Introduction
The importance of the Hedgehog (Hh) signaling
pathway in human ovarian cancer is related to its roles in cell
invasion and differentiation (1),
cellular apoptosis (2) and having
an effect on patient prognosis (1,3). The
Hh pathway is critically important in the maintenance of human
cancer stem cells, in part to the positive transcription factor,
GLI1 (4–6). One of the key phenotypic
characteristics of cancer stem cells is a high level of drug
resistance (1–3,7,8),
which may include resistance to platinum compounds (3,9).
Activator protein 1 (AP1), is the positive
transcriptional regulator for ERCC1 and other genes of nucleotide
excision repair and base excision repair (9,10).
Inhibition of AP1 leads to inhibition of nucleotide excision repair
(11,12) and sensitization of cells to the
anti-cancer agents cisplatin, carboplatin and oxaliplatin (11–14).
GLI1 has an important role in regulating C-JUN (15), which participates with C-FOS in the
formation of AP1 (16). The role
of GLI1 in regulating C-JUN function was first described in
keratinocytes by Laner-Plamberger and colleagues (15). They showed that GLI1 and GLI2
directly regulate the expression of C-JUN by binding to a
GLI-binding site (GBS) in the C-JUN promoter.
We have investigated the potential role of GLI1, as
a determinant of cellular resistance to cisplatin (9). When GLI1 is inhibited by use of an
anti-GLI1 shRNA, the following molecular sequence is observed.
There is downregulation of mRNA and protein of GLI1 and Sonic
Hedgehog, but not GLI2. The C-JUN molecular cascade is switched
from a Ser 63/73 cascade to a Thr 91/93 cascade, the latter of
which is proapoptotic (17). The
normal cisplatin-induced upregulation of DNA repair genes is
blocked, resulting in reduced mRNA and protein levels of ERCC1, XPD
and XRCC1 (9). Cisplatin-DNA
adduct repair is blocked and cells are sensitized to cisplatin by a
factor of six (9).
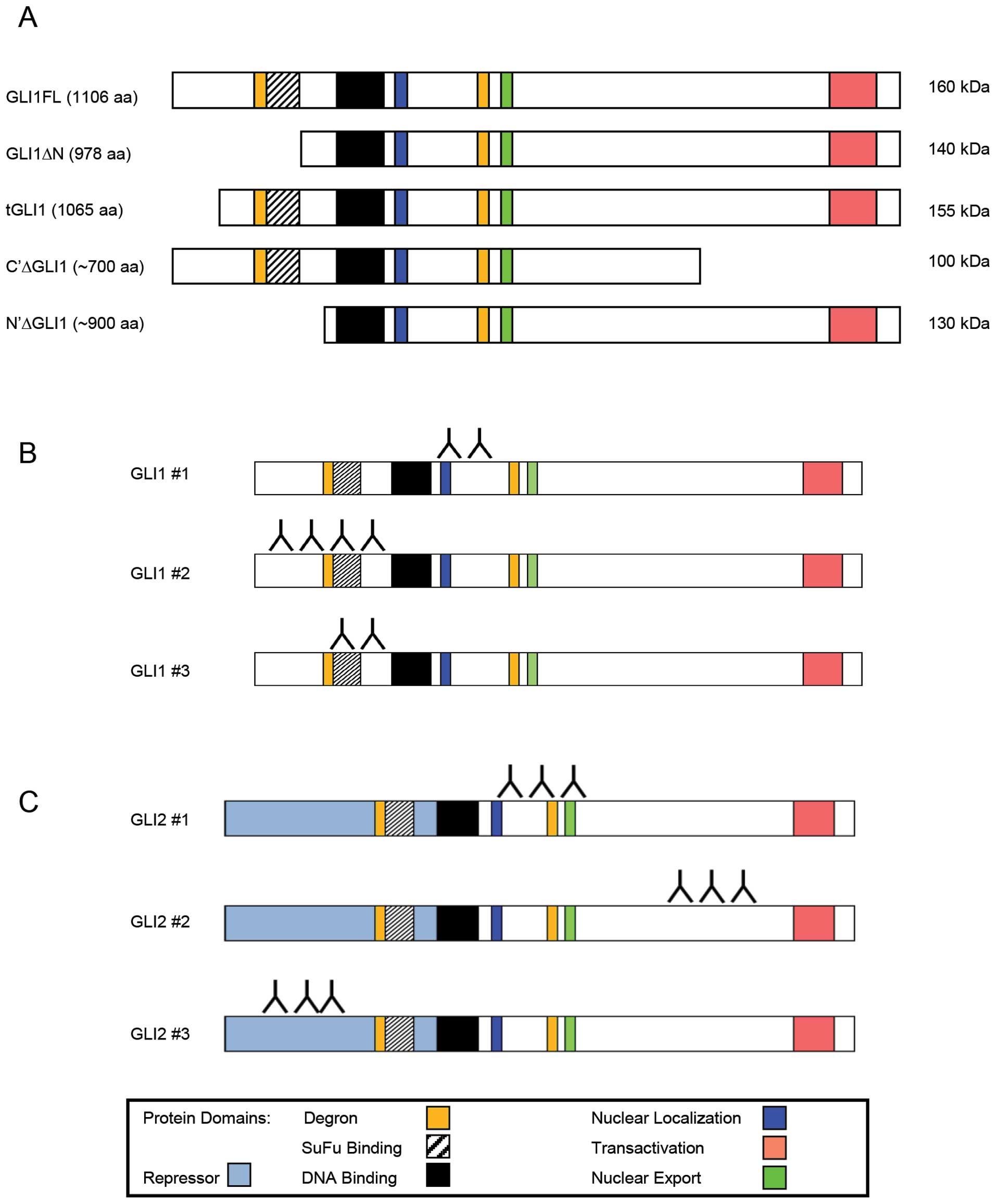
There are five currently known isoforms of the GLI1
protein, shown in Fig. 1A. These
isoforms range from ∼700 to 1106 amino acids in length; and, range
in molecular weight from 100 to 160 kDa. Two are splice variants of
the full-length mRNA. At least one isoform is a post-translational
N-terminal truncation of the full-length protein (18). We sought to investigate which
isoform of GLI1 is responsible for regulating C-JUN, a known
