Introduction
Colorectal cancer (CRC) is a cancer from
uncontrolled cell growth in the lining of the large intestine, such
as colon and rectum. CRC is the fourth most common cancer in the
world, but it is more common in developed countries. CRC is the
third most common cancer and the fourth leading cause of
cancer-related death in Korea and the incidence of CRC have been
increased rapidly over the past few decades (1,2).
While surgical resection cures >50% of CRC patients, 40–50% of
these subjects eventually experience recurrences, leaving only a
minority amenable to re-operation (3). Because of unsatisfactory treatment
options for CRC, there is an urgent need to develop novel
preventive treatment approaches for this malignancy.
Progressive inhibition or evasion of apoptosis has
been found during the transformation of colorectal epithelium to
carcinoma (4), indicating that
dysfunction of apoptosis has important roles in colorectal
carcinogenesis. The cytotoxic action of most chemotherapeutic drugs
is often mediated by the activation of apoptotic pathways (5). Recent progress in understanding the
molecular mechanisms and role of apoptosis in CRC development has
provided novel targets for therapy.
Nuclear factor-κB (NF-κB), a transcription factor,
controls the expression of genes involved in tumor cell growth,
proliferation, angiogenesis, invasion and survival. NF-κB is
composed of two subunits, p65 and p50, and is normally sequestered
in the cytosol by an inhibitory protein, IκBα. Exposure of cells to
a variety of extracellular stimuli leads to the rapid
phosphorylation, ubiquitination, and ultimately proteolytic
degradation of IκBα, resulting in the release of NF-κB from its
inhibitory protein to translocate to the nucleus where it regulates
transcription of various genes (6). Increased NF-κB activity has been
demonstrated in colon cancer, which is believed to enhance cancer
cell survival by inhibiting apoptosis. In addition, the
inflammatory colon diseases such as Crohn’s disease and ulcerative
colitis are associated with the constitutive activation of NF-κB
(7). Inhibition of NF-κB in cancer
cells converts inflammation-induced tumor growth to
inflammation-induced tumor regression mediated by TNF-α and TRAIL
(8). Therefore, inhibition of
NF-κB signaling pathway provides attractive targets for new
chemopreventive and chemotherapeutic approaches.
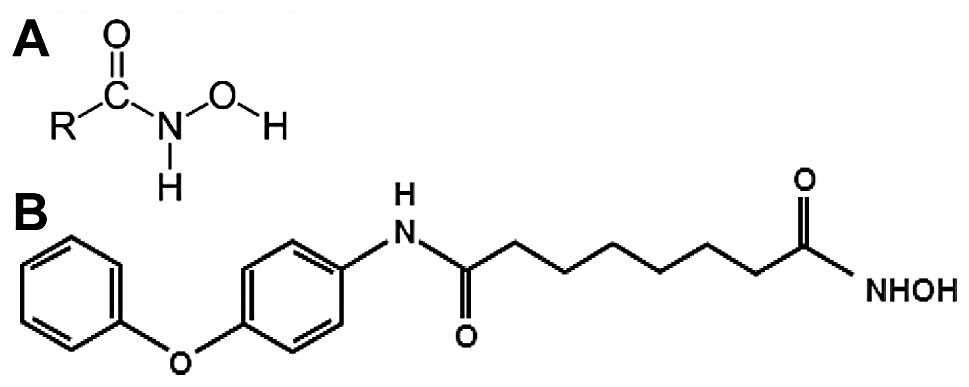
Hydroxamic acids are known as iron chelators and
microbial siderophores that show diverse biological activities such
as antibacterial, antifungal, antitumor and anti-inflammatory
properties (9). Some hydroxamates,
such as suberoylanilide hydroxamic acid, has been used clinically
for the treatment of cancer (10).
For the purpose of creating more effective antitumor drugs many
derivatives that possess the hydroxamic acid functional group were
synthesized (11). The newly
designed hydroxamic acid derivatives have been shown to have
anticancer effect in various cancer cells (12). MHY218
[N1-hydroxy-N8-(4-phenoxyphenol)octanedianide]
was synthesized and it was a novel hydroxamic acid derivative.
MHY218 has inhibitory activity of histone deacetylase and
anticancer effects against human ovarian cancer cells (13) and tamoxifen-resistant MCF-7 breast
cancer cells (14). The chemical
structures of hydroxamic acids and MHY218 are shown in Fig. 1. However, the effects of MHY218
were not studied in HCT116 human colon cancer cells in previous
reports. Therefore, the main purpose of this study was to focus on
investigating the effects of MHY218 on cell cycle and apoptosis in
HCT116 human colon cancer cells.
Materials and methods
Chemicals
The simplified code name and structure of MHY218
[N1-hydroxy-N8-(4-phenoxyphenol)octanedianide]
used in this study is shown in Fig.
1B. Detailed method for the design and synthesis of this
compound is described elsewhere (13). This was dissolved in sterile
dimethyl sulfoxide (DMSO) to generate 10 mM stock solution. The
solutions were stored at −80°C. Subsequent dilutions were made in
RPMI-1640 (Hyclone, Logan, UT, USA). The maximal concentration of
DMSO did not exceed 0.1% (v/v) in the treatment range, where there
was no influence on the cell growth. All other chemicals with the
highest purity available were from Sigma-Aldrich Co. (St. Louis,
MO, USA).
Cell culture
HCT116 human colon cancer cells (p53 wild-type) were
obtained from American Type Culture Collection (Mansssas, VA, USA)
and were cultured in RPMI-1640 (Hyclone) supplemented with 10%
fetal bovine serum (FBS, Hyclone), 2 mM glutamine (Sigma-Aldrich),
100 U/ml penicillin (Hyclone), and 100 μg/ml streptomycin
(Hyclone) at 37°C in a humidified 5% CO2.
Cell viability assay
Cell viability was determined by MTT assay. For the
MTT assay, HCT116 cells were seeded in a 24-well culture plate at a
density of 4×104 cells/well, cultured for 24 h in the
growth media, and then treated with or without various reagents for
the indicated concentrations. The cells were incubated with 0.5
mg/ml 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
(MTT, Sigma-Aldrich) at 37°C for 2 h. The formazan granules
generated by the live cells were dissolved in DMSO, and the
absorbance at 540 nm was monitored by using a multi-well
reader.
Nuclear staining with Hoechst 33342
Cells were washed with phosphate-buffered saline
(PBS) and fixed with 3.7% paraformaldehyde (Sigma-Aldrich) in PBS
for 10 min at room temperature. Fixed cells were washed with PBS
and stained with 4 μg/ml Hoechst 33342 for 20 min at room
temperature. The cell were washed two more times with PBS and
analyzed via a fluorescent microscope.
DNA fragmentation assay
Cells were lysed in a buffer, containing 5 mM
Tris-HCl (pH 7.5), 5 mM EDTA, and 0.5% Triton X-100, for 30 min on
ice. Lysates were vortexed and cleared by centrifugation at 14,000
rpm for 20 min. Fragmented DNA in the supernatant was treated with
RNase, followed by proteinase K digestion,
phenol:chloroform:isoamyl alcohol mixture (25:24:1) extraction and
isopropanol precipitation. DNA was separated through a 1.5% agarose
gel, was stained with 0.1 μg/ml ethidium bromide, and was
visualized by UV source.
