Introduction
Prostate cancer is one of the most frequently
diagnosed non-cutaneous malignancies and the sixth leading cause of
cancer death in males, accounting for 14% (903,500) of the total
new cancer cases and 6% (258,400) of the total cancer deaths in
males, in 2008 (1). Incidence and
mortality rates are rising in Asian countries. Older age, race and
family history remain the only well-established risk factors and
there are no established preventable risk factors for prostate
cancer (2). Mutations of tumor
suppressor genes in individuals suffering from prostate cancer may
lead its specificity among selected group of individuals. For
example, it has been found that putative tumor suppressor gene
(PTEN/MMAC1) on 10q23 is one of the most frequently deleted
chromosomal regions in human prostate cancer (3,4).
Therefore, genome-based therapies are potentially ideal options for
prostate cancer therapy.
Cytoskeleton plays essential roles in the mitosis,
migration and invasion of carcinoma cells (5). Many cytoskeleton-associated proteins
of prostate cancer cells have been intensively investigated
(6–10). SCIN is a calcium dependent actin
filament serving and capping protein (11). It could regulate vesicle transport
and exocytosis by organizing the cytoskeleton underneath the plasma
membrane (12,13). It also regulated cell
differentiation through the MAP kinases P38 and ERK1/2 mediated
signaling pathway (14). The
reduction in expression of Scinlb, the homolog of SCIN in
zebrafish, was found to be associated with increased cell death
(15). Previous studies have found
SCIN is abnormal in certain kind of carcinomas and revealed that
SCIN is involved in many processes of carcinoma cells, such as
proliferation and tumor resistance. In the megakaryoblastic
leukemia cell lines, SCIN is downregulated and overexpression of
SCIN in these cells inhibited proliferation and tumorigenesis
(16). In the T cell lysis
resistant tumor cells, SCIN is overexpressed and the knockdown of
SCIN significantly attenuated the resistance to cytolytic T
lymphocytes killing (17).
However, the function of SCIN in prostate cancer is largely
unknown.
In the present study, we found that SCIN is
overexpressed in prostate cancer specimens. To investigate the
physiological function of SCIN in prostate cancer, we took
advantage of lentivirus-mediated RNAi to suppress the expression of
SCIN in the prostate cancer PC3 cells. We found that SCIN
suppression reduced the proliferation and colony formation ability
of prostate cancer cells. Furthermore, SCIN silencing was
associated with G0/G1 cell cycle arrest. After SCIN silencing, the
expression of p21 and p16 was significantly increased and the
expression of cyclin A2 was reduced. Taken together, our study
indicates that SCIN is essential for the proliferation of prostate
cancer and lentivirus mediated SCIN silencing is a promising
therapeutic method for the treatment of prostate cancer.
Materials and methods
Immunohistochemistry
Tumor specimens from 193 patients who underwent
surgery for primary breast cancer from 2008 to 2012 at Fuzhou
General Hospital (Fuzhou, China), were evaluated in this study. The
samples were used with the written informed consent from patient
and the approval of the ethics committee of Fuzhou General
Hospital. The mean age of the patients at the time of surgery was
71.1 years. All tumors were from patients with newly diagnosed
prostate cancer who had received no therapy before sample
collection.
Immunoperoxidase staining was performed on 2
μm paraffin sections. All sections were treated with 0.3%
H2O2 to exhaust endogenous peroxidase
activity. Avidin/biotin blocking solutions were used to prevent the
non-specific binding of possible endogenous biotin- or
avidin-binding proteins. Blocking with 10% normal goat serum was
performed before applying the primary antibody against SCIN (1:150,
Cat# HPA024264, Sigma-Aldrich). Biotinylated goat anti-rabbit IgG
was used as the secondary antibody. The immunoreactions were
detected by staining with 3,3′-diaminobenzidine (DAB). For all
cases, representative pictures (×20 magnification) of selected
regions in the SCIN-positive sections were taken with a microscope.
Negative controls were performed by omitting the primary
antibody.
The slides were evaluated in a double-blinded manner
by three pathologists independently, and the scores were supplied
by the proportion of positive tumor cells and the intensity of the
coloring. The result of the tissues was determined from at least
1,000 cells that were counted systematically at ×400 magnification
in ten visual fields selected at random. The proportion of positive
tumor cells were recorded according to the following
classification: 0, no cells stained; 1, <1/3 of cells stained;
2, 1/3–2/3 of cells stained and 3, >2/3 of cells stained.
However, the groups could also be classified into the following 4
groups by the intensity of the coloring: 0, no coloring; 1,
stramineous; 2, buffy; and 3, dark brown. The two scores were
combined to obtain the final one: scores equal to 0 indicate
negative (−), whereas scores from 2 to 3 indicate weakly positive
(+), 4 indicate positive (++), and 5 to 6 indicate
strongly-positive (+++).