regulator of several genes within nucleotide excision repair and
base excision repair. We present in this report our findings, which
suggest that only one of the five known isoforms of GLI1 binds the
GBS in the promoter of C-JUN.
Materials and methods
Cell culture
Cisplatin-sensitive A2780 human ovarian cancer cells
and cisplatin-resistant A2780-CP70 and A2780-CIS human ovarian
cancer cells were retrieved from frozen stock and used between
passages 5 and 30. RPMI-1640 media (Gibco/Invitrogen, Carlsbad, CA,
USA) was used to culture cells with the following additives: 10%
fetal bovine serum (Gibco), l-glutamine (Gibco), insulin
(Sigma-Aldrich, St. Louis, MO, USA) and penicillin/streptomycin
(Gibco).
Electrophoretic mobility shift assay
(EMSAs)
Nuclear lysates for electrophoretic mobility shift
assays (EMSAs) were prepared from A2780-CP70 cells and the protein
concentration determined as described previously (9). Oligonucleotides containing the C-JUN
promoter GLI-binding-site (GBS) were purchased with and
without a 5′-biotin label: forward, 5′-CTCA ACGTGGGGGGCCGACTCTCG-3′ and
reverse, 5′-CGAGAGTCGGCCCCCCACGTTGAG-3′.
Double-stranded DNA (dsDNA) probes were generated by adding 1
μM of the forward and reverse compliment oligonucleotides in
annealing buffer (10 mM Tris, 1 mM EDTA, 50 mM NaCl, pH 8.0),
heated to 95°C and then cooled at a rate of 1°C/min to room
temperature.
The DNA-binding reaction was carried out using 20
fmol of biotin-labeled dsDNA, 1 μg poly(dI-dC) and 20
μg nuclear lysate protein in a 20 μl volume of
reaction buffer (40 mM HEPES, 25 mM KCl, 10 mM MgCl2, 10
mM ZnSO4, 500 μM EDTA, 10% glycerol, pH 7.8) on
ice for 30 min. In competition experiments, excess unlabeled C-JUN
GBS or unlabeled consensus (forward, 5′-CTCAACGGACCACCCAGACTAT
CG-3′; reverse, 5′-CGATAGTCTGGGTGGTCCGTTGAG-3′) dsDNA were
incubated concurrently at levels 50-, 100- and 200-fold in excess
in the binding reaction. In steric hindrance experiments, GLI1
antibodies (using GLI1 nos. 2 and 3 listed below in western
blotting) and nuclear lysate were incubated on ice for 30 min prior
to the DNA-binding reaction. DNA-protein complexes were separated
on a native polyacryl-amide gel, transferred to a positively
charged PVDF membrane (BrightStar Plus, Ambion, Austin, TX, USA)
and probe binding visualized using the Light Shift Chemiluminescent
EMSA kit (Thermo Pierce, Rockford, IL, USA).