Cell cycle analysis
The DNA content was measured following the staining
of the cells with propidium iodide. The cells were treated under
the appropriate conditions for 24 h, subsequently trypsinized,
washed once in cold PBS, and then fixed in 70% ethanol at 4°C
overnight. The fixed cells were pelleted and stained in cold
propidium iodide (PI, Sigma-Aldrich) solution (50 μg/ml in
PBS) at room temperature for 30 min in the dark. Flow cytometry
analysis was performed on a FACScan flow cytometry system
(Becton-Dickinson, San Jose, CA, USA).
Caspase activity assay
The cells were harvested and washed with cold PBS.
Total cells were lysed with the lysis buffer [40 mM Tris (pH 8.0),
120 mM, NaCl, 0.5% NP-40, 0.1 mM sodium orthovanadate, 2
μg/ml aprotinin, 2 μg/ml leupeptin and 100
μg/ml phenymethylsulfonyl fluoride (PMSF)] at 4°C for 30
min. Cell lysate protein (100 μg) was mixed in assay buffer
in a final volume of 100 μl, followed by addition of 10
μl of 2 mM of the substrate caspase-8 (Z-IETD-pNA),
caspase-9 (Ac-LEHD-pNA), or caspase-3 (Z-DEVD-pNA) for the
respective caspase assay. The reaction mixture was incubated at
37°C for 30 min and liberated p-nitroaniline (pNA) was measured at
405 nm using a multi-well reader.
Preparation of cytosolic and nuclear
protein extracts
The cells were washed with cold PBS and resuspended
in Buffer A [10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM
KCl, 0.5 mM DTT, 0.5 mM PMSF and Protease inhibitor cocktail
(Sigma-Aldrich)] and incubated on ice. After 15 min, 0.5% Nonidet P
(NP)-40 was added to lyse the cells, which were vortexed for 10
sec. Cytosolic extracts were obtained after centrifuging at 12,000
rpm for 60 sec at 4°C. Nuclear extracts were resuspended in Buffer
C [20 mM HEPES (pH 7.9), 1.5 mM MgCl2, 300 mM NaCl, 0.2
mM EDTA, 20% v/v glycerol, 0.5 mM DTT, 0.5 mM PMSF and protease
inhibitor cocktail] and incubated on ice for 20 min with gentle
vortexing every 5 min. Nuclear cell extracts were recovered after
centrifugation for 10 min at 12,000 rpm at 4°C. Protein
concentration was determined by Bradford protein assay reagent
(Bio-Rad, Hercules, CA, USA).
Western blot analysis
The cells were treated with the appropriate
conditions, harvested, and washed with cold PBS. Total cells
lysates were lysed in lysis buffer [40 mM Tris (pH 8.0), 120 mM,
NaCl, 0.5% NP-40, 0.1 mM sodium orthovanadate, 2 μg/ml
aprotinin, 2 μg/ml leupeptin and 100 μg/ml PMSF]. The
supernatant was collected and protein concentrations were then
measured with protein assay reagents (Pierce, Rockford, IL, USA).
Protein extracts were denatured by boiling at 100°C for 5 min in
sample buffer (0.5 M Tris-HCl, pH 6.8, 4% SDS, 20% glycerol, 0.1%
bromophenol blue, 10% β-mercaptoethanol). Equal amount of the total
proteins were subjected to 6-15% SDS-PAGE and transferred to PVDF.
The membranes were blocked with 5% non-fat dry milk in
Tris-buffered saline with Tween-20 buffer (TBS-T) (20 mM Tris, 100
mM NaCl, pH 7.5 and 0.1% Tween-20) for 1 h at room temperature.
Then, the membranes were incubated overnight at 4°C with primary
antibodies. The membranes were washed once for 10 min, 4 times with
TBS-T buffer and incubated for 1 h with horseradish
peroxidase-conjugated anti-rabbit or anti-mouse immunoglobin (Santa
Cruz Biotechnology Inc., Santa Cruz, CA, USA). The membranes were
washed again for 10 min, 4 times with TBS-T buffer.
Antigen-antibody complexes were detected by the enhanced
chemiluminescence (ECL) detection system (Amersham Biosciences
Corp., Little Chalfont, Bucks, UK).
Reverse transcriptase-polymerase chain
reaction (RT-PCR) analysis
For RT-PCR analysis, total RNA was extracted from
cultured cells using a TRIzol reagent as described by the
manufacturer (Invitrogen), PCR amplification cDNA was prepared
using a Bioneer RT/PCR PreMix containing 1 U Taq DNA polymerase,
250 μM dNTPs, 10 mM Tris-HCl, 40 mM KCl, 1.5 mM
MgCl2 (Bioneer, Korea). The assay was carried out in a
20 μl reaction mixture containing 1.0 μg of total
RNA, 30 pmol of each primer, 1 μg Oligo dT (Bioneer), using
a PCR Thermal Cycler Dice Takara TP600 (Takara, Otsu, Japan). The
mRNAs were amplified with the primers indicated in Table I. GAPDH served as an internal
control. The cycling conditions were as follows: cDNA synthesis at
42°C for 60 min, RTase inactivation at 94°C for 5 min, 1 ×
denaturation (94°C for 30 sec), 30 × annealing (58°C for 30 sec),
and 1 × extension (72°C for 1 min) for 30 cycles. PCR products were
analyzed by electrophoresis on 1% agarose gel (Bio Basic Inc.,
Markham, Ontario, Canada) in the presence of ethidium bromide, and
were visualized with a UV transilluminator (MultiImage™ Light
Cabinet, Alpha Innotech Co., San Leandro, CA, USA).
 | Table I.Primer sequences for RT-PCR. |
Table I.
Primer sequences for RT-PCR.
| Gene | | Sequence of primers
(5′→3′) |
|---|
| Cox-2 | Sense | AGA TCA TCT CTG CCT
GAG TAT CTT |
| Antisense | TTC AAA TGA GAT TGT
GGG AAA ATT GCT |
| GAPDH | Sense | CGG AGT CAA CGG ATT
TGG TCG TAT |
| Antisense | AGC CTT CTC CAT GGT
GGT GAA GAC |
Luciferase reporter assay for NF-κB
activity
The activity of NF-κB was examined using a
luciferase plasmid DNA, pTAL-NF-κB that contains a specific binding
sequence for NF-κB (BD Biosciences Clontech, CA, USA). Transfection
was carried out using TransIT-LT1 transfection reagent (Promega,
Madison, WI, USA). Briefly, HCT116 cells were seeded in 6-well
plates. When cultured cells reached ∼50% confluence, cells were
treated with 2 μg DNA/6 μl transfection complexes in
a total volume of media with 2 ml for 24 h. Subsequently, various
concentrations of MHY218 were treated and incubated for 1 h, and
then 10 ng/ml TNF-α was treated and incubated for 6 h. Cells were
washed with PBS and added by Stead-Glo Luciferase Assay System
(Promega) to the plate. Luciferase activity was measured by a
luminometer (GENious, Tecan, Salzburg, Austria). The obtained raw
luciferase activities were normalized by protein concentration in
each well.
