Introduction
In breast cancer, HER2 proto-oncogene is notably
overexpressed in about 25–30% and the patients and show poor
prognosis, with a lower disease-free survival rate and a shorter
time to relapse (1–6). HER2 has been proven to be an
excellent target for therapy as HER2-driven mechanism has been
linked to tumor growth, resistance to chemotherapy and metastasis
(7,8). While Herceptin is being used as a
first-line drug treatment for HER2-positive breast cancer, about
52% of breast cancer patients fail to respond to the initial
Herceptin treatment or develop resistance to the antibody therapy
within one year (9–11). Currently much effort is going into
uncovering cellular factors that elicit the drug resistance in
HER2-positive cancer. To date, the mechanism for this incurred
resistance is poorly understood.
We have previously identified a specific
upregulation of the transcription factor STAT3 from the
HER2-overexpressing and ER-positive human breast cancer cell line
MCF7-HER2 (unpublished data). The screen was performed with 44,000
genome-wide microarray of MCF7 cancer cells where STAT3 mRNA
expression was increased by 3.62-fold in MCF7-HER2 cells compared
with MCF7 wild-type. STAT3 is a latent cytoplasmic transcription
factor that conveys signals from the cell surface to the nucleus by
cytokines or growth factors (12).
Clinically, increase in serum IL-6 levels has been correlated with
a poor prognosis in breast cancer patients (13). STAT3 has been reported
constitutively activated in about 50–60% of the primary breast
tumors and activated through the phosphorylation of Y705 by
cytoplasmic non-receptor tyrosine kinases (14–17).
Recent studies have revealed the potential roles of
STAT3 in breast cancer. It was shown that STAT3-RANTES autocrine
signaling is essential for tamoxifen resistance in breast cancer
(18). Another group has
demonstrated that the JAK2/STAT3 signaling was specifically
required for the growth of CD44+/CD24− stem
cell-like breast cancer cells in human tumors (19). It was also reported that HER2
overexpression elicited IL6 secretion and activated STAT3,
enforcing an autocrine loop of HER2-IL6-STAT3 expression (20). These data suggest that STAT3
signaling may play a crucial role for the chemotherapeutic
resistance in human breast cancer.
In this work, we report that HER2 overexpression
leads to activation of phosphorylation of STAT3 with subsequent
upregulation of stem cell markers that may eventually lead to
resistance to Herceptin treatment. Using HER2/ER-positive breast
cancer model, we have demonstrated the reduction in the epithelial
characteristics and subsequent increase in stem cell-like
characteristics. Testing combined treatments of STAT3 inhibitor and
Herceptin resulted in a significant inhibition of growth for
HER2-overexpressing cancer cells. More importantly, HER2/ER/STAT3
activation signaling held true in human breast cancer patient
tissues. Our results suggest that a combined treatment of STAT3
inhibitor with Herceptin may help overcome the incurred resistance
derived from the enriched cancer stem-like cells in breast tumors.
Considering that breast tumors co-express HER2 and ER, these
results have important implications for targeted therapy.
Materials and methods
Breast cancer cell lines
BT474 wild-type, SKBR3 wild-type and MCF7 wild-type
cells were obtained from the American Type Culture Collection
(ATCC). They were maintained in a monolayer culture in DMEM/F12
(Dulbecco’s modified Eagle’s medium) with 10% fetal bovine serum,
2.5% L-glutamine and 0.5% penicillin/streptomycin. The MCF7-HER2
(MCF7 cells transfected with HER2) cell line was a generous gift of
Dr C. Kent Osborne (Baylor College of Medicine, Houston, TX).
MCF7-HER2 cells were maintained in a monolayer culture in DMEM 1X
with 10% fetal bovine serum, 2.5% L-glutamine, 0.5%
penicillin/streptomycin, and G418 (400 μg/ml).
Tumorsphere formation assay
Matrigel (BD, Cambridge, MA), 200 μl was
spread as a thick layer on a 24-well plate and allowed to
polymerize at 37°C for 15 min, then 2×104 cells grown as
monolayer were trypsinized to single cells and plated on top of the
pre-coated Matrigel. Plates were incubated at 37°C to allow cells
to fully settle down before media was replaced with appropriate
growth media containing 5% Matrigel. Cells were grown for 15 days;
fresh growth media with Matrigel was replenished every two days.
Images of representative fields were taken.
Short hairpin interfering RNA
transfection
The HER2 short hairpin RNA (shRNA) and control shRNA
were purchased from the Ori Gene (Rockville, MD). The transfection
was performed by using Lipofectamine 2000 (Invitrogen) reagent
following the manufacturer’s instructions. The STAT3 shRNA was a
kind gift of Dr Hyung-Gyoo Kang at the Children’s Hospital in Los
Angeles. The HER2 and STAT3 shRNA knockdown expressions after shRNA
transfection were determined by a Western blot analysis at 48–72 h
of transfection.
Western blot analyses
Monolayer cultures of respective cell lines at an
80–90% confluence were lysed using 100 μl of RIPA (Thomas
Scientific Co.) buffer. Tris-glycine (Bio-Rad) gels were loaded
with 50–100 μg of lysates. After running gel
electrophoresis, the gel was transferred to a nitrocellulose
membrane for 2 h. The membrane was blocked for 1 h in 5% BSA or 5%
skim milk at 4°C. The membrane was then washed 3 times with 1X TTBS
and incubated overnight with the primary antibody at 4°C. Primary
antibodies of Oct-4, Sox-2, STAT3, pSTAT3, E-cadherin, vimentin,
slug and β-actin were purchased from Cell Signaling Technology
(Danvers, MA).
Boyden chamber invasion assay
Mouse fibroblasts (NIH-3T3) were used as a
chemo-attractant, and grown in a 24-well plate in 2 ml of the same
media as MCF7-HER2 cells. MCF7 WT and MCF7-HER2 experimental cells
were synchronized to an equal number (125,000 cells) in a 6-well
plate and were serum starved overnight. Boyden chambers were
prepared with 25 μl of 1:6 diluted Matrigel and allowed to
incubate for about 2 h to solidify. After cell synchronization,
invasion was allowed to occur for 40 h. The cells were then fixed
with 0.5% glutaraldehyde and stained with 5% toluidine blue for
cell counting. Three different microscope fields of ×40 were used
to quantify the invasion statistics when counting cells.
FACS profile analysis
Approximately 500,000 cells of MCF7 WT or MCF7-HER2
cells were washed with 1X PBS, trypsinized and then transferred to
a 15-ml aliquot tube. Cell suspensions were centrifuged down,
re-suspended in 2 ml 1X PBS and then divided into two tubes 0.5 ml
each for staining. One tube was used as an unstained control and
the other one as stained with 10 μl CD44 antibody (FITC
Green; BD Biotech). The tubes were vortexed briefly and incubated
at room temperature for 15 min in the dark. Each tube was then
washed with 3.5 ml 1X PBS and then centrifuged for 6 min. After
aspirating the supernatant, the cells were re-suspended in 3 ml 1X
PBS and subjected to the FACS profiling at UCLA FACS Core
Laboratory.