Western and southwestern blotting
Nuclear and whole cell lysates were isolated as
previously described (9). The
laboratory of Dr Rodney Rocconi kindly provided protein lysates
from the human ovarian cancer cell lines: SKOV3, OV-90, ES-2 and
TOV-112D. A random sample of seven human ovarian cancer and three
non-cancer ovary samples were obtained from the Mitchell Cancer
Institute Bio-Bank. Protein was isolated using TRIzol (Invitrogen)
according to the manufacturer’s instructions. Approximately 10
μg of patient samples protein lysate was loaded per lane on
each gel.
The following primary antibodies were used:
α-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), GLI1
[Cell Signaling (Danvers, MA, USA) 2553; R&D (Minneapolis, MN,
USA) AF3455; BioLegend (San Diego, CA, USA) 642401] or GLI2 [Santa
Cruz Biotechnology sc-20291; Santa Cruz Biotechnology sc-28674;
Abcam (Cambridge, MA, USA) ab26506]. The next day, protein bands
were visualized by chemiluminescence (Thermo-Pierce Super Signal
West Dura Extended Duration Substrate) using the following
HRP-secondary antibody: anti-goat (Promega, Madison, WI, USA),
anti-rabbit, or anti-mouse (Cell Signaling). Membrane images were
recorded using a Fuji LAS-3000 Intelligent Darkbox Digital
Imager.
Protein bands were quantitated by densitometry using
the Fuji Image Gauge Software and GLI1 values of each sample were
normalized to α-tubulin. For patient samples, the values were
normalized to an A2780-CP70 protein standard and expressed as
relative ratio to account in differences in each western blotting.
The results were then analyzed by two-sided t-test using GraphPad
Prism software (GraphPad, La Jolla, CA, USA).
For southwestern blotting, a combination of western
and Southern blotting techniques was employed to characterize the
protein-DNA interactions of the C-JUN GBS based on the method of
Cheng et al (19).
Approximately 60 μg of nuclear lysate was loaded per lane
and transferred to a PVDF membrane. Each lane was individually cut
from the membrane and subjected either to western blotting with
GLI1 or GLI2 antibodies, or southwestern blotting with the C-JUN
dsDNA biotin-labeled probe. Proteins were renatured by incubating
the membrane in 5% non-fat milk/TNED buffer (10 mM Tris, 50 mM
NaCl, 0.1 mM EDTA, 1 mM DTT, pH 7.8) and incubated overnight with 5
nM C-JUN GBS in 5% milk/TNED at 4°C. The protein-DNA bands were
visualized using Streptavidin-HRP Conjugates using the Light Shift
Chemiluminescent EMSA kit.
Plasmids, transfections and
immunoprecipitation
Full-length GLI1 cDNA was obtained in plasmid form
as a gift of Bert Vogelstein (Addgene plasmid no. 16419, Cambridge,
MA, USA). GLI1 was MYC-tagged (EQKLISEEDL) on the C-terminal end of
the protein by PCR using the primers: forward,
5′-AAAAAAAAAAGCTTATGTTCAACTCGAT GACCCCA-3′; reverse,
5′-AAAAAAAAAAAGCTTCTACAGATCTTCTTCAGAAATAAGTTTTTGTTCGGCACTAGAG
TTGAGGAATTC-3′. The resulting fragment was digested with
HindIII and cloned into the pLNCX vector (Clonetech,
Mountainview, CA, USA). Verification of the insert and orientation
was done by sequencing.