Zymography
Zymography was used to semiquantitatively determine
the gelatinolytic activity of matrix metalloproteinase-9 (MMP-9)
secreted into culture media. Equal amount of conditioned culture
media from equal number cells were applied to SDS-PAGE containing
0.25% gelatin. The gel was incubated with the renaturing buffer
(2.5% Triton X-100) with gentle agitation for 30 min at room
temperature. The gel was washed once for 10 min, 2 times with
distilled water and incubated overnight at 37°C with the Developing
buffer [1 M Tris-HCl (pH 7.5), 1 M CaCl2, 10%
NaN3, 1 M NaCl]. The gel was stained with 0.5% (w/v)
Coomassie Blue R-250 for 30 min at room temperature and then
destained with the destaining solution (methanol:acetic
acid:distilled water = 50:10:40). Areas of protease activity
appeared as clear bands against a dark blue background where the
protease had digested the substrate. The product of zymography was
visualized with a White light transilluminator
(BioSpectrum® Imaging System, Upland, CA, USA).
Statistical analysis
Results were expressed as the mean ± SD of three
separate experiments and analyzed by Student’s t-test. Means were
considered significantly different at p<0.05 or p<0.01.
Results
MHY218 inhibits the growth of HCT116
cells
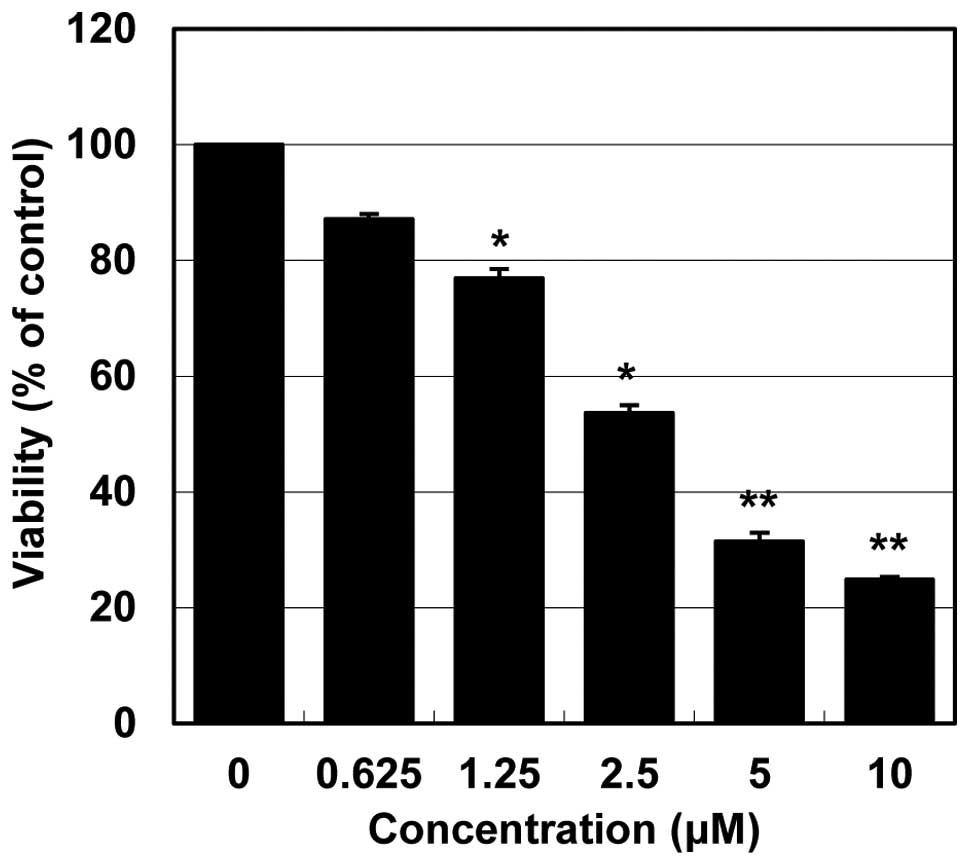
To investigate the effects of MHY218 on the
viability of HCT116 cells, the MTT assay was performed. As shown in
Fig. 2. MHY218 showed
concentration-dependent cytotoxicity on HCT116 cells. The
IC50 value of MHY218 on HCT116 cells was ∼3.0
μM.
MHY218 modulates the cell cycle in HCT116
cells
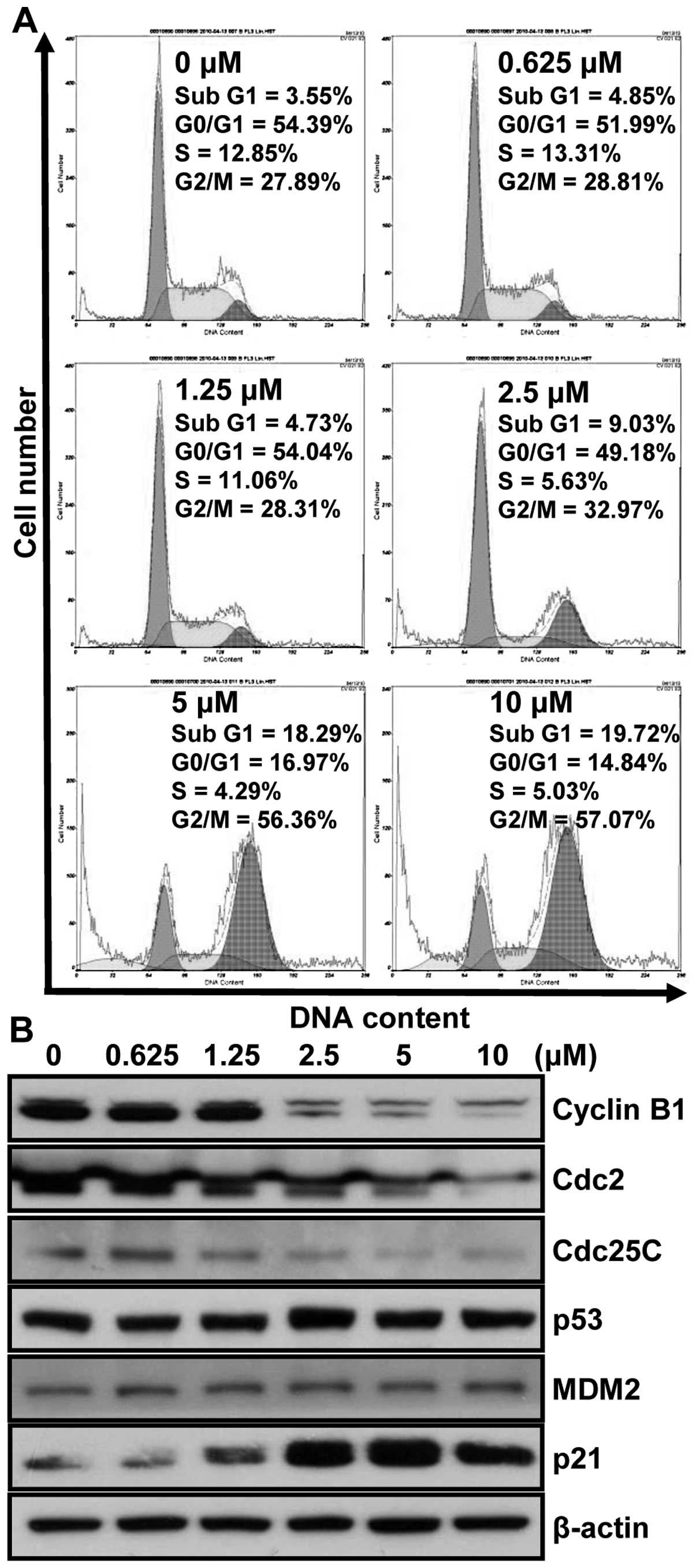
To investigate whether the inhibition of HCT116 cell
growth was mediated, at least in part, by regulating the cell
cycle, flow cytometry analysis of PI-stained HCT116 nuclei was
performed. This flow cytometry analyses data showed that MHY218
treatment induced the accumulation of cells in G2/M phase of the
cell cycle, with the occurrence of sub-G1 peak, indicating DNA
degradation through either necrosis or apoptosis (Fig. 3A). G2/M phase arrest by MHY218
treatment reached the maximum percentage at 24 h. After 24 h
incubation with different concentrations of MHY218, the population
of the cells at the G2/M phase increased from 27.89% (vehicle
alone) to 57.07% (10 μM MHY218) (Fig. 3A). As shown in Fig. 3A, the increase of cell population
in G2/M phase consequently occurred with the decrease in G0/G1
cells as compared to those of control. In addition, we also
observed the appearance of the peak corresponding to a population
of cells with sub-G1 DNA content. This peak represented
MHY218-induced DNA degradation either by necrosis or by apoptosis
in HCT116 cells. After 24 h incubation of 10 μM MHY218, as
shown in Fig. 3A, the fractions of
sub-G1 peak increased from 3.55% (vehicle alone) to 19.72% (10
μM MHY218). These result supported that the MHY218 treatment
for 24 h mainly induced the inhibition of cell growth via G2/M
phase arrest in the cell cycle.