Stattic (STAT3 inhibitor) treatment
All inhibitors were prepared as 200 mmol/l DMSO
stocks for in vitro use. MCF7-HER2 cells that were at least
60–70% confluent were washed with 1X PBS and treated with 5
μM of Stattic (STAT3 Inhibitor V; Santa Cruz Biotechnology)
dissolved in 4 ml of 1X DMEM media. Untreated Stattic MCF7-HER2
cells were used as a control. Treatment lasted 24 h and cells were
harvested for further analysis.
MTT assay for dose response curve
MCF7-HER2 cells were cultured and counted and each
test was conducted in triplicate. After mixing well, media/cell
solution was diluted to 4,000 cells per 100 μl. Cells were
then plated in a 96-well plate: 6 wells for each treatment dosage,
4,000 cells per well (100 μl). Cells were treated with
Stattic (0 μM control, 1, 5 and 10 μM). Treatments
lasted 48 h and then 50 μl of MTT buffer reagent was added
into each well. After incubation for 4 h at 37°C, 100 μl of
DMSO was added and gently agitated for 15 min. The plate was then
read at 560 nm absorbance.
Combined treatment of Stattic and
Herceptin
The initial set up was similar to the one described
in MTT assay dose response curve. This time, however, 10,000 cells
were plated per 100 μl in 8 wells per treatment. Cells were
allowed to grow until about 50% confluence, and then treated with
the following dosage treatments: untreated control, Stattic (5
μM), Herceptin (10 μg/ml, a gift of Genentech) and
Stattic (5 μM) + Herceptin (10 μg/ml) combination.
Treatments lasted 72 h and cell growth was monitored.
Co-immunoprecipitation assay
To monitor the protein-protein interactions among
HER2, ER and STAT3 proteins, ER specific monoclonal antibody (Santa
Cruz Biotechnology, sc-7207) was added to the cancer cell lysates
and incubated overnight. The immune-pellets were spun at 3,000 rpm
for 5 min and washed three times with RIPA buffer (Thomas
Scientific Co). The pellets were then ran on the PAGE gel and
immunoblotted for HER2 and STAT3 proteins, respectively.
Tissue microarray analyses
Human breast cancer tissue micro-array slides
(BRC961) were purchased from US Biomax Inc. (Ijamsville, MD, USA).
Breast cancer tissue microarray slides were stained using standard
IHC methods with an IHC-validated phospho-specific STAT3 antibody
(Tyr705; Cell Signaling Technology, D3A7).
Statistics
For determining statistical significance in all
quantifications, Student’s t-test was used; the data are presented
as the mean ± SD, and considered significant at p<0.05.
Results
Activation of STAT3 in the
HER2-overexpressing and ER-positive breast cancer model
Initially we tested the effect of STAT3 in
HER2-positive breast cancer. We wished to determine whether the
co-expression of HER2 and ER induced STAT3 phosphorylation, and
whether pSTAT3 promotes stem-like cell phenotype in the breast
cancer model. To this end, the basal expression of STAT3 and stem
cell markers were examined in various human breast cancer cell
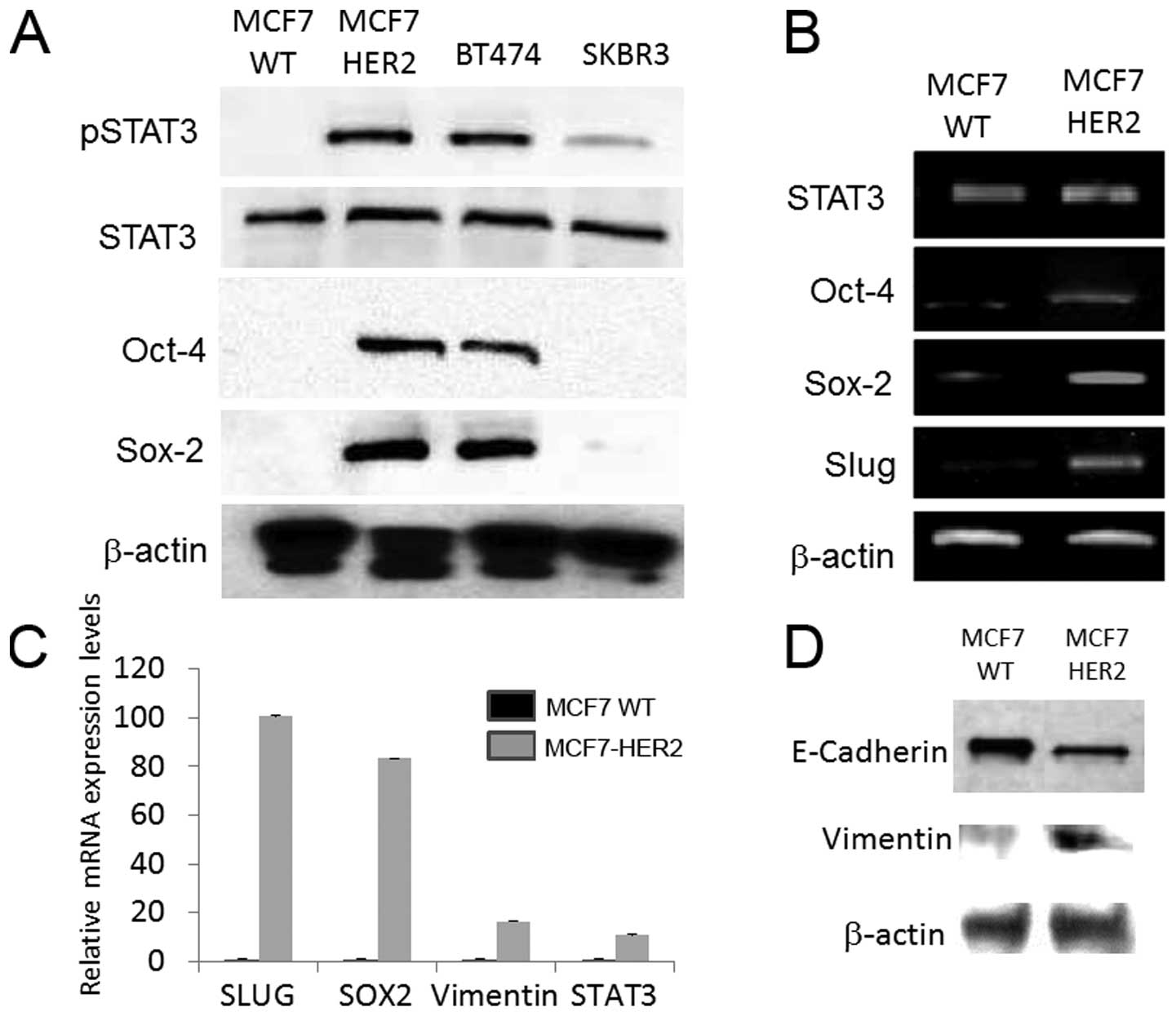
lines by western blot analysis and real-time PCR. STAT3 was
phosphorylated in MCF7-HER2 cells, but not in MCF7 wild-type
(Fig. 1A). In addition, STAT3 was
phosphorylated in BT474 as well as at very low level in SKBR3. MCF7
wild-type lacks HER2 amplification and SKBR3 lacks ER. This is
consistent with previous data in which MCF7 WT did not typically
show phosphorylation of STAT3 (21,22).