GLI1-MYC was transfected into A2780-CP70 cells using
FuGENE6 (Roche, Indianapolis, IN, USA). At 24 h post-transfection,
GLI1-MYC transfected cells were lysed in 1% Triton X-100, 50 mM
Tris-HCl pH 7.2, 150 mM NaCl, protease inhibitor cocktail (Sigma)
and PhosSTOP (Roche). Approximately, 1,000 μg GLI1-MYC
protein lysate was immunoprecipitated overnight using Protein A/G
Plus-Agarose beads (Santa Cruz Biotechnology, sc-2003) and 4
μg MYC antibody (Santa Cruz Technology, 9E10). The next day,
the immunoprecipitation was washed and eluted by heating the sample
for 5 min at 95°C in 20 μl of 2X Laemmli sample buffer
(Bio-Rad, Hercules, CA, USA).
Chromatin immunoprecipitation assays
(ChIPs)
Cells were seeded at 1.8×106 in
10-cm2 dishes and transfected with the pLNCX-GLI1-MYC
plasmid the following morning. ChIP assays were performed 24 h
after transfection using the Thermo Pierce Agarose ChIP kit
according to the manufacturer’s instructions. Normal rabbit IgG
antibody was used for mock control (provided). Two antibodies were
used for GLI1: the Cell Signal GLI1 antibody listed in western
methods and a goat polyclonal to GLI1 (C-18, Santa Cruz
Biotechnology) which was previously described by Laner-Plamberger
et al (15). Additionally,
the MYC antibody was used to identify the full-length GLI1 and
130-kDa isoform. Real-time PCR was performed on the isolated DNA
using Fast Sybr Green Master Mix (Applied Biosystems, Foster City,
CA, USA). Primer sequences for amplifying the C-JUN promoter were
described previously (15). Fold
enrichment was determined by first calculating the non-specific
adjustment by subtracting the Ct of the mock from the Ct of each
immunoprecipitation (DDCt). Fold enrichment was then determined
using the equation 2−ΔΔCt. The results shown are from
five independent experiments, with each immunoprecipitation ran in
triplicate for real-time PCR. Results were analyzed by one-way
ANOVA and Tukey’s post test using GraphPad Prism Software.
Results
GLI1, not GLI2, is responsible for
binding the GBS in the C-JUN promoter
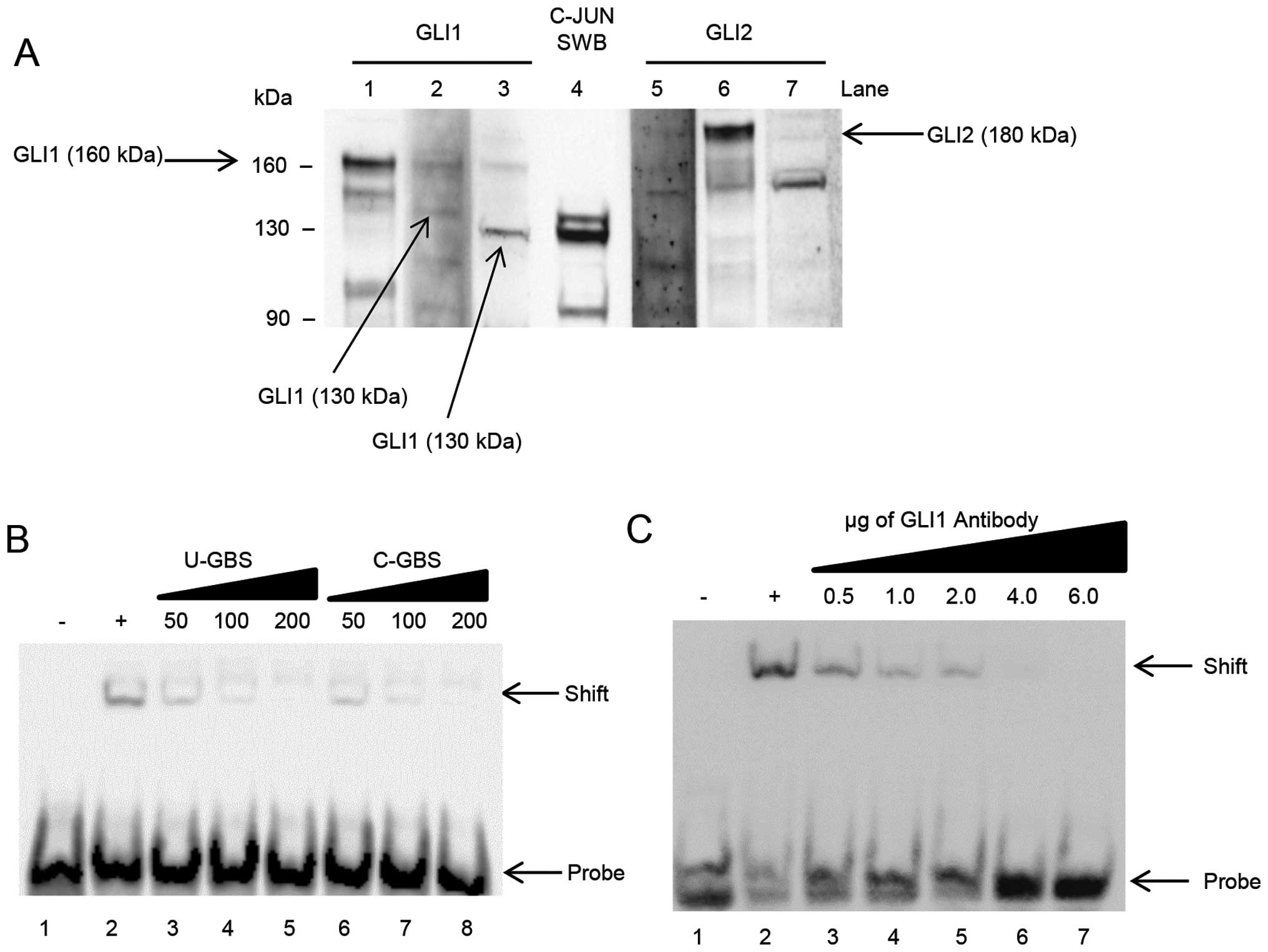
First, we performed simultaneous western and
southwestern blot analyses on A2780-CP70 nuclear lysate, shown in
Fig. 2A. Three different GLI1
antibodies (Fig. 1B) were used,
shown in lanes 1–3, to detect the different isoforms via western
blotting. Additionally, lanes 5–7 are western blot analyses using
three GLI2 antibodies (Fig. 1C).
