BT, Perez I, Barnard DC,
Thompson AB and Patton JG: PSF and p54nrbbind a
conserved stem in U5 snRNA. RNA. 8:1334–1347. 2002.
|
|
39.
|
Emili A, Shales M, McCracken S, Xie W,
Tucker PW, Kobayashi R, Blencowe BJ and Ingles CJ: Splicing and
transcription-associated proteins PSF and p54nrb/nonO
bind to the RNA polymerase II CTD. RNA. 8:1102–1111. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Xing Y, Johnson CV, Dobner PR and Lawrence
JB: Higher level organization of individual gene transcription and
RNA splicing. Science. 259:1326–1330. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Yang YS, Hanke JH, Carayannopoulos L, et
al: NonO, a non-POU-domain-containing, octamer-binding protein, is
the mammalian homolog of Drosophila nonAdiss. Mol Cell Biol.
13:5593–5603. 1993.PubMed/NCBI
|
|
42.
|
Bosserhoff AK: Melanoma inhibitory
activity (MIA) an important molecule in melanoma development and
progression. Pigment Cell Res. 18:411–416. 2005.PubMed/NCBI
|
|
43.
|
Schmid R, Schiffner S, Opolka A, Grässel
S, Schubert T, Moser M, Bosserhoff AK, et al: Enhanced cartilage
regeneration in MIA/CD-RAP deficient mice. Cell Death Dis.
11:e972010. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Schiffnert S, Zimarat N and Bosserhoff AK:
p54nrbis a new regulator of progression of malignant
melanoma. Carcinogenesis. 32:1176–1182. 2011.
|
|
45.
|
Carriero MV, Del Vecchio S, Capezzoli M,
Franco P, et al: Urokinase receptor interacts with
αvβ5vitronectin receptor, promoting
urokinase-dependent cell migration in breast cancer. Cancer Res.
59:5307–531. 1999.
|
|
46.
|
Campbell MJ, Esserman LJ, Zhou Y,
Shoemaker M, et al: Breast cancer growth prevention by statins.
Cancer Res. 66:8707–8714. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Sassano A, Katsoulidis E, Antico G, Altman
JK, Redig AJ, et al: Suppressive effects of statins on acute
promyelocytic leukemia cells. Cancer Res. 67:4524–4532. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Buommino E, Baroni A, Canozo N,
Petrazzuolo M, Nicoletti R, Vozza A and Tufano MA: Artemisinin
reduces human melanoma cell migration by down-regulating
αvβ3integrin and reducing metalloproteinase 2
production. Invest New Drugs. 274:12–418. 2009.PubMed/NCBI
|