Moreover, we found that the stem cell markers, Oct-4 and Sox-2,
were expressed in MCF7-HER2 and BT474 cells, but not in MCF7
wild-type and SKBR3 (Fig. 1A).
Real-time PCR analyses confirmed the upregulation of stem cell
markers in HER2-overexpressing, ER-positive cancer cells (Fig. 1B). Our results support the
hypothesis that HER2 over-expression and ER positivity promote
STAT3 phosphorylation and induces the stem cell-like phenotype.
Transcription factor slug, which has been implicated
as a driver for the epithelial-mesenchymal transition (EMT) was
upregulated in MCF7-HER2 cells (Fig.
1B), leading us to the hypothesis that, HER2 induced stem cell
marker expression and slug upregulation promoted the EMT phenotype
in MCF7 cells. Real-time PCR analyses showed the gene expression
pattern matching with the mesenchymal microenvironment, showing
significant upregulation in vimentin, slug and concurrent
downregulation of E-cadherin in MCF7-HER2 compared with control
MCF7 wild-type (Fig. 1C). In line
with the PCR data, western blot analyses revealed that epithelial
marker E-cadherin was downregulated, while mesenchymal maker
vimentin was upregulated in HER2 transfected MCF7 cells (Fig. 1D). These results indicate that the
sequential activations elicited by HER2 amplification converge into
the HER2-pSTAT3-stem cell markers - early EMT characteristics in
signal pathways.
STAT3 inhibitor abolished both the stem
cell marker and EMT marker expressions
To uncover Herceptin resistance mechanism in
HER2-overexpressing cancer cells, HER2/ER-positive cancer cells
were treated with STAT3 inhibitor and examined for cancer stem cell
phenotype. Stattic is a non-peptidic small molecule that inhibits
STAT3 activation by selectively inhibiting dimerization and nuclear
translocation of STAT3 (23). For
the purpose of proving the effect of Stattic on inhibiting STAT3
and to investigate its effects on the HER2-ER-STAT3, we treated
cells with 5 μM Stattic for 24 h and monitored its effects
on EMT and stem cell marker expression.
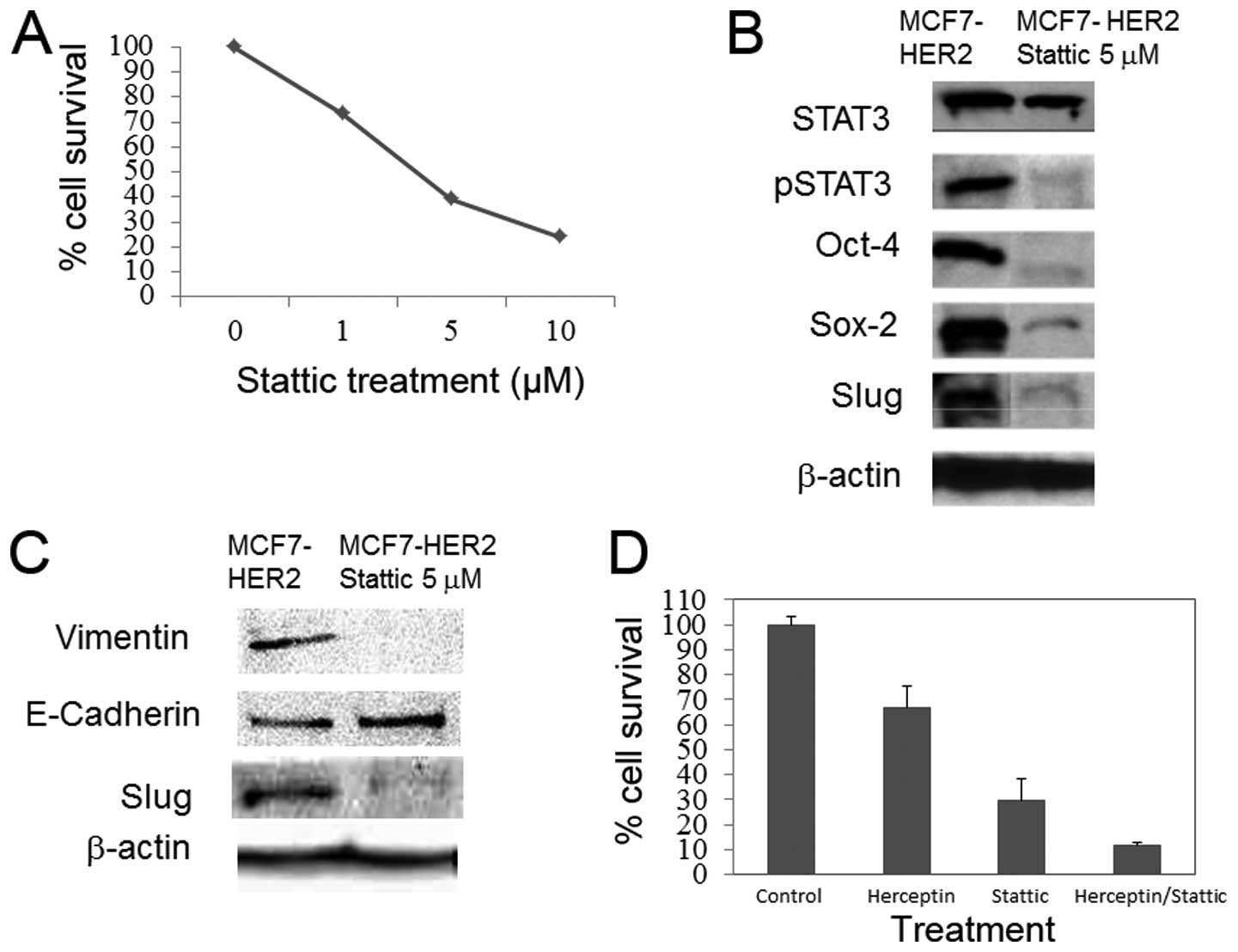
To begin with, dose response to Stattic was
monitored in MCF7-HER2 cells (Fig.
2A). Stattic treatment of 5 μM resulted in 28% of cell
survival. Western blots of the MCF7-HER2 cells treated with 5
μM of Stattic illustrated inhibition of STAT3 activation, as
expected, but also showed significantly diminished Sox-2, Oct-4,
and slug expression to the point of abrogation (Fig. 2B). While total STAT3 was almost
intact with the Stattic treatment, pSTAT3 expression was also
abolished simultaneously. For EMT marker expression, we monitored
expression of E-cadherin, vimentin and slug protein. In agreement
with cancer stem cell markers, the expression levels of EMT
markers, vimentin and slug, were clearly decreased upon Stattic
treatment (Fig. 2C). Our data
suggest that STAT3 activation is responsible for the stem cell
marker expression in HER2-overexpressing breast cancer cells.
Combined treatment of Stattic and
Herceptin show synergistic anti-growth effects
Given Stattic’s effect on reducing stem cell-like
characteristics, we designed a combined treatment regimen
consisting of Stattic, STAT3 inhibitor, and Herceptin, HER2
monoclonal antibody. The goal was to demonstrate that
HER2-positive, ER-positive cancer cells were more sensitive to
Herceptin treatment when also exposed to Stattic, due to the
inhibition of STAT3. This would also help prove the presence of
cross-talk between HER2/ER-STAT3 pathways through the synergistic
effect of the combined treatment. Thus, we hypothesized that
Stattic could help delay or even reverse incurred resistance
associated with HER2 overexpression.