MHY218 modulates cell cycle regulatory
proteins in HCT116 cells
To assess the effect of MHY218 on the intracellular
protein expression levels of G2/M phase in the cell cycle, we
performed western blot analysis. As shown in Fig. 3B, the expression levels of cyclin
B1, Cdc25C and Cdc2 were decreased by MHY218 treatment as compared
to the basal levels in a concentration-dependent manner. The
induction of p21WAF1/CIP1 causes subsequent
arrest in the G0/G1 or G2/M phase of the cell cycle by binding of
the cyclin-cyclin-dependent kinase (CDK) complex. Thus further
studies were performed to elucidate whether
p21WAF1/CIP1 was induced by the MHY218 treatment
either via a 53-dependent or a p53-independent pathway in HCT116
cells. The results show that p53 was not significantly changed and
p21WAF1/CIP1 was increased without the change in
the level of MDM2 by MHY218 treatment in HCT116 cells (Fig. 3B). Therefore, these results suggest
that MHY218 treatment induces G2/M phase arrest in cell cycle by
downregulating expressions of cyclin, CDKs, and induction of
p21WAF1/CIP1 via p53-independent pathway.
MHY218 induces morphological changes and
apoptosis in HCT116 cells
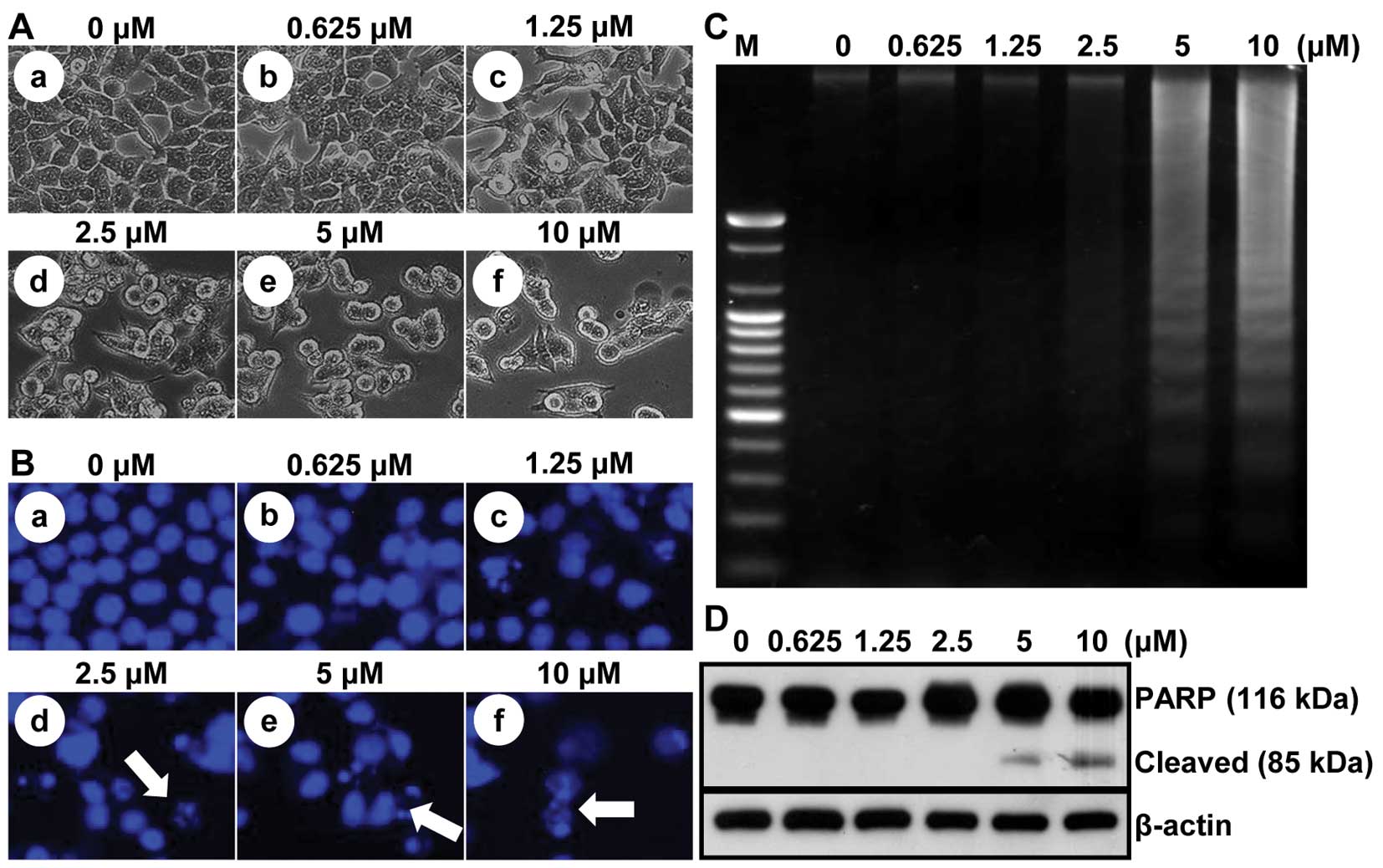
To assess whether there are any morphological
changes in MHY218-treated HCT116 cells, we examined the cells by
phase-contrast light microscopy after 24 h of incubation with or
without MHY218. Under the light microscope, untreated HCT116 cells
spread regularly in the culture plate and grew to near confluence
(Fig. 4Aa). In contrast,
MHY218-treated HCT116 cells were shrunken and changed to round
form. Additionally, cell numbers were decreased in a
concentration-dependent manner (Fig.