Cell culture
Human prostate cancer PC3 cells and human embryonic
kidney HEK293T cells are provided by the Cell Bank of Chinese
Academy of Sciences (Shanghai, China). PC3 and HEK293T cells were
maintained in DMEM medium (HyClone) supplemented with 10% fetal
bovine serum (FBS), L-glutamine, penicillin and streptomycin at
37°C in a humidified 5% CO2 atmosphere.
Construction of SCIN shRNA plasmid
A 67 bp shRNA targeted SCIN (NM_033128) was designed
(5′-CTAGCCG
GGCAAGTGTCCTAAAGTGCAAATTCAAGAGATTTGCACTTTAGGACACTTGCTTTTTAAT-3′).
Another 67 bp random shRNA
(5′-CTAGCCCGGTTCTCCGAACGTGTCACGTATCTCGAGATACGTGACACGTTCGGAGAATTTTT
TTAAT-3′) was used as negative control. Both shRNA were liagated
into lentiviral pFH-L plasmid (Shanghai Hollybio, Shanghai,
China).
Lentivirus packaging and infection
For lentivirus packaging, HEK 293T cells were
transfected with pFH-L-SCIN shRNA or control shRNA together with
two helper plasmids (pVSVG-I and pCMVΔR8.92, Shanghai Hollybio)
using Lipfectamine 2000 (Invitrogen) according to the
manufacturer’s instructions. Two days after transfection,
supernatant containing packaged lentivirus was collected and passed
through 0.45 μm filters. To infect PC3 cells, lentivirus
particles were added to the culture medium at a MOI of 50. Cells
were then cultured in the incubator at 37°C.
RNA extraction and real-time PCR
(RT-PCR)
After lentivirus infection, PC3 cells were washed by
PBS and collected. Total RNA was extracted using TRIzol reagent
(Invitrogen). cDNA was then retro-transcribed with oligo dT using
M-MLV reverse transcriptase (Promega). The expression level of SCIN
was measured by RT-PCR with primers: forward,
5′-ATTGTGGAGGTTGATGTTGATG-3′; and reverse,
5′-AGTGGTGAGGTCTGGTAGTC-3′. The primers of actin, used as
endogenous control, are: forward, 5′-GTGGACATCCGCAAAGAC-3′ and
reverse 5′-AAAGGGTGTAACGCAACTA-3′. The PCR reaction system was 10
μl 2X SYBR premix ex-taq, 0.8 μl primers (2.5
μM), 5 μl cDNA and ddH2O up to final 20
μl volume. Probe amplification was performed as follows: 1
min at 95°C; 40 cycles of 95°C for 5 sec and at 60°C for 20 sec.
The expression level of SCIN was determined by cycle threshold (Ct)
normalized with that of actin using 2−ΔΔCt formula. All
experiments were repeated at least three times.
Western blot analysis
Lentivirus-transduced cells were washed twice with
ice-cold PBS and suspended in a lysis buffer (2% mercaptoethanol,
20% glycerol, 4% SDS in 100 mM Tris-HCl buffer, pH 6.8). After 15
min of incubation on ice, cells were disrupted by ultrasound on
ice. Total cell lysates were then centrifuged (12,000 × g, 15 min,
4°C) and the supernatants were employed for further processing. The
protein concentration was determined by using the BCA protein assay
kit. Equal amount of proteins was loaded and separated by SDS-PAGE,
and was then transferred onto PVDF membrane (Schleicher &
Schuell Co., Keene, NH) using an electro-blotting apparatus (Tanon,
Shanghai, China). The membrane was blocked with 5% non-fat milk in
TBST solution for 1 h at room temperature, and incubated overnight
at 4°C with specific antibody tocsin at a dilution 1:1,500. After
the three washes with TBST solution, the membrane was incubated
with horseradish peroxidase-conjugated secondary antibody diluted
with TBST solution at room temperature for 2 h. The signals of
detected proteins were visualized on ECL plus Western blotting
detection system (Amersham Biosciences Inc., Piscataway, NJ). GAPDH
protein levels were used as a loading control.
MTT assay
PC3 cells were seeded into a 96-well plate at an
initial concentration of 2,000 cells/well and proceeded to MTT
assay according to the manual. In brief, MTT solution was added to
wells and incubated at 37°C for 4 h at different time points after
lentivirus infection (1, 2, 3, 4, 5 days). Then the converted dye
solubilized in acidic isopropanol (10% SDS, 5% isopropanol and 0.01
M HCl) was used and incubated at 37°C for 10 min. The optical
density was measured using microplate reader at the wavelength of
595 nm. The experiment was repeated at least three times.
BrdU incorporation assay
Cell proliferation was also quantified by measuring
BrdU incorporation during DNA synthesis using the BrdU Cell
Proliferation ELISA kit. The experiment was performed according to
the manufacturer’s protocol. Briefly, 10 μl/well of BrdU
labeling solution was added to cells at 24 or 72 h after culture.