Based upon the dose response curve, it was estimated
that 5 μM would be the optimal dosage for the combined
treatment. In a second MTT to determine the effectiveness of
Stattic and Herceptin combined treatment, a control group, a
solitary 10 μg/ml Herceptin treatment group, a solitary 5
μM Stattic treatment group and a combined 10 μg/ml
Herceptin with 5 μM Stattic treatment group were compared to
determine which treatment would provide the highest apoptosis rate.
It was observed that while all treatments lead to significant
reduction in the cell population based upon the control population,
the Herceptin and Stattic combination treatment showed the highest
rate of apoptosis (Fig. 2D). The
combination treatment displayed significant effect (cell survival
11%) when compared to the solitary Herceptin and Stattic
treatments.
Targeted knockdown of STAT3 and HER2
expression significantly reduces the CD44+ subcellular
population
STAT3 specific inhibitor Stattic treatment
convincingly abolished the stem cell phenotype. To test whether
STAT3 drives the stem cell phenotype in breast cancer cells, we
knocked down STAT3 gene by shRNAs and examined the CD44+
subpopulation by fluorescence activated cell sorting (FACS).
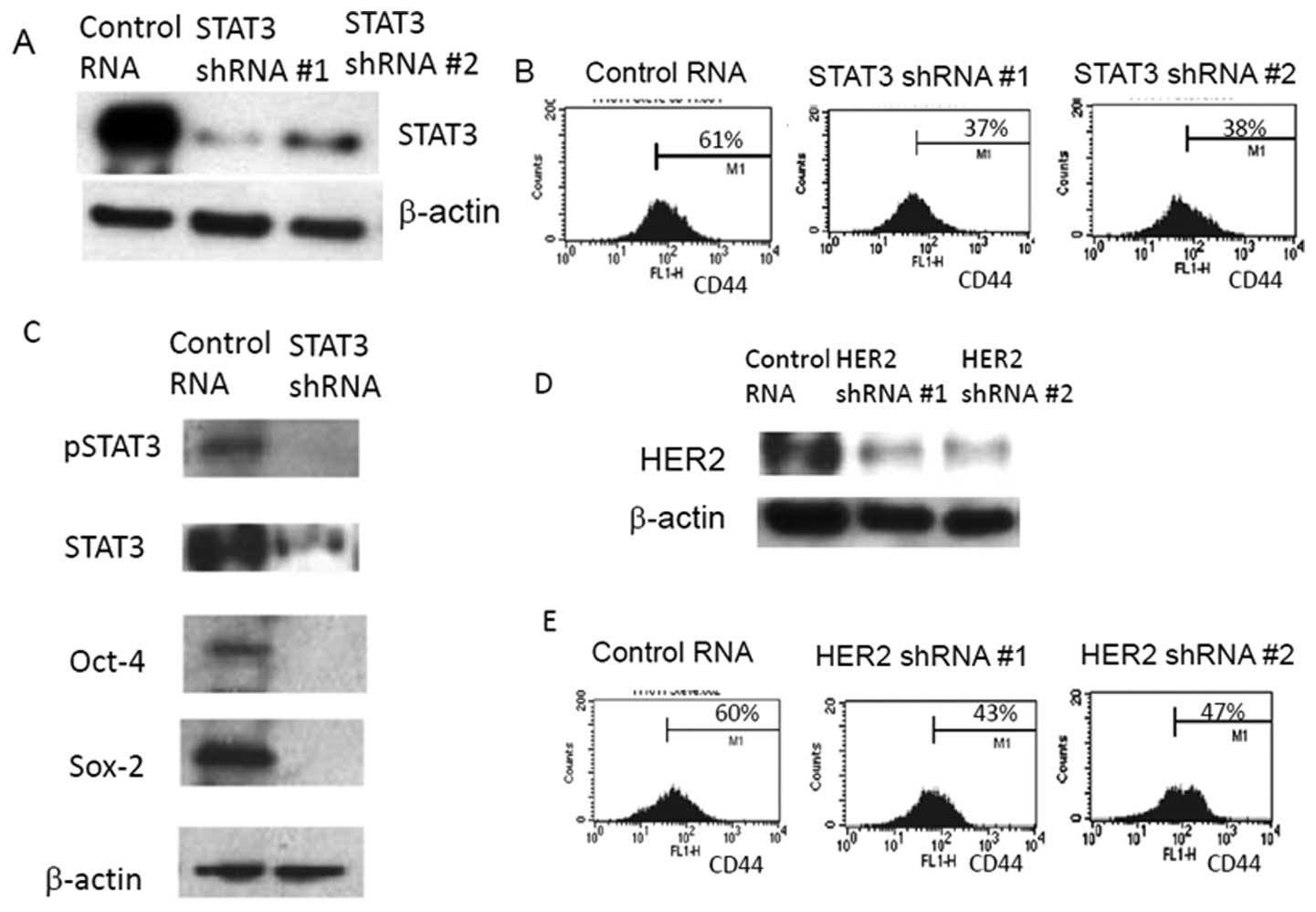
Targeted STAT3 knockdown was confirmed by western blot analysis and
subsequently applied to FACS analysis (Fig. 3A). When STAT3 expression was
knocked down with two independent shRNAs, the CD44(+) subpopulation
has clearly reduced from 61.21% (CD44+ of the control
RNA) to 37.22% and 38.23%, respectively (Fig. 3B). In agreement with FACS data,
western blot analyses of STAT3 knockdown cells also showed
downregulation of stem cell marker expressions (Fig. 3C). These results suggest that STAT3
plays a clear role in inducing the expression of stem cell markers
Oct-4, Sox-2 and CD44.
Not only the expression level of stem cell markers
of Oct-4 and Sox-2 was increased, but CD44(+) cell population was
also increased in HER2-overexpressing, ER-positive breast cancer
cells. To establish the potential link between HER2 amplifi cation
and the stem cell phenotype, we knocked down the HER2 gene by
shRNAs and examined CD44(+) FACS profile (Fig. 3D). The CD44(+) subcellular
population was reduced from 60.77% to a range of 43.17-47.16%, when
HER2 expression was knocked down by two independent HER2 shRNAs
(Fig. 3E). This confirms that HER2
is also a driver for induction of the cancer stem cell-like
phenotype in MCF7 cells.