Southwestern blotting was performed using biotin labeled dsDNA
probe to the GBS found in the C-JUN promoter (lane 4).
The southwestern blotting in lane 4 shows three
bands that clearly bind the GBS: a double band at ∼130 kDa and a
third band at ∼90 kDa. Anti-GLI1 antibody no. 1 (lane 1); did not
recognize a 130-kDa protein in nuclear lysate. The second GLI1
antibody, lane 2, recognizes a band at ∼130 kDa. The western
blotting with the third GLI1 antibody, lane 3, recognizes a band at
130-kDa. The two 130-kDa proteins recognized by the second and
third GLI1 antibodies, correspond to the 130-kDa double band seen
in lane 4. No full-length 160-kDa GLI1 binds the GBS of the C-JUN
promoter in A2780-CP70 cells (lane 4).
Western blot analyses using three GLI2 antibodies
were performed shown in Fig. 2A,
lanes 5–7. None of the three GLI2 antibodies recognized a 130- or
90-kDa molecular weight protein such as those shown in lane 4.
Although GLI2 can be a positive transcriptional regulator for C-JUN
(15), there is no protein band in
lanes 5–7, that correspond to the positive double band at 130-kDa
in lane 4.
Next, we performed experiments to confirm a GLI
protein in A2780-CP70 cells specifically binds the GBS in the C-JUN
promoter. We performed two different EMSA approaches to address
this question. The first EMSA approach was to perform DNA-binding
competition experiments, using unlabeled oligonucleotides to the
GBS in the C-JUN promoter, as well as consensus GBS
oligonucleotides (15). The second
EMSA approach was to sterically inhibit binding of GBS to nuclear
lysate protein, using antibodies to GLI1.
In Fig. 2B, we
demonstrate successful competition for the GBS in A2780-CP70 cells.
We used the GBS from the C-JUN promoter and separately, we used the
consensus GBS sequence. In our negative control, lane 1, no nuclear
lysate was added with the biotin-labeled C-JUN probe. When nuclear
lysate is added we see a band shift, indicating that a GLI protein
binds the GBS in the C-JUN promoter. Increasing concentrations of
unlabeled C-JUN GBS results in decreased band shift signal,
demonstrated in lanes 3–5. As the ratio of unlabeled DNA to labeled
DNA increases, signal is reduced in a stepwise fashion. In lanes
6–8, the increase of unlabeled consensus GBS also successfully
competed for GLI binding. When unlabeled consensus GBS is increased
in a stepwise fashion, the band shift signal is reduced.