After overnight incubation, cells were fixed with 200
μl/well of fix solution for 30 min in the dark at room
temperature, and then incubated with peroxidase-conjugated
anti-BrdU antibody for 90 min. A substrate solution was then added
into each well, and absorbance was measured using a microplate
reader (Bio-Rad 680) at a wavelength of 450 nm with a reference
wavelength of 630 nm. The number of proliferating cells is
represented by the level of BrdU incorporation which directly
correlates to the absorbance values. Growth rate (R) was calculated
by the following equation: R = (A72h −
A24h)/A24h ×100, where A72h and
A24h indicate the absorbance at 450 nm after 24 and 72 h
of incubation.
Colony formation assay
Colony formation assay was performed according to
the literature (18). In brief,
three days after lentivirus infection, about 200 cells of PC3 cells
were seeded into each well of a 6-well plate. Cells were cultured
in the 37°C incubator for about 9 days until most single clones had
more than 50 cells. Cells were washed by PBS and fixed by 4% PFA.
Then, Giemsa staining was performed and images were captured by a
fluorescence microscope. The experiments were performed at least
three times.
Flow cytometry analysis
Cell cycle of PC3 cells was analyzed by flow
cytometry. In brief, four days after lentivirus infection, PC3
cells were collected, washed by PBS and fixed by 75% ethanol. Cells
were stained with propidium iodide (PI) and RNase overnight at 4°C.
Samples were then analyzed by Cell Lab Quanta Beckman Coulter. The
experiment was repeated at least three times.
Statistical analysis
The results of immunohistochemistry staining were
evaluated by χ2 test and the other data were evaluated
by Student’s t-test and expressed as the mean ± SD. Statistical
analyses were performed using GraphPad Prism 5 and P<0.05 was
considered to be statistically significant.
Results
SCIN is highly expressed in prostate
cancer
Previous studies indicate that the expression of
SCIN is changed in certain carcinoma cells (17,19,20).
To investigate the function of SCIN in prostate cancer, we
evaluated the protein level in 173 prostate cancer specimens using
immunohistochemical staining. Of the 173 prostate cancer samples, 9
(5.2%) are strongly-positive, 106 (60.7%) are positive and 44
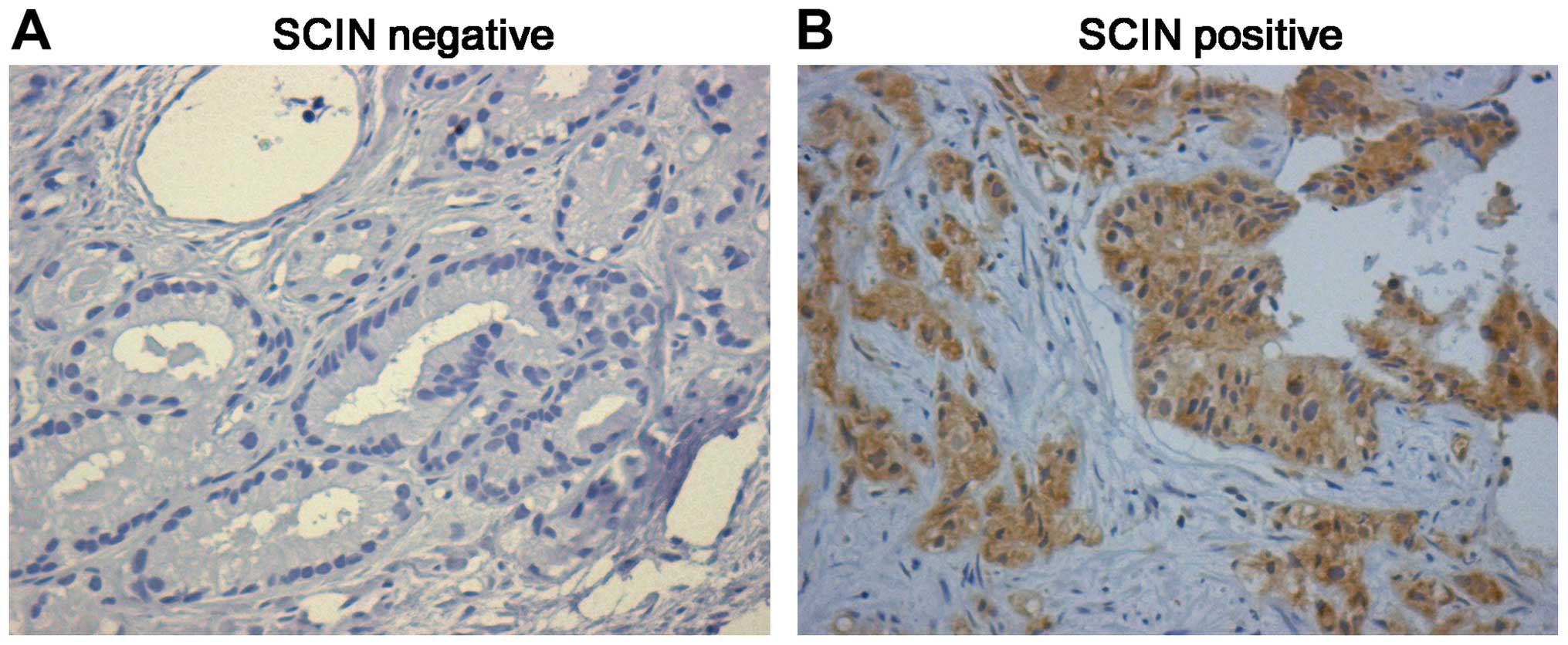
(25.4) are weak positive. Representative immunohistochemical
staining is shown in Fig. 1, and
the entire dataset is illustrated in Table I. These results suggest that the
high expression level of SCIN in prostate cancer may be involved in
the pathogenesis of prostate cancers. Previous studies suggest that
prostate cancer is associated with age and gender of patients
(21,22). To investigate the association of
SCIN expression with these two factors, we analyzed the SCIN
expression level in prostate cancer tissues with different factors.