HER2 overexpression causes the increased
cell invasiveness of breast cancer cells
Cancer stem cell characteristics are associated with
enhanced cell invasiveness. To determine whether forced HER2
overexpression leads to STAT3 activation and functionally enhances
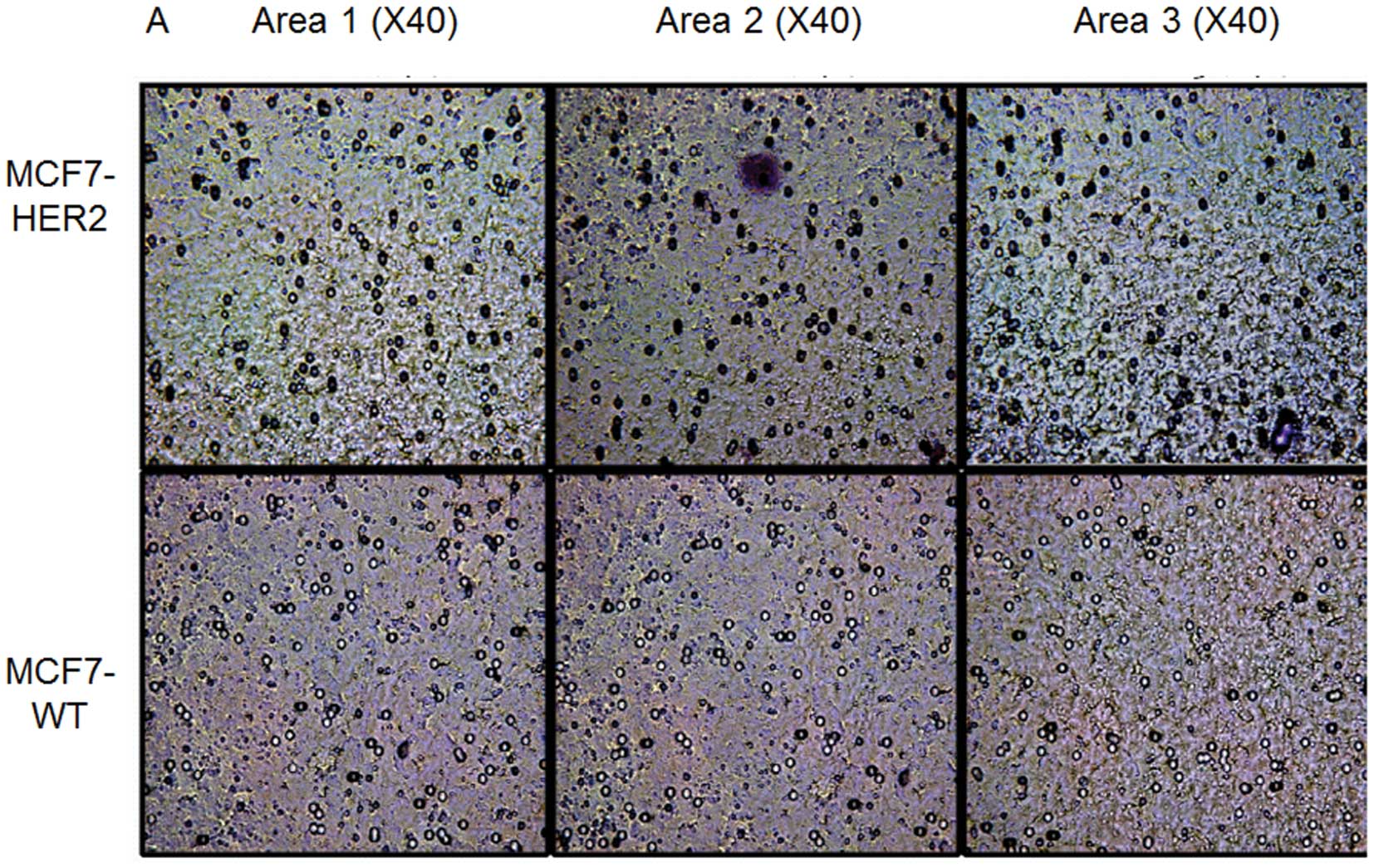
the invasiveness of MCF7 cells, an in vitro Boyden chamber
invasion assay was implemented. As expected, MCF-7 wild-type
control cells showed minimal invasion (8% of invasion population)
through the Matrigel, even in the presence of NIH3T3 mouse
fibroblasts as a chemoattractant (Fig.
4A). In contrast, MCF7-HER2 cells were 4 times more invasive
(31%, Fig. 4B), as they averaged
23 more invaded cells per high powered field (HPF, ×40). Although
MCF7 is typically known for being a non-invasive cell line, HER2
overexpression leading to STAT3 activation resulted in an increase
of the invasiveness of the cell line.
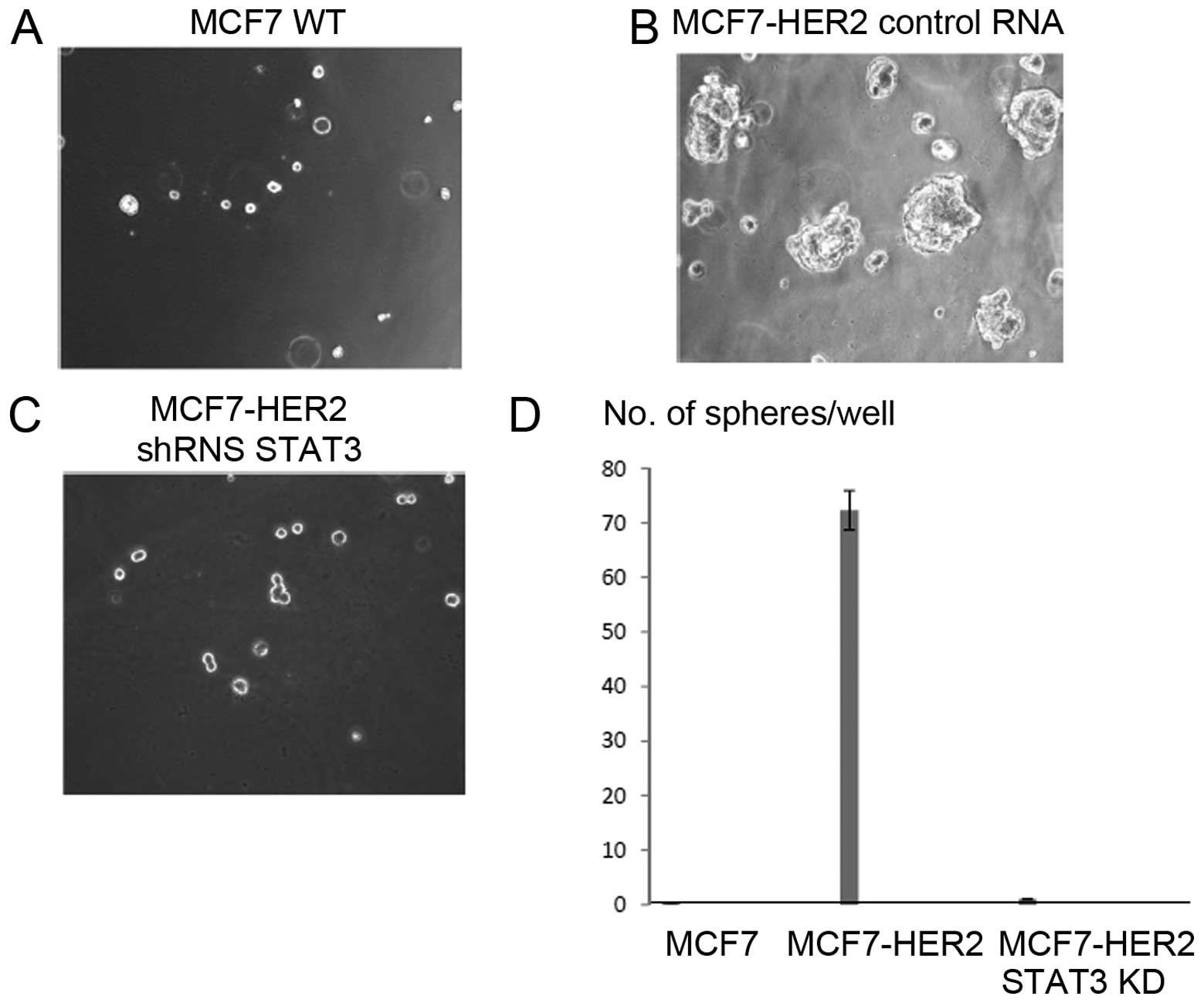
HER2 overexpression promotes tumorsphere
formation whereas knockdown of STAT3 abolishes the sphere
formation
Formation of spheres by tumor cells is often
directly correlated with cancer stem cell phenotype. Having
considered Oct-4, Sox-2 and CD44 expression data, we wished to
determine the tumorsphere formation capacity with
HER2-overexpressing breast cancer. Approximately 20,000 cells were
seeded on each Matrigel 3D well and tumorsphere formation was
observed up to 15 days. While MCF7 WT did not form any spheres,
HER2-overexpressing MCF7 cells formed stable tumorspheres at day 6
(Fig. 5A and B). To verify that
the tumorsphere forming capacity is due to STAT3, we repeated the
3D culture with the targeted STAT3 knockdown cells. As shown in
Fig. 5C, the figure, STAT3
knockdown cells did not form any spheres. The tumorspheres were
counted for each well and presented as a graph (Fig. 5D). These results are consistent
with the STAT3 knockdown effects on expression of stem cell markers
Oct-4, Sox-2 and CD44. STAT3 is necessary for tumorsphere formation
in HER2-overexpressing cancer.