In Fig. 2C, we used
antibodies to GLI1, to conduct competition experiments to block
binding of the DNA probe to nuclear lysate protein. In these
experiments, we used a combination of GLI1 antibodies nos. 2 and 3
which corresponds to the doublet bands seen in the southwestern
blotting in Fig. 2A. These
antibodies are directed to the N-terminal region of the protein, in
the area where the GBS is expected to bind (Fig. 1B). Once bound, the antibodies
should occupy the protein and thereby inhibit the binding of GLI1
to the GBS of the C-JUN promoter. In lanes 3–7 the nuclear lysates
were pre-incubated with increasing amounts of GLI1 antibodies. As
shown in Fig. 2C, when increasing
amounts of GLI1 antibody are added, the ability to bind the C-JUN
GBS decreases in a stepwise fashion indicating competitive blockage
of the DNA-binding domain of GLI1. This suggests GLI1 binds the
C-JUN GBS in A2780-CP70 cells.
The 130-kDa isoform is produced from
full-length GLI1 and binds the C-JUN promoter
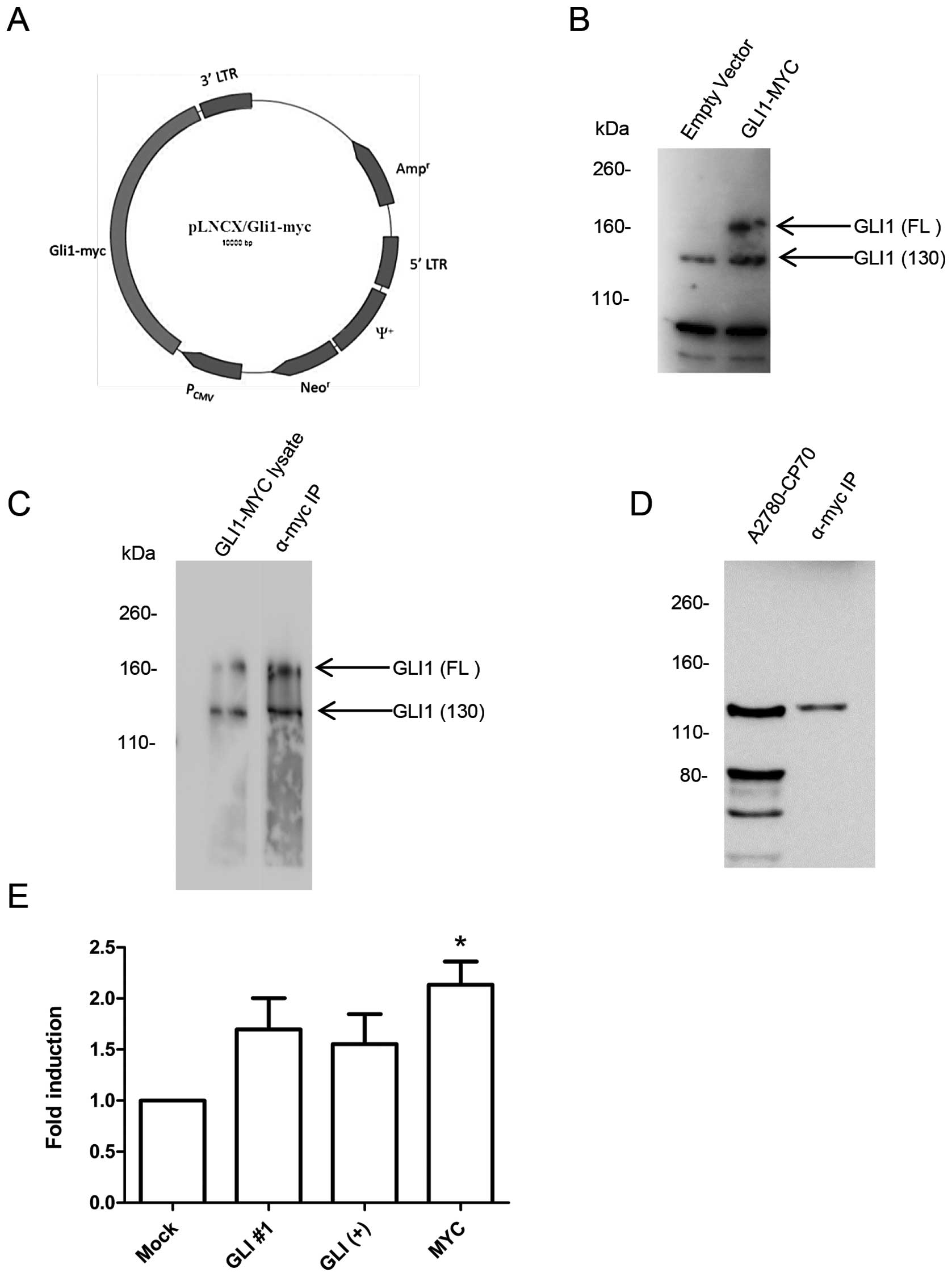
We generated an expression plasmid containing the
full-length GLI1 cDNA fused with a C-terminal MYC tag by PCR, shown
in Fig. 3A. Transfection of
A2780-CP70 cells with the C-terminal MYC tagged GLI1 overexpress
full-length GLI1 and the 130-kDa isoform, as compared with empty
vector transfected cells, shown in Fig. 3B.