We found that SCIN expression level is not associated with gender
and age (data not shown). We also determined the relationship
between the immunohistochemical expression level of SCIN and the
degree of differentiation of cancers. We found that SCIN protein is
related to the degree of differentiation of prostate malignancy
(Table I, p<0.05, χ2
test).
 | Table I.Expression pattern of SCIN in prostate
cancer tissues with a different degree of differentiation. |
Table I.
Expression pattern of SCIN in prostate
cancer tissues with a different degree of differentiation.
| Type of tissues | No. of cases | SCIN expression
| P-value |
|---|
| Negative (−) | Weakly positive
(+) | Positive (++) | Strongly-positive
(+++) |
|---|
|
Well-differentiated | 10 | 4 | 2 | 4 | 0 | |
| Moderately
differentiated | 89 | 6 | 32 | 47 | 4 | 0.0004* |
| Poorly
differentiated | 74 | 5 | 10 | 54 | 5 | |
Lentivirus-mediated RNAi efficiently
knocks down endogenous SCIN expression in PC3 cells
To investigate the physiological function of SCIN in
prostate cancer cells, we took advantage of lentivirus-mediated
RNAi technology, a powerful method to knock down the endogenous
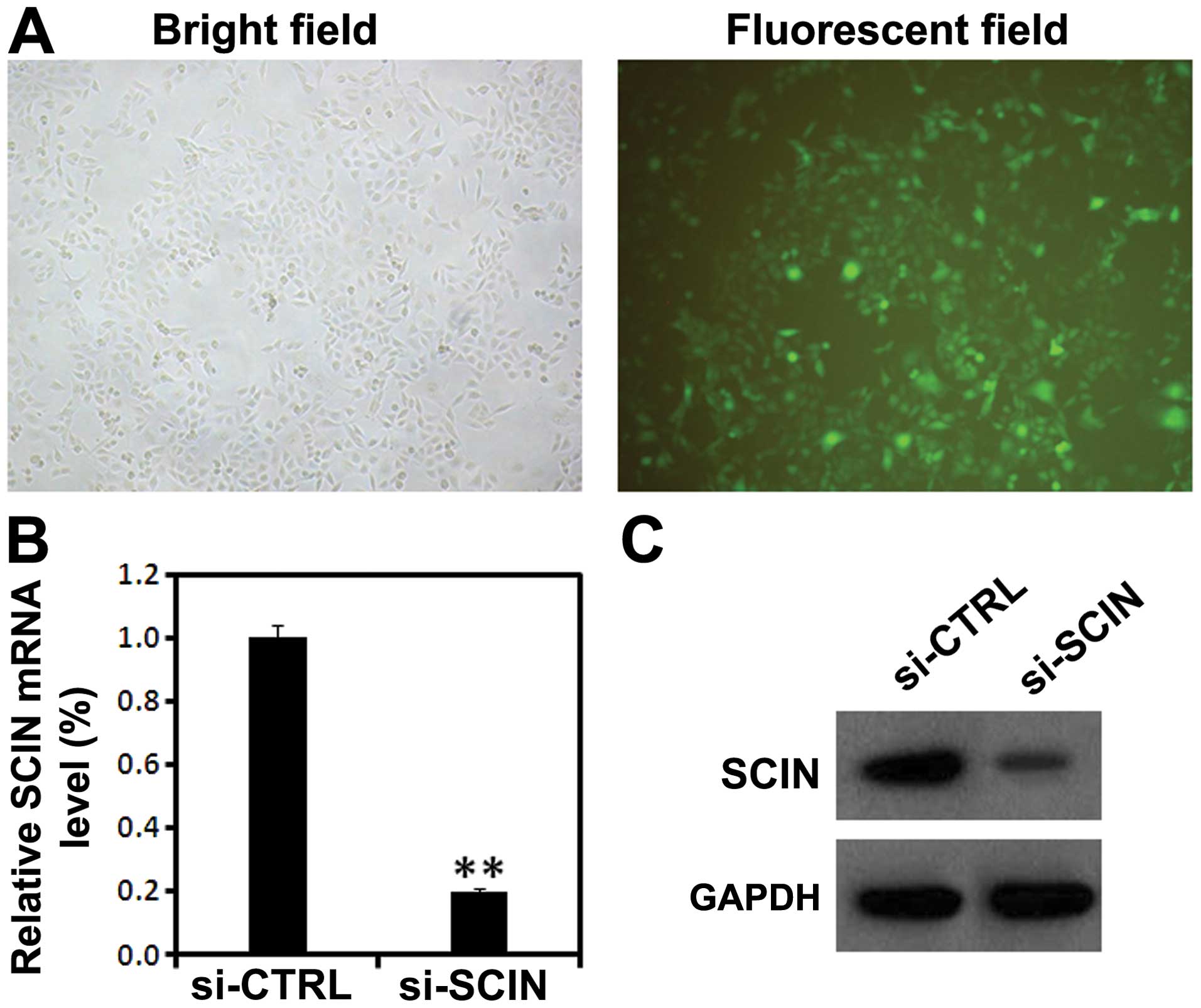
gene expression in vivo. Five days after lentivirus
infection, GFP fluorescence showed high percentage of PC3 cells
being infected by lentivirus (Fig.