4Ab–f). To investigate whether the growth inhibitory effects of
MHY218 were due to the induction of apoptosis in HCT116 cells, the
morphological changes were assessed with Hoechst 33342 staining. As
shown in Fig. 4B, nuclei with
chromatin condensation and formation of apoptotic bodies, which are
characteristics of apoptosis, were seen in cells cultured with
MHY218 in a concentration-dependent manner (Fig. 4Bb–4f), whereas the control cells
maintained nuclear structure intact (Fig. 4Ba). We also analyzed whether DNA
fragmentation, another hallmark of apoptosis, was induced by MHY218
treatment on HCT116 cells. Following agarose gel electrophoresis of
HCT116 cells treated with MHY218 for 24 h, a typical ladder pattern
of internucleosomal fragmentation was observed in a
concentration-dependent manner (Fig.
4C). Polypeptide degradation, including poly(ADP-ribose)
polymerase (PARP), was examined to see the possible involvement of
apoptosis-associated protease activity during the growth inhibition
of the colon cancer cells. PARP cleavage was evident by the
appearance of the p85 PARP cleavage fragment (Fig. 4D) and clearly observed in the 5
μM and 10 μM of MHY218 treatment.
MHY218 modulates the expression levels of
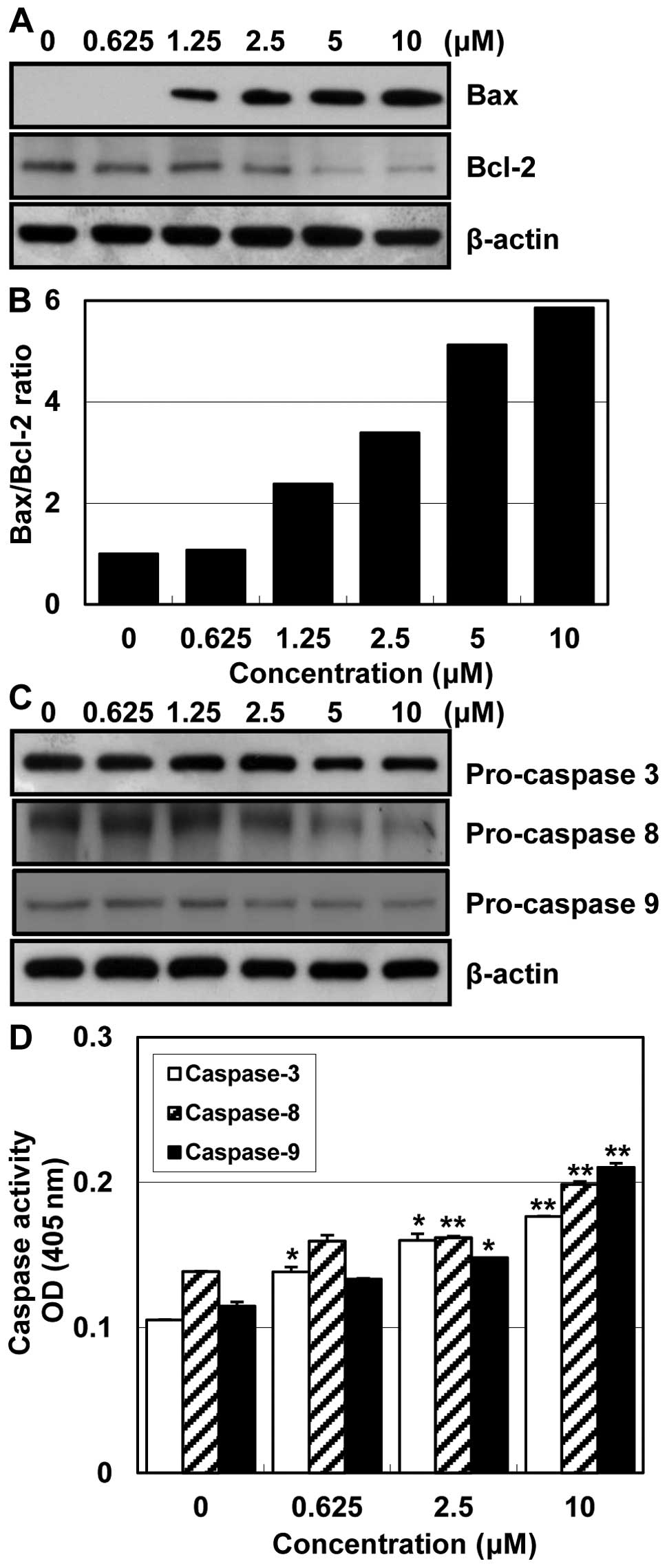
apopotosis-related proteins in HCT116 cells
To determine whether the expression levels of
apoptosis-related proteins were modulated by MHY218, western blot
analysis was performed. The expression level of Bax protein was
markedly upregulated, but Bcl-2 was downregulated in a
concentration-dependent manner (Fig.
5A). The Bax/Bcl-2 ratio was also significantly increased with
increase of MHY218 concentration (Fig.
5B). These data suggest that MHY218 induces apoptosis by the
alterations in expression levels of Bax/Bcl-2 protein. The
expression levels of pro-caspase-3, -8 and -9 were decreased,
indicating the activation of these caspases (Fig. 5C). In an attempt to further
characterize the mechanisms of apoptosis, the activity of
caspase-3, -8 and -9 was determined by colorimetric assay. The
activity of caspase-3, -8 and -9 was increased with the treatment
of MHY218 in a concentration-dependent manner (Fig. 5D). These results, taken all
together, imply that MHY218 seems to induce apoptosis through the
internal and external pathway in HCT116 cells.