Using protein lysate from transfected cells,
immunoprecipitation was performed using the MYC tag antibody.
Fig. 3C shows the western blotting
comparing whole-cell lysate transfected with GLI1-MYC and the
lysate immunoprecipitated with a MYC antibody. The
immunoprecipitation of the transfected lysate shows both the
full-length and the 130-kDa isoforms of GLI1 were isolated using
the MYC antibody. The 130-kDa protein is produced by N-terminal
cleavage of the full-length GLI1 (18).
The southwestern blotting was repeated using the MYC
immunoprecipitation of transfected cells. Shown in Fig. 3D, no full-length GLI1,
corresponding to the 160-kDa protein, was observed to bind the
C-JUN GBS. However, a band corresponding to the 130-kDa isoform of
GLI1 observed in the nuclear lysate was similarly observed in the
MYC immunoprecipitation. This suggests that only the 130-kDa GLI1
isoform binds the GBS of the C-JUN promoter.
The in vivo binding of the GLI1 isoform was
assessed by performing ChIP assays. Cells were transfected with
GLI1-MYC cDNA and three antibodies were used to immunoprecipitate
GLI1. The first two antibodies were specific to GLI1 and they
recognized both endogenous and transfected GLI1 and GLI1 isoforms.
The third antibody was the MYC antibody, which only recognized the
transfected GLI1-MYC and the 130-kDa GLI1 isoform but does not
recognize any endogenous GLI1 isoforms. The ChIP results are shown
in Fig. 3E.
The ChIP using the GLI1 antibody no. 1, which
recognized both endogenous and transfected GLI1 and isoforms, had a
fold induction of 1.69 over the mock control. The second GLI1
antibody was used as a positive control based on a previous ChIP
demonstrating GLI1 binds the C-JUN promoter (15). The ChIP with the GLI1+
antibody, which recognized both the endogenous and transfected GLI1
isoforms, had a fold induction of 1.55. The anti-MYC antibody only
recognized the transfected GLI1-MYC and the 130-kDa isoform as
shown previously in Fig. 3C. The
MYC ChIP had the highest fold induction of 2.13 and was
statistically significant (P<0.05). Taken together with the
results from the southwestern blot analyses, the data strongly
suggest that only the 130-kDa isoform recognizes the C-JUN GBS.