2A). Furthermore, using RT-PCR and western blot analysis, we
found that after si-SCIN infection, the mRNA level of SCIN was
significantly reduced compared with the si-CTRL infected group
(Fig. 2B and C). These results
suggest that lentivirus-mediated RNAi was able to significantly
knock down the endogenous SCIN expression in PC3 cells.
SCIN silencing reduces the proliferation
and colony formation ability of PC3 cells
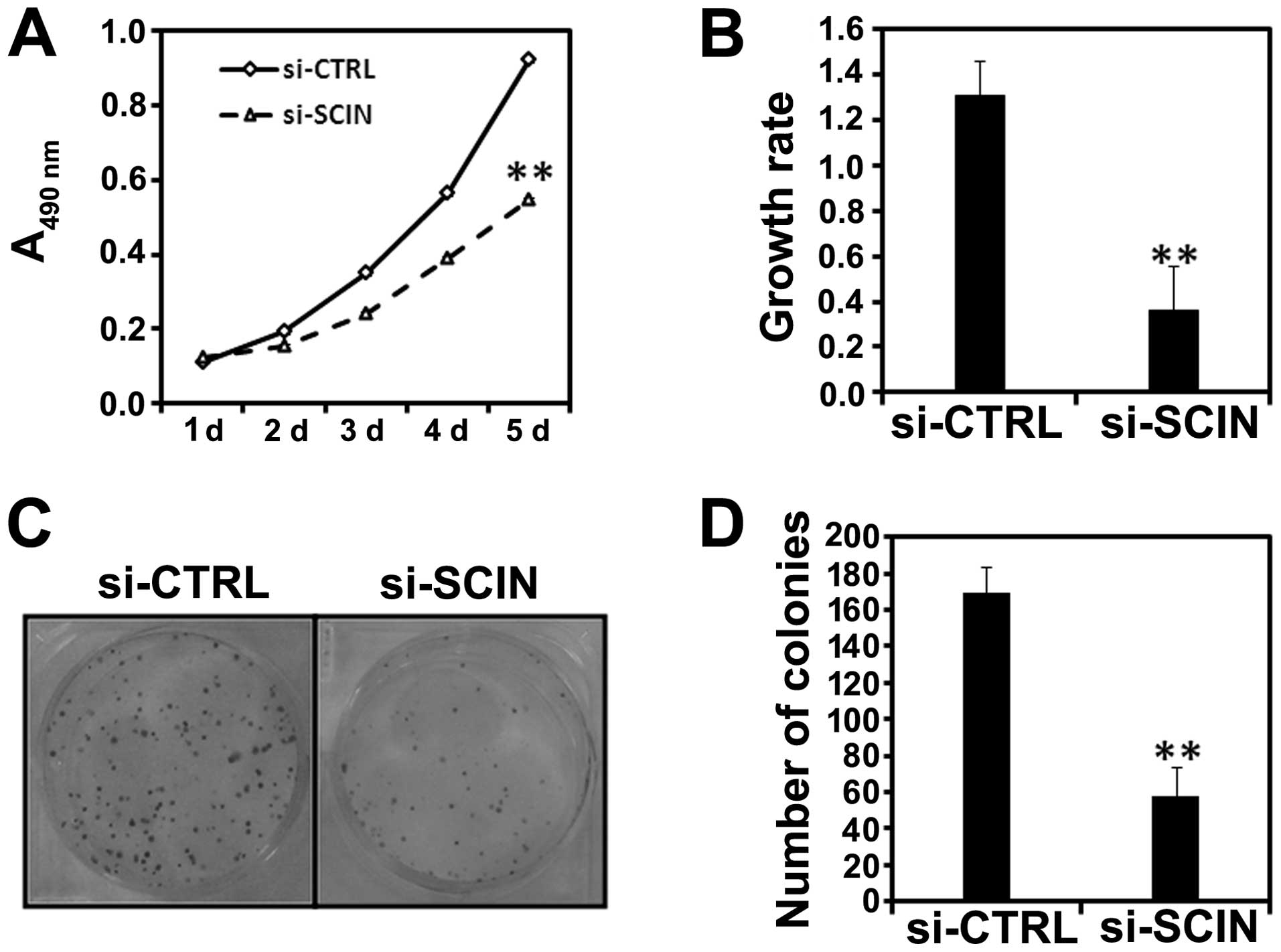
Using MTT assay, we examined the impact of SCIN
silencing on the proliferation ability of PC3 cells. We found that
after si-SCIN infection, cell growth was dramatically reduced
compared with the si-CTRL infected PC3 cells (Fig. 3A). Cell proliferative activity was
then assessed by BrdU incorporation into cellular DNA. Fig. 3B shows a significant decrease in
DNA synthesis as demonstrated by decreased BrdU incorporation in
the si-SCIN group in comparison to si-CTRL after 72 h of lentivirus
transduction (**P<0.01). Furthermore, we found the
colony numbers were reduced in si-SCIN infected PC3 (Fig. 3C and D) cells. In summary, SCIN
silencing was able to inhibit the proliferation and colony
formation ability of prostate cancer cells.