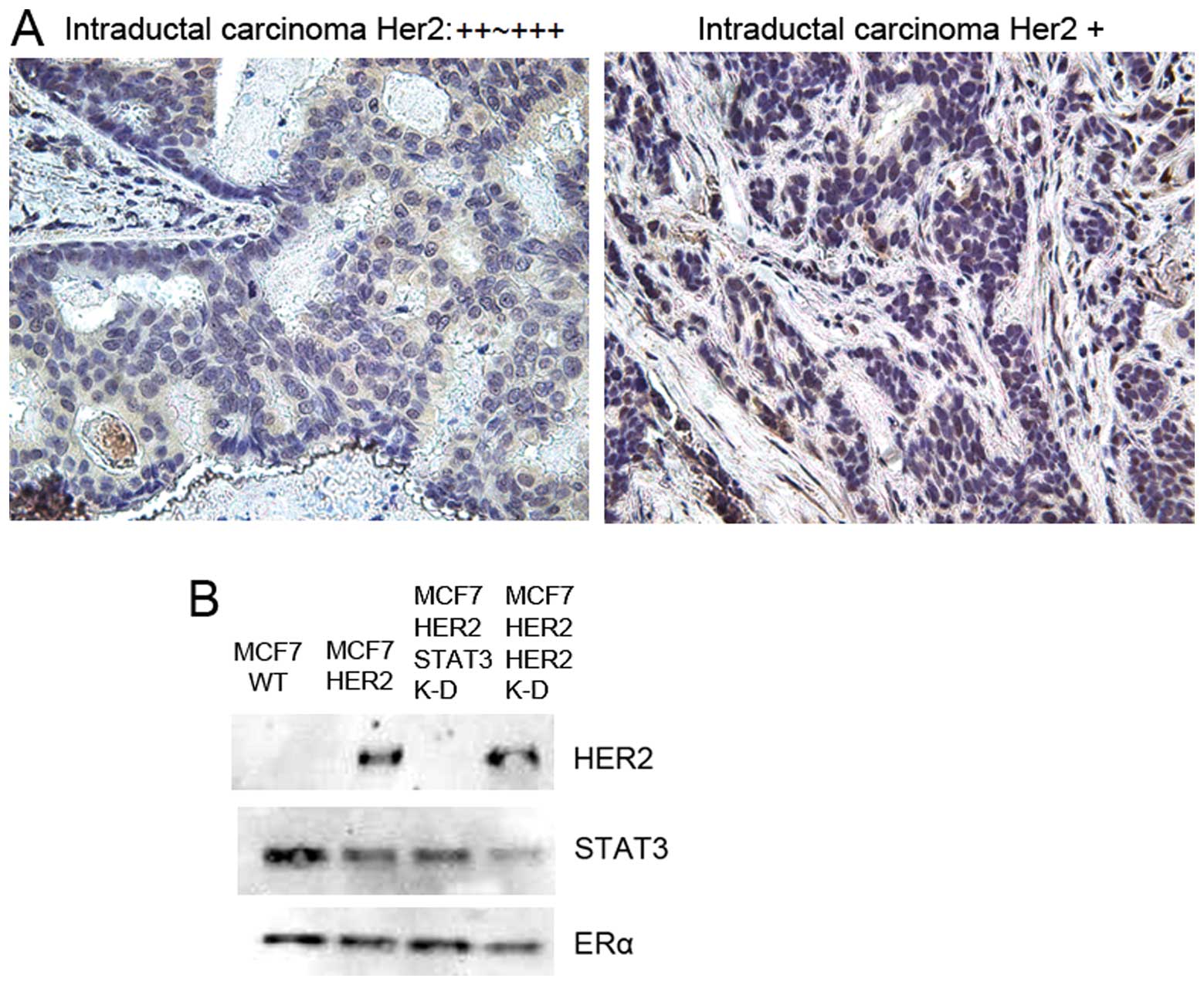
Phosphorylated STAT3 is identified in
HER2-overexpressing, ER-positive human breast cancer tissues
Recently, we found that STAT3 was upregulated by
3.64-fold in MCF7 cells transfected with HER2 compared to a MCF7
wild-type (12). However, the
relationship between STAT3 in primary tumors and HER2
overexpression was not described. Thus, we hypothesized that
HER2-positive, ER-positive breast cancer cells expressed
phosphorylated STAT3. To test this, we have examined tissue
microarrays of 71 primary breast tumors with previously confirmed
ER, PR and HER2 expressions. We observed the phosphorylated STAT3
expression from these breast tumors by immunohistochemistry. We
found that pSTAT3 was expressed only in HER2(+) and ER(+) primary
breast tumors (Fig. 6A). As shown
in the figure, 9 out of 17 HER2+/ER+ tumors
expressed pSTAT3 and 6 out of 9 HER2++ or
HER2+++/ER+ tumors showed pSTAT3 expression
(Table I), moreover, 2 out of 4
showed pSTAT3 in ER/PR/HER2 triple negative patient tissues. This
pSTAT3 activation is possibly through other STAT3 activation
signaling axis in triple negative breast tumors such as Src or
non-RTK phosphorylation mechanisms.
 | Table I.Summarized pSTAT3 staining from the
tissue microarray of human breast tumors (US biomax,
BRC961).a |
Table I.