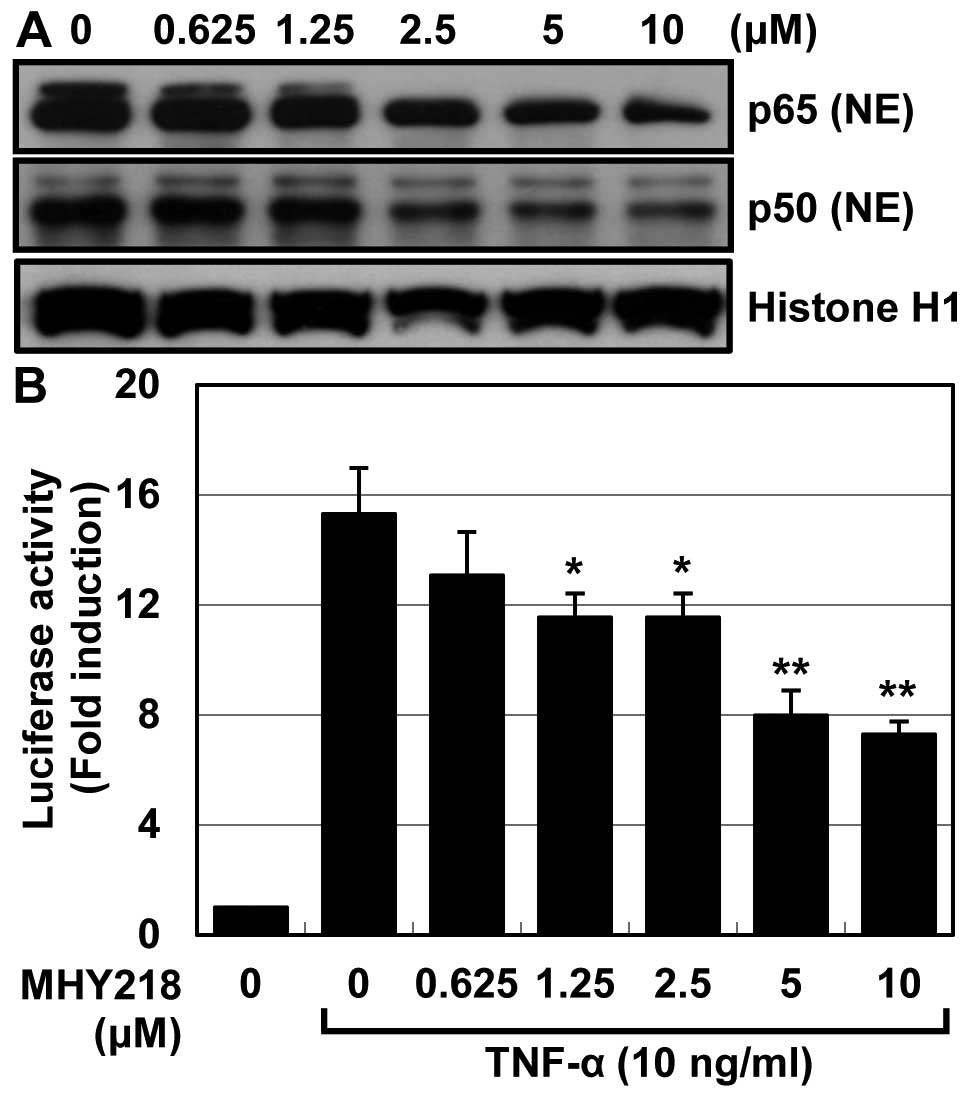
MHY218 modulates the expression levels of
NF-κB, COX-2, MMP-9 and 5-LOX protein in HCT116 cells
NF-κB regulates the expression of a wide variety of
genes involved in tumor cell growth and survival. We examined
whether MHY218 has the potential to inhibit NF-κB activation in
MHY218-treated HCT116 cells. After 24 h exposure with the indicated
concentration of MHY218, the levels of nuclear NF-κB p65 and NF-κB
p50 were examined, with western blot analysis, because the nuclear
translocation of the NF-κB subunits p65 and p50 is essential for
NF-κB activation. As shown in Fig.
6A, treatment with MHY218 decreased nuclear translocations of
p65 and p50, the functionally active subunits of NF-κB, to the
nucleus, in a concentration-dependent manner. Next to determine the
effect of MHY218 on the TNF-α-induced NF-κB transcriptional
activity, HCT116 cells were transiently transfected with the
NF-κB-regulated luciferase reporter construct, and the transfected
cells were then stimulated with TNF-α alone or with a combination
of TNF-α and MHY218. As shown in Fig.
6B, MHY218 significantly suppressed TNF-α-induced activation of
NF-κB as compared to untreated cells. These data indicated that
MHY218 treatment resulted in a significant inhibition of NF-κB
activation.
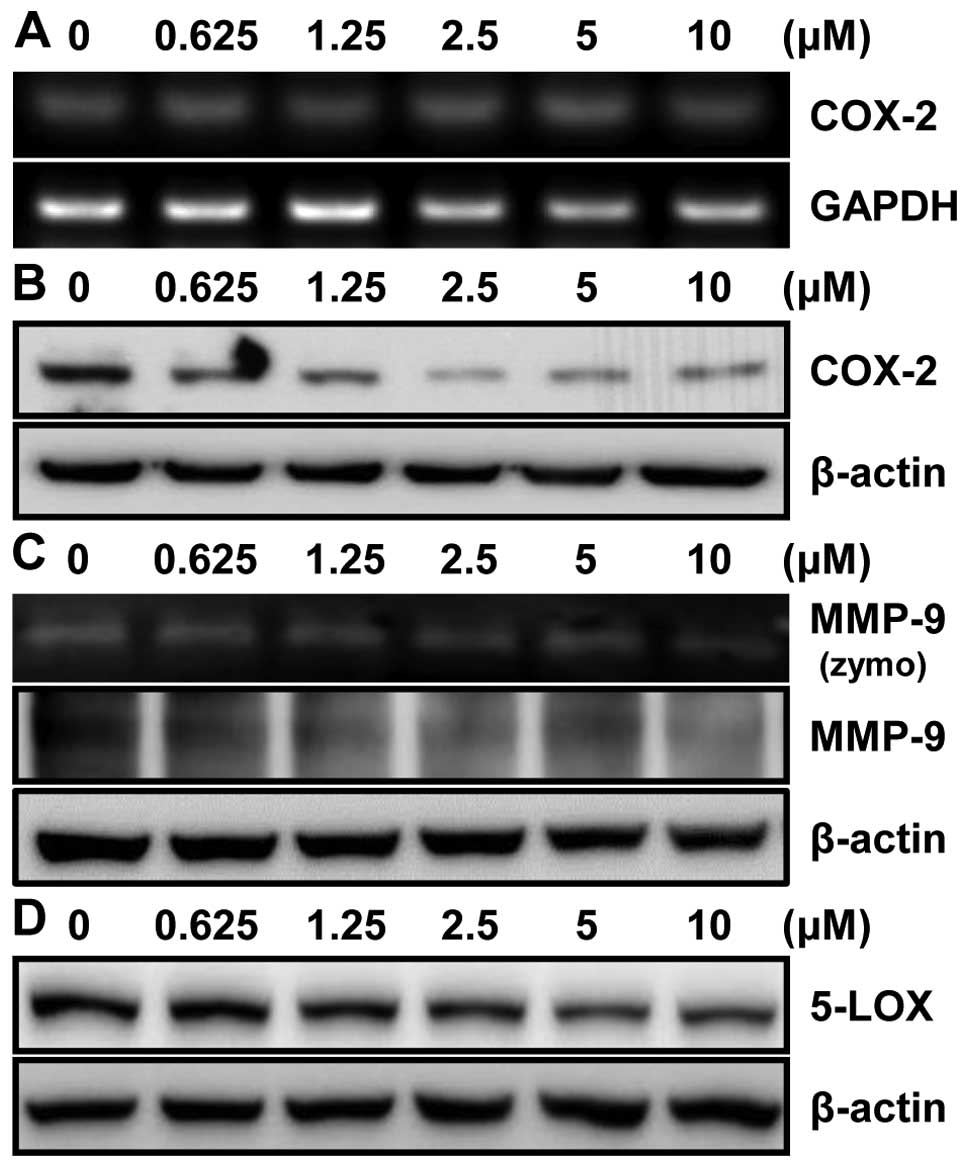
To further investigate the effect of MHY218 on
expression level of the COX-2 protein, an NF-κB downstream target
gene, in HCT116 cells, the RT-PCR and western blot analyses showed
no change in COX-2 mRNA (Fig. 7A)
but a significant decrease in COX-2 protein expression after MHY218
treatment in a concentration-dependent manner (Fig. 7B). These data suggested that the
inhibition of the COX-2 expression is consistent with the results
that MHY218 inhibited the NF-κB activation. We next examined the
effect of MHY218 on MMP-9 activity in HCT116 cells. Gelatin
zymography showed that MMP-9 activity was significantly inhibited
by MHY218 in a concentration-dependent manner (Fig. 7C). In order to determine the MMP-9
protein level in cells after treatment with MHY218, western blot
analysis was performed. Being consistent with the observed
alteration of activity, MMP-9 protein expression was significantly
reduced in HCT116 cells treated with MHY218 in a
concentration-dependent manner (Fig.