SCIN silencing leads to G0/G1 cell cycle
arrest
To investigate the inhibition of the mechanisms of
SCIN silencing induced cell proliferation, flow cytometry analysis
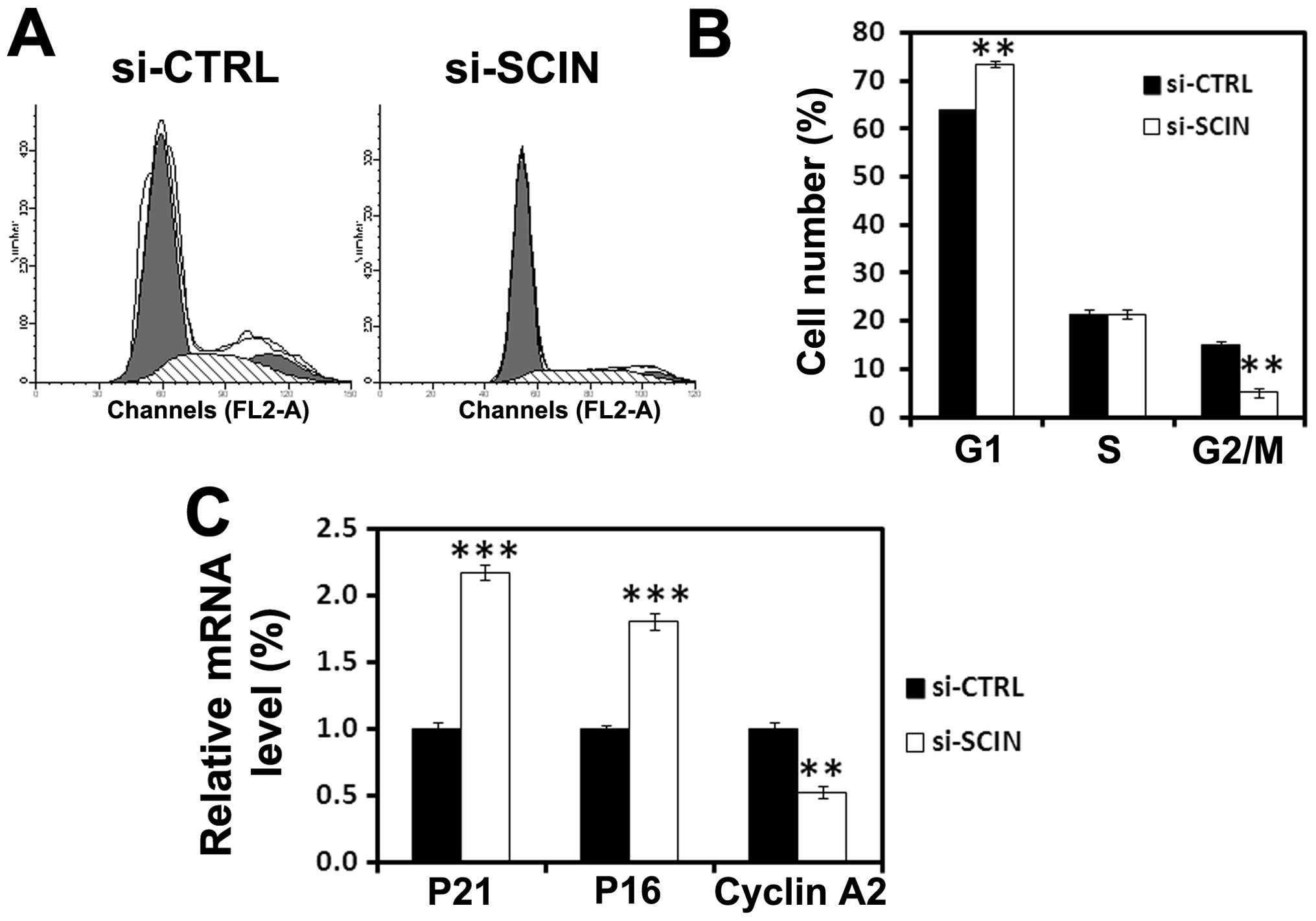
of cell cycle was performed. We found that after si-SCIN infection,
73% of PC3 cells are in the G0/G1 phase and 4% are in the S phase
of the cell cycle, while 64% of si-CTRL infected cells are in the
G0/G1 and 18% are in the S phase of the cell cycle (Fig. 4A and B). Furthermore, to obtain
mechanistic insight into a role for SCIN in cell cycle arrest, we
performed real-time RT-PCR analysis to assess the mRNA level of
cell cycle-related molecules. After 3 days of transduction,
RNAi-mediated SCIN silencing induced upregulation of
p21waf1/cip1 and p16 and downregulation of cyclin A2 in
PC3 cells (Fig. 4C). The results
demonstrated that SCIN may be involved in regulating the
cyclins-cyclin-dependent kinases (CDKs)-CDK inhibitor (CDKI)
expression in PC3 cells.
Discussion
Prostate cancer is one of the most common causes of
cancer-related death. Based on the cell types, prostate cancer
could be classified to two main types, small cell prostate cancer
and non-small cell prostate cancer. Different therapeutic methods
have been used clinically for the treatment of prostate cancer,
including palliative care, surgery, chemotherapy and radiation
therapy. However, only 15% survive for five years after diagnosis
(23,24). New therapeutic gene targets and
methods are needed. Lentivirus mediated gene silencing could
specifically suppress target genes in vivo. The specificity,
higher infection rate and stability of this method gives it high
potential as a therapeutic method for treatment of cancer. In the
present study, using this method, we specifically suppress
endogenous SCIN expression in prostate cancer cell lines and
significantly inhibit the proliferation of PC3 cells.
In carcinoma cells, the cytoskeleton is abnormal and
the expression of some cytoskeleton-associated proteins is aberrant
(6). In the present study, we
found that SCIN is highly expressed in the prostate cancer samples
and about 7% prostate cancer samples have aberrantly strong SCIN
expression. A previous study found that T cell lysis resistant
tumor cells have aberrant strong SCIN expression and the knockdown
of SCIN significantly attenuated the resistance to cytolytic T
lymphocytes killing. Knockdown of SCIN expression in vivo
may facilitate prostate cancer cells being killed by T lymphocytes.
Thus, lentivirus-mediated SCIN silencing in vivo, could have
stronger effects.
Deregulated cell cycle progression is one of the
primary characteristics of cancer cells (25). Cell cycle progression involves
sequential activation of CDKs whose association with corresponding
regulatory cyclins is necessary for their activation (26,27).
Cyclin-dependent kinase inhibitor 2A (CDKN2A, p16Ink4A)
also known as multiple tumor suppressor-1 (MTS-1), is a tumor
suppressor protein, that in humans is encoded by the CDKN2A gene.
P16 plays an important role in regulating the cell cycle, and
mutations in p16 increase the risk of developing a variety of
cancers. p21 is a potent cyclin-dependent kinase inhibitor (CKI).
The p21 (CIP1/WAF1) protein binds to and inhibits the activity of
cyclin-CDK2 or -CDK1 complexes, and thus functions as a regulator
of cell cycle progression at G1. The protein encoded by cyclin A2
binds and activates CDC2 or CDK2 kinases, and thus promotes both
cell cycle G1/S and G2/M transitions. In the present study SCIN
silencing caused a marked increase in expression of p16 and
p21waf1/cip1 and decreased expression of cyclin A2 in
PC3 cells. These observations suggest that SCIN may play an
important role in cell cycle progression of human prostate cancer
by regulating the cyclins-CDKs-CDKI expression.
In conclusion, our results suggest that SCIN
silencing by lentivirus-mediated RNAi was able to reduce the
proliferation and colony formation ability of prostate cancer
cells, possibly due to the cell cycle arrest at G0/G1 phase. Our
studies provide a potential therapeutic gene target for the
treatment of prostate cancer.
Acknowledgements
This study was supported by grants
from the National Natural Science Foundation of China
(81272247).
References
|
1.
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2.
|
Gronberg H: Prostate cancer epidemiology.
Lancet. 361:859–864. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Dong JT, Li CL, Sipe TW and Frierson HF
Jr: Mutations of PTEN/MMAC1 in primary prostate cancers from
Chinese patients. Clin Cancer Res. 7:304–308. 2001.PubMed/NCBI
|
|
4.
|
Li J, Yen C, Liaw D, et al: PTEN, a
putative protein tyrosine phosphatase gene mutated in human brain,
breast, and prostate cancer. Science. 275:1943–1947. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Hall A: The cytoskeleton and cancer.