Summarized pSTAT3 staining from the
tissue microarray of human breast tumors (US biomax,
BRC961).a
| Total no. of cancer
tissues | HER2/ER
IHC-positive tissue numbers | Nuclear pSTAT3
IHC-positive tissue numbers |
|---|
|
HER2+/ER+ | 71 | 17 (23.9% of
71) | 9 (52.9% of
17) |
|
HER2++-HER2+++/ER+ | 71 | 9 (12.6% of
71) | 6 (66.6% of 9) |
|
HER2+-HER2+++/ER− | 71 | 24 (33.8% of
71) | 0 (0% of 24) |
|
HER2−/ER−/PR− | 71 | 7 (9.8% of 71) | 2 (28.5% of 7) |
We also determined whether there are physical
connections between HER2, ER and STAT3. Our next hypo thesis was
that ER is bound to both HER2 receptor and STAT3 to transduce the
phosphorylation signals from HER2 to STAT3. We used the MCF7-HER2
cell line for our model system of HER2(+)/ER(+) breast cancer. We
immunoprecipitated the cell lysates with antibody against ERα and
subsequently immuno blotted for HER2 and STAT3 in MCF7 wild-type,
MCF7-HER2, MCF7-HER2 STAT3 knockdown and MCF7-HER2 HER2 knockdown
cells (Fig. 6B). ERα was bound to
both HER2 and STAT3 simultaneously in MCF7-HER2 cancer cells. When
STAT3 is knocked down, HER2 is still bound to ER, when HER2 is
knocked down, ER is still bound to STAT3. The tissue microarray and
immunoprecipitation data suggest, for the first time, the existence
of HER2-ER-STAT3 signaling axis in HER2-positive breast
cancers.
Discussion
We demonstrated herein that HER2 overexpression
plays an essential role in inducing STAT3 phosphorylation, which
further leads to increased expression of stem cell markers in human
breast cancer cells. In particular, we show that co-expression of
HER2 and ER induces phosphorylation of STAT3 in human breast
tumors. This first established a signal transduction pathway of
HER2-ER-STAT3 in HER2-positive breast cancer. Moreover, we
demonstrated that HER2-ER-STAT3 activation led to the enhanced stem
cell marker and EMT marker expressions. In relation to the acquired
stem cell phenotype, Kong et al have shown that the
expression of Sox-2 and Oct-4 are important indicators for cancer
progression to metastasis and drug resistance (24). This supports the notion that
HER2/ER overexpression activates STAT3 which leads to an increase
in cancer stem cell markers, causing overexpression of HER2 and
cells become resistant to chemotherapy.
Upon treatment with Stattic, we observed a
significant reduction in the stem cell marker expression. In
addition, when we knocked down the STAT3 gene, CD44+
subpopulation was reduced, suggesting the pivotal role of STAT3 in
the cancer stem cell transition in HER2 amplified environment. More
importantly, we found that MCF7-HER2 cells that were treated with
Stattic, were more sensitive to Herceptin than MCF7 cells that were
only treated with Herceptin or Stattic. This is a reflection of
previous studies indicating that inhibited STAT3 has been
correlated with increased apoptosis in cancer cells, increased
chemosensitivity, suppressed tumor growth, reduced invasiveness,
and decreased angiogenesis (25–27).
Although STAT3 may play a vital role in early embryogenesis, its
presence in the vast majority of adult cells is largely expendable,
thus make it an attractive target for certain cancer therapies
(28,29).
Our study, as well as others, suggests that STAT3
plays a crucial role in metastasis and therapeutic resistance in
solid tumors. For example, STAT3 has been considered a fundamental
component of resistant tumor growth in breast cancer (12,13),
head and neck squamous cell carcinoma (27) and lung cancer (30,31)
due to induction of an invasive EMT-like phenotype. There have been
numerous studies linking the EMT phenomenon to the expression of
stem cell-like characteristics to the point where they seem to
overlap (32–34). Thus, given that EMT is associated
with lasting tumor aggressiveness, invasion, and angiogenesis, it
is considered a prime suspect for driving cancer stem-like
cells.
Most of the reports arguing for EMT and cancer stem
cell correlation focus on the idea that an EMT phenotype drives a
cancer stem cell microenvironment that is characterized as
CD44hi/CD24low in breast cancer, which is
associated with therapeutic resistance, tumor invasion and poor
prognosis (35,36). As we observed in the current study,
HER2 overexpression leading to STAT3 activation resulted in
upregulation of CD44 expression. Furthermore, a recent study by
Oliveras-Ferraros et al concluded that a mesenchymal
CD44+/CD24− microenvironment in HER2
overexpressed breast cancer was linked to resistance to Herceptin
treatment (20).
We conclude that HER2 overexpression in ER-positive
breast cancer results in STAT3 activation, further causing stem
cell-like characteristics and resistance to Herceptin. We have
found a model for commonly incurred Herceptin resistance. After an
extended period of time constitutively activated STAT3, from HER2
and ER expression, may induce more and more resistant stem
cell-like characteristics. While we used the specific STAT3
activation inhibitor Stattic in our study, other STAT3 activation
inhibitors and shRNA should be explored for further effectiveness
with Herceptin treatment.
Acknowledgements
We thank Dr Hemlata Sukhija and Dr May
Ong (Division of Cancer Research and Training, Charles Drew
University) for helpful suggestions. We would like to thank Dr Y.
Elshimali for experimental assistance in tissue microarray
analysis. We also acknowledge all the members of the Division of
Cancer Research and Training for helpful discussions. This work was
supported in full by the grant from NIH/National Cancer Institute
1U54CA14393-01 awarded to J.V.V.
References
|
1.
|
Wu Y, Elshimali Y, Sarkissyan M, Mohamed
H, Clayton S and Vadgama JV: Expression of FOXO1 is associated with
GATA3 and Annexin-1 and predicts disease-free survival in breast
cancer. Am J Cancer Res. 2:104–115. 2012.PubMed/NCBI
|
|
2.
|
Hartman Z, Yang X, Glass O, Lei G, Osada
T, Dave SS, et al: HER2 overexpression elicits proinflammatory IL-6
autocrine signaling loop that is critical for tumorigenesis. Cancer
Res. 71:4380–4391. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Slamon DJ, Clark GM, Wong SG, Levin WJ,
Ulrich A and McGuire WL: Human breast cancer: correlation of
relapse and survival with amplification of the HER2/neu oncogene.
Science. 235:177–182. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Hynes NE and Stern DF: The biology of
erbB-2/neu/HER2 and its role in cancer. Biochim Biophys Acta.
1198:165–184. 1994.PubMed/NCBI
|
|
5.
|
Yarden Y: Biology of HER2 and its
importance in breast cancer. Oncology. 61:1–13. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Pegram MD, Konecny G and Slamon DJ: The
molecular and cellular biology of HER2/neu gene
amplification/overexpression and the clinical development of
Herceptin (trastuzumab) therapy for breast cancer. Cancer Treat
Res. 103:57–75. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Zhang W, Ding W, Chen Y, Feng M, Ouyang Y,
Yu Y and He Z: Up-regulation of breast cancer resistance protein
plays a role in HER2-mediated chemoresistance through PI3K/Akt and
nuclear factor-kappa B signaling pathways in MCF-7 breast cancer
cells. Acta Biochim Biophys Sin (Shanghai). 43:647–653. 2011.
View Article : Google Scholar
|
|
8.
|
Siddiqa A, Long L, Li L, Marciniak R and
Kazhdan I: Expression of HER-2 in MCF-7 breast cancer cells
modulates anti-apoptotic proteins Survivin and Bcl-2 via the
extracellular signal-related kinase (ERK) and phosphoinositide-3
kinase (PI3K) signaling pathways. BMC Cancer. 8:1292008. View Article : Google Scholar
|
|
9.
|
Gemmete JJ and Mukherji SK: Trastuzumab
(herceptin). Am J Neuroradiol. 32:1373–1374. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Harbeck N, Pegram MD, Rüschoff J and Möbus
V: Targeted therapy in metastatic breast cancer: the HER2/neu
oncogene. Breast Care (Basel). 5(Suppl 1): 3–7. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Hubalek M, Brunner C, Matthä K and Marth
C: Resistance to HER2-targeted therapy: mechanisms of trastuzumab
resistance and possible strategies to overcome unresponsiveness to
treatment. Wien Med Wochenschr. 160:506–512. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Cheng GZ, Zhang W, Sun M, Wang Q, Coppola
D, Mansour M, et al: Twist is transcriptionally induced by
activation of STAT3 and mediates STAT3 oncogenic function. J Biol
Chem. 283:14665–14673. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Lin L, Hutzen B, Zuo M, Ball S, Deangelis
S and Foust E: Novel STAT3 phosphorylation inhibitors exhibit
potent growth-suppressive activity in pancreatic and breast cancer
cells. Cancer Res. 70:2445–2454. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Armanious H, Gelebart P, Mackey J, Ma Y
and Lai R: STAT3 upregulates the protein expression and
transcriptional activity of β-catenin in breast cancer. Int J Clin
Exp Pathol. 3:654–664. 2010.
|
|
15.
|
Zhong Z, Wen Z and Darnell J: Stat3: a
STAT3 family member activated by tyrosine phosphorylation in
response to epidermal growth factor and interleukin-6. Science.
264:95–98. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Hart JR, Liao L, Yates J and Vogt P:
Essential role of Stat3 in PI3K-induced oncogenic transformation.
Proc Natl Acad Sci USA. 108:13247–13252. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Yang Z, Cai J, Xie S, Li G, Song W, Yan Q,
Yan L and Zhang F: Therapeutic effects of signal transducer and
activator of transcription 3 siRNA on human breast cancer in
xenograft mice. Chin Med J. 124:1854–1861. 2011.PubMed/NCBI
|
|
18.
|
Yi EH, Lee CS, Lee JK, Lee YJ, Shin MK,
Cho CH, et al: STAT3-RANTES autocrine signaling is essential for
tamoxifen rsistance in human breast cancer cells. Mol Cancer Res.
11:31–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Marotta LC, Almendro V, Marusyk A,
Shipitsin M, Schemme J, Walker SR, et al: The JAK2/STAT3 signaling
pathway is required for growth of CD44+CD24−
stem cell-like breast cancer cells in human tumors. J Clin Invest.
121:2723–2735. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Oliveras-Ferraros C, Vazquez-Martin A,
Martin-Castillo B, Cufi S, Del Barco S, Lopez-Bonet E, et al:
Dynamic emergence of the mesenchymal CD44pos/CD24neg/low phenotype
in HER2-gene amplified breast cancer cells with de novo resistance
to trastuzumab (Herceptin). Biochem Biophys Res Commun. 397:27–33.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Fang X, Cai Y, Liu J, Wang Z, Wu Q, Zhang
Z, et al: Twist2 contributes to breast cancer progression by
promoting an epithelial-mesenchymal transition and cancer stem-like
self-renewal. Oncogene. 30:4707–4720. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Berishaj M, Gao SP, Ahmed S, Leslie K,
Al-Ahmadie H, Gerald W, et al: Stat3 is tyrosine-phosphorylated
through the interleukin-6/glycoprotein 130/Janus kinase pathway in
breast cancer. Breast Cancer Res. 9:R322007. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Schust J, Sperl B, Hollis A, Mayer T and
Berg T: Stattic: a small-molecule inhibitor of STAT3 activation and
dimerization. Chem Biol. 13:1235–1242. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Kong D, Li Y, Wang Z and Sarkar FH: Cancer
stem cells and epithelial-to-mesenchymal transition
(EMT)-phenotypic cells: are they cousins or twins? Cancers (Basel).
3:716–729. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Sato T, Neilson LM, Peck AR, Liu C, Tran
T, Witkiewicz A, et al: Signal transducer and activator of
transcription-3 and breast cancer prognosis. Am J Cancer Res.
1:347–355. 2011.PubMed/NCBI
|
|
26.
|
Aggarwal BB, Sethi G, Ahn KS, Sandur SK,
Panday MK, Kunnunmakkara AB, et al: Targeting
signal-transducer-and-activator-of-transcription-3 for prevention
and therapy of cancer: modern target but ancient solution. Ann NY
Acad Sci. 1091:151–169. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Masuda M, Wakasaki T, Suzui M, Toh S, Joe
AK and Weinstein IB: Stat3 orchestrates tumor development and
progression: the Achilles’ heel of head and neck cancers? Curr
Cancer Drug Targets. 10:117–126. 2010.PubMed/NCBI
|
|
28.
|
Takeda K, Noguchi K, Shi W, et al:
Targeted disruption of the mouse STAT3 gene leads to early
embryonic lethality. Proc Natl Acad Sci USA. 94:3801–3804. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Akira S: Roles of STAT3 defined by
tissue-specific gene targeting. Oncogene. 19:2607–2611. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Ho JN, Kang GY, Lee SS, Kim J, Bae IH,
Hwang SG and Um HD: Bcl-XL and STAT3 mediate malignant actions of
gamma-irradiation in lung cancer cells. Cancer Sci. 101:1417–1423.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Hsu H-S, Lin JH, Hsu TW, Su K, Wang CW,
Yang KY, et al: Mesenchymal stem cells enhance lung cancer
initiation through activation of IL-6/JAK2/STAT3 pathway. Lung
Cancer. 75:167–177. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Floor S, van Staveren WC, Larsimont D,
Dumont JE and Maenhaut C: Cancer cells in epithelial-to-mesenchymal
transition and tumor propagating-cancer stem cells: distinct,
overlapping or same populations. Oncogene. 30:4609–4621. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A and Zhou AY: The epithelial-mesenchymal transition generates
cells with properties of stem cells. Cell. 133:704–715. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Takebe N, Warren RQ and Ivy SP: Breast
cancer growth and metastasis: interplay between cancer stem cells,
embryonic signaling pathways and epithelial-to-mesenchymal
transition. Breast Cancer Res. 13:2112011. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Al-Ejeh F, Smart CE, Morrison BJ,
Chenevix-Trench G, Lopez JA and Lakhani SR: Breast cancer stem
cells: treatment resistance and therapeutic opportunities.
Carcinogenesis. 32:650–658. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Blick T, Hugo H, Widodo E, Waltham M,
Pinto C and Mani SA: Epithelial mesenchymal transition traits in
human breast cancer cell lines parallel the CD44(hi/)CD24(lo/) stem
cell phenotype in human breast cancer. J Mammary Gland Biol
Neoplasia. 15:235–252. 2010. View Article : Google Scholar : PubMed/NCBI
|