7C). Another downstream target gene of NF-κB is 5-lipoxygenase
(5-LOX). We next examined the effect of MHY218 on 5-LOX protein
level in HCT116 cells. 5-LOX protein level was also significantly
reduced in HCT116 cells treated with MHY218 in a
concentration-dependent manner (Fig.
7D). These data suggested that MHY218 inhibited TNF-α-induced
NF-κB activation through the suppression of nuclear translocation
of NF-κB, leading to reduced expression of NF-κB-regulated gene
products.
Discussion
This study was conducted to investigate the effects
of MHY218 on HCT116 human colon cancer cells. MHY218 induced cell
cycle arrest and apoptosis in HCT116 human colon cancer cells.
MHY218 also modulated the activity of NF-κB and downregulated the
expression levels of COX-2, MMP-9 and 5-LOX protein in HCT116
cells.
The treatment of HCT116 cells with MHY218 resulted
in growth inhibition concentration-dependently. Flow cytometric
analysis revealed that MHY218 induced G2/M phase arrest. Different
classes of cyclins and their CDK control cell cycle progression.
G2/M transition provides an effective checkpoint in the cell cycle
progression that is regulated by cyclin B1, Cdc2 and Cdc25C
(15). In this study, treatment of
HCT116 cells with MHY218 resulted in arrest of cells in G2/M phase
and is associated with a decrease in the protein levels of cyclin
B1, Cdc2 and Cdc25C.
p21WAF1/CIP1, a cyclin-dependent
kinase inhibitor, is commonly associated with the G1 checkpoint and
G2/M phase, its association with inhibiting the expression of the
Cdc2/cyclin B1 complex has also been demonstrated (16,17).
p21WAF1/CIP1 transcription can be regulated
through p53-dependent (18) and
p53-independent pathways (19). In
the present study, the protein level of p53 was not significantly
increased, whereas p21WAF1/CIP1 was increased
without a change on the expression of MDM2 by MHY218 in HCT116
cells. These results suggested that MHY218 activated
p21WAF1/CIP1 expression and that this induced
G2/M phase arrest of the p53-independent pathway. Therefore, the
upregulation of p21WAF1/CIP1, and the
downregulation of cyclin B1, Cdc2 and Cdc25C may be one of the
molecular mechanisms by which MHY218 inhibited HCT116 cells growth
and induced cell cycle arrest.
The treatment of MHY218 also induced apoptosis as
demonstrated by the formation of apoptotic bodies and DNA
fragmentation. Apoptosis (programmed cell death), is an important
process requited for homeostasis (20). Apoptosis occurs through two broad
pathways: the intrinsic pathway (the mitochondrial pathway) and
extrinsic pathway (the death receptor pathway) (21). Caspases are key players in the
extrinsic pathway. Another group of key players in the intrinsic
pathway of apoptosis is the Bcl-2 family, which consists of >20
members of pro-apoptotic proteins (including Bax, Bak, Bok, Bad and
Bid), and anti-apoptotic proteins (including Bcl-2,
Bcl-XL, Mcl-1 and Bfl-1/A1) (22).
The activation of effector caspase-3, in response to
MHY218 treatment also resulted in cleavage of PARP in HCT116 cells.
The ratio between Bcl-2 and Bax has been suggested as a primary
event in determining the susceptibility to apoptosis through
maintaining the integrity of the mitochondria and inhibiting the
activation of caspase cascade (23). In this study, MHY218 treatment
resulted in a significant increase in pro-apoptotic protein Bax and
decrease in anti-apoptotic protein Bcl-2 resulting in a shift in
Bax/Bcl-2 ratio in favor of apoptosis. MHY218 treatment also
increased the activation of initiator caspase-8 and -9 and the
downstream effector caspase-3 (24).
Certain chemopreventive agents for CRC, such as
aspirin and other non-steroidal anti-inflammatory drugs (NSAIDs),
have been shown to induce apoptosis through a suppression of NF-κB
activation (25). NF-κB acts as
the ‘first responder’ to various types of cellular stress, such as
free radicals, cytokines, ultraviolet radiation and bacterial
components. It plays an important role in inflammation and
carcinogenesis by regulating the expression of downstream target
genes, including COX-2 (26),
MMP-9 (27) and 5-LOX (28). The present study investigated the
effect of MHY218 on the NF-κB pathway. Treatment of MHY218 to
HCT116 cells resulted in inhibition of nuclear translocation of
NF-κB concentration-dependently. NF-κB transcription factors can
block apoptosis induced by TNF-α (29). TNF-α normally has less or no
cytotoxic effect unless NF-κB activation or protein synthesis is
blocked (30). In line with this,
luciferase reporter assay revealed significant suppression of
TNF-α-induced NF-κB transcriptional activity in MHY218-treated
HCT116 cells in a concentration-dependent manner.
COX-2 is the inducible form of cyclooxygenase that
catalyzes the rate limiting step in prostaglandin synthesis from
arachidonic acid and plays an important role in cancer and tumor
promotion (31). It has been
suggested that COX-2 induction mediated by NF-κB pathway could lead
to malignant cell proliferation and invasion (32). These carcinogenic effect of COX-2
can be reversed by NSAIDs, elucidating the important of COX-2
inhibition in cancer therapy (33). It has been shown to inhibit COX-2
expression by repressing degradation of the inhibitory unit
inhibitor IκBα and hindering the nuclear translocation of the
functionally active subunit of NF-κB, thereby blocking improper
NF-κB activation (34). The
western blot analysis data also revealed that treatment of cells
with MHY218 markedly inhibited COX-2, MMP-9 and 5-LOX protein
expression in a concentration-dependent manner. However, RT-PCR
analysis data showed no change in COX-2 mRNA. These results suggest
that MHY218 may also be effective against colon cancer cells
through suppression of NF-κB activity and NF-κB gene products.
In conclusion, MHY218 suppressed growth of HCT116
cells by causing G2/M cell cycle arrest and apoptosis. These
results suggest that MHY218-induced cell cycle arrest and apoptosis
are associated with inhibition of NF-κB pathway. Taken together,
these results suggested that the novel compound MHY218 may be
useful in the chemoprevention and/or treatment of colon cancer.
Acknowledgements
This study was supported by the
National Research Foundation of Korea (NRF) grant funded by the
Korea government (MSIP) (No. 2009-0083538). We thank Aging Tissue
Bank for providing research information.