Cancer Metastasis Rev. 28:5–14. 2009. View Article : Google Scholar
|
|
6.
|
Bernal SD, Baylin SB, Shaper JH, Gazdar AF
and Chen LB: Cytoskeleton-associated proteins of human lung cancer
cells. Cancer Res. 43:1798–1808. 1983.PubMed/NCBI
|
|
7.
|
Chang WT, You BJ, Yang WH, Wu CY, Bau DT
and Lee HZ: Protein kinase C delta-mediated cytoskeleton remodeling
is involved in aloe-emodin-induced photokilling of human lung
cancer cells. Anticancer Res. 32:3707–3713. 2012.PubMed/NCBI
|
|
8.
|
Niu J, Mo Q, Wang H, Li M, Cui J and Li Z:
Invasion inhibition by a MEK inhibitor correlates with the
actin-based cytoskeleton in lung cancer A549 cells. Biochem Biophys
Res Commun. 422:80–84. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Chen QY, Xu LQ, Jiao DM, et al: Silencing
of Rac1 modifies lung cancer cell migration, invasion and actin
cytoskeleton rearrangements and enhances chemosensitivity to
antitumor drugs. Int J Mol Med. 28:769–776. 2011.PubMed/NCBI
|
|
10.
|
Chen CH, Lin H, Chuang SM, Lin SY and Chen
JJ: Acidic stress facilitates tyrosine phosphorylation of HLJ1 to
associate with actin cytoskeleton in lung cancer cells. Exp Cell
Res. 316:2910–2921. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Marcu MG, Zhang L, Elzagallaai A and
Trifaro JM: Localization by segmental deletion analysis and
functional characterization of a third actin-binding site in domain
5 of scinderin. J Biol Chem. 273:3661–3668. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Dumitrescu Pene T, Rose SD, Lejen T, Marcu
MG and Trifaro JM: Expression of various scinderin domains in
chromaffin cells indicates that this protein acts as a molecular
switch in the control of actin filament dynamics and exocytosis. J
Neurochem. 92:780–789. 2005.
|
|
13.
|
Trifaro JM, Gasman S and Gutierrez LM:
Cytoskeletal control of vesicle transport and exocytosis in
chromaffin cells. Acta Physiol (Oxf). 192:165–172. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Nurminsky D, Magee C, Faverman L and
Nurminskaya M: Regulation of chondrocyte differentiation by
actin-severing protein adseverin. Dev Biol. 302:427–437. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Jia S, Nakaya N and Piatigorsky J:
Differential expression patterns and developmental roles of
duplicated scinderin-like genes in zebrafish. Dev Dyn.
238:2633–2640. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Zunino R, Li Q, Rose SD, et al: Expression
of scinderin in megakaryoblastic leukemia cells induces
differentiation, maturation, and apoptosis with release of
plateletlike particles and inhibits proliferation and
tumorigenesis. Blood. 98:2210–2219. 2001. View Article : Google Scholar
|
|
17.
|
Abouzahr S, Bismuth G, Gaudin C, et al:
Identification of target actin content and polymerization status as
a mechanism of tumor resistance after cytolytic T lymphocyte
pressure. Proc Natl Acad Sci USA. 103:1428–1433. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Franken NA, Rodermond HM, Stap J, Haveman
J and van Bree C: Clonogenic assay of cells in vitro. Nat Protoc.
1:2315–2319. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Thorsen K, Schepeler T, Oster B, et al:
Tumor-specific usage of alternative transcription start sites in
colorectal cancer identified by genome-wide exon array analysis.
BMC Genomics. 12:5052011. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Miura N, Takemori N, Kikugawa T, Tanji N,
Higashiyama S and Yokoyama M: Adseverin: a novel
cisplatin-resistant marker in the human bladder cancer cell line
HT1376 identified by quantitative proteomic analysis. Mol Oncol.
6:311–322. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Harichand-Herdt S and Ramalingam SS:
Gender-associated differences in lung cancer: clinical
characteristics and treatment outcomes in women. Semin Oncol.
36:572–580. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Cepeda OA and Gammack JK: Cancer in older
men: a gender-based review. Aging Male. 9:149–158. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Reddy C, Chilla D and Boltax J: Lung
cancer screening: a review of available data and current
guidelines. Hosp Pract (Minneap). 39:107–112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Bach PB, Mirkin JN, Oliver TK, et al:
Benefits and harms of CT screening for lung cancer: a systematic
review. JAMA. 307:2418–2429. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Grana X and Reddy EP: Cell cycle control
in mammalian cells: role of cyclins, cyclin dependent kinases
(CDKs), growth suppressor genes and cyclin-dependent kinase
inhibitors (CKIs). Oncogene. 11:211–219. 1995.PubMed/NCBI
|
|
26.
|
Schafer KA: The cell cycle: a review. Vet
Pathol. 35:461–478. 1998. View Article : Google Scholar
|
|
27.
|
Molinari M: Cell cycle checkpoints and
their inactivation in human cancer. Cell Prolif. 33:261–274. 2000.
View Article : Google Scholar : PubMed/NCBI
|