The 130-kDa GLI1 isoform is expressed at
higher levels in ovarian cancer
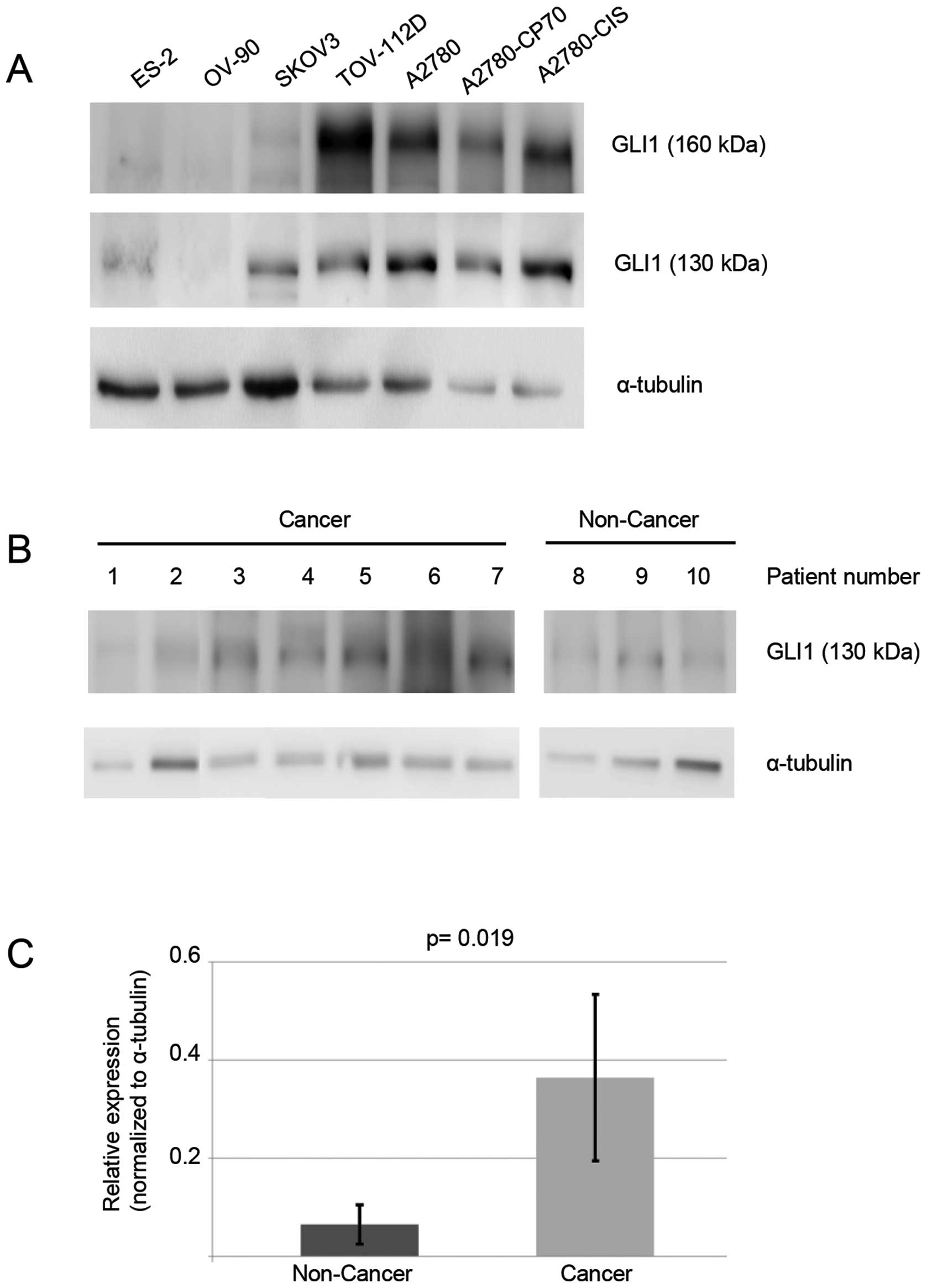
In Fig. 4A, six
additional human ovarian cancer cell lines were studied for the
presence of the 130-kDa GLI1 protein: ES-2, OV-90, SKOV-3,
TOV-112D, A2780, A2780-CP70 and A2780-CIS. The cisplatin-resistant
cell lines, A2780-CP70 and A2780-CIS, express a higher level of the
130-kDa GLI1 isoform when compared to the parental
cisplatin-sensitive A2780 cells. A2780-CP70 cells have a 1.6-fold
higher and A2780-CIS a 2.7-fold higher protein level of the 130-kDa
GLI1 isoform.
In Fig. 4B, we
performed western blot analyses on ten ovary tissue samples from
ten separate patients. Seven were cancer specimens, three were
non-cancer specimens. The 130-kDa GLI1 isoform was quantitated and
non-cancer ovarian samples had an average signal of 0.066. Ovarian
cancer samples had an average signal of 0.365, a 6-fold difference
(Fig. 4C, P=0.019). Thus, the