References
|
1.
|
Hwang HJ, Kang YJ, Hossain MA, et al:
Novel dihydrobenzofuro[4,5-b][1,8]naphthyridin-6-one derivative,
MHY-449, induces apoptosis and cell cycle arrest in HCT116 human
colon cancer cells. Int J Oncol. 41:2057–2064. 2012.
|
|
2.
|
National Cancer Information Center: Cancer
incidence and death. Goyang . http://www.cancer.go.kr/mbs/cancer.
Accessed July 21, 2013.
|
|
3.
|
Kelly C and Cassidy J: Chemotherapy in
metastatic colorectal cancer. Surg Oncol. 16:65–70. 2007.
View Article : Google Scholar
|
|
4.
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar
|
|
5.
|
Sun SY, Hail N Jr and Lotan R: Apoptosis
as a novel target for cancer chemoprevention. J Natl Cancer Inst.
96:662–672. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Aggarwal BB and Shishodia S: Suppression
of the nuclear factor-kappaB activation pathway by spice-derived
phytochemicals: reasoning for seasoning. Ann NY Acad Sci.
1030:434–441. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Horst D, Budczies J, Brabletz T, Kirchner
T and Hlubek F: Invasion associated up-regulation of nuclear factor
kappaB target genes in colorectal cancer. Cancer. 115:4946–4958.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Luo JL, Maeda S, Hsu LC, Yagita H and
Karin M: Inhibition of NF-kappaB in cancer cells converts
inflammation-induced tumor growth mediated by TNFalpha to
TRAIL-mediated tumor regression. Cancer Cell. 6:297–305. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Nandy P, Lien EJ and Avramis VI:
Inhibition of ribonucleotide reductase by a new class of isoindole
derivatives: drug synergism with cytarabine (Ara-C) and induction
of cellular apoptosis. Anticancer Res. 19:1625–1633.
1999.PubMed/NCBI
|
|
10.
|
Choudhary C, Kumar C, Gnad F, et al:
Lysine acetylation targets protein complexes and co-regulates major
cellular functions. Science. 325:834–840. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Walkinshaw DR and Yang XJ: Histone
deacetylase inhibitors as novel anticancer therapeutics. Curr
Oncol. 15:237–243. 2008.PubMed/NCBI
|
|
12.
|
Mottet D and Castronovo V: Histone
deacetylases: target enzymes for cancer therapy. Clin Exp
Metastasis. 25:183–189. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Jeon HS, Ahn MY, Park JH, et al:
Anticancer effects of the MHY218 novel hydroxamic acid-derived
histone deacetylase inhibitor in human ovarian cancer cells. Int J
Oncol. 37:419–428. 2010.PubMed/NCBI
|
|
14.
|
Park JH, Ahn MY, Kim TH, et al: A new
synthetic HDAC inhibitor, MHY218, induces apoptosis or
autophagy-related cell death in tamoxifen-resistant MCF-7 breast
cancer cells. Invest New Drugs. 30:1887–1898. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Molinari M: Cell cycle checkpoints and
their inactivation in human cancer. Cell Prolif. 33:261–274. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Niculescu AB 3rd, Chen X, Smeets M, Hengst
L, Prives C and Reed SI: Effects of p21(Cip1/Waf1) at both the G1/S
and the G2/M cell cycle transitions: pRb is a critical determinant
in blocking DNA replication and in preventing endoreduplication.
Mol Cell Biol. 18:629–643. 1998.PubMed/NCBI
|
|
17.
|
Baus F, Gire V, Fisher D, Piette J and
Dulic V: Permanent cell cycle exit in G2 phase after DNA damage in
normal human fibroblasts. EMBO J. 22:3992–4002. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
el-Deiry WS, Tokino T, Velculescu VE, et
al: WAF1, a potential mediator of p53 tumor suppression. Cell.
75:817–825. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Gartel AL and Tyner AL: Transcriptional
regulation of the p21((WAF1/CIP1)) gene. Exp Cell Res. 246:280–289.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Iannolo G, Conticello C, Memeo L and De
Maria R: Apoptosis in normal and cancer stem cells. Crit Rev Oncol
Hematol. 66:42–51. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Lorenzo HK and Susin SA: Therapeutic
potential of AIF-mediated caspase-independent programmed cell
death. Drug Resist Updat. 10:235–255. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Guo B, Godzik A and Reed JC: Bcl-G, a
novel pro-apoptotic member of the Bcl-2 family. J Biol Chem.
276:2780–2785. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Harris MH and Thompson CB: The role of the
Bcl-2 family in the regulation of outer mitochondrial membrane
permeability. Cell Death Differ. 7:1182–1191. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Lahiry L, Saha B, Chakraborty J, et al:
Theaflavins target Fas/caspase-8 and Akt/pBad pathways to induce
apoptosis in p53-mutated human breast cancer cells. Carcinogenesis.
31:259–268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Kopp E and Ghosh S: Inhibition of NF-kappa
B by sodium salicylate and aspirin. Science. 265:956–959. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Spehlmann ME and Eckmann L: Nuclear
factor-kappa B in intestinal protection and destruction. Curr Opin
Gastroenterol. 25:92–99. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Bond M, Fabunmi RP, Baker AH and Newby AC:
Synergistic upregulation of metalloproteinase-9 by growth factors
and inflammatory cytokines: an absolute requirement for
transcription factor NF-kappa B. FEBS Lett. 435:29–34. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Chopra A, Ferreira-Alves DL, Sirois P and
Thirion JP: Cloning of the guinea pig 5-lipoxygenase gene and
nucleotide sequence of its promoter. Biochem Biophys Res Commun.
185:489–495. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Nagaki M, Naiki T, Brenner DA, et al:
Tumor necrosis factor alpha prevents tumor necrosis factor
receptor-mediated mouse hepatocyte apoptosis, but not fas-mediated
apoptosis: role of nuclear factor-kappaB. Hepatology. 32:1272–1279.
2000. View Article : Google Scholar
|
|
30.
|
Wajant H, Pfizenmaier K and Scheurich P:
Tumor necrosis factor signaling. Cell Death Differ. 10:45–65. 2003.
View Article : Google Scholar
|
|
31.
|
Mann JR and DuBois RN: Cyclooxygenase-2
and gastrointestinal cancer. Cancer J. 10:145–152. 2004. View Article : Google Scholar
|
|
32.
|
Kim JH, Lee KW, Lee MW, Lee HJ, Kim SH and
Surh YJ: Hirsutenone inhibits phorbol ester-induced upregulation of
COX-2 and MMP-9 in cultured human mammary epithelial cells:
NF-kappaB as a potential molecular target. FEBS Lett. 580:385–392.
2006. View Article : Google Scholar
|
|
33.
|
Claria J and Romano M: Pharmacological
intervention of cyclooxygenase-2 and 5-lipoxygenase pathways.
Impact on inflammation and cancer. Curr Pharm Des. 11:3431–3447.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Surh YJ, Chun KS, Cha HH, et al: Molecular
mechanisms underlying chemopreventive activities of
anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS
through suppression of NF-kappa B activation. Mutat Res.
480–481:243–268. 2001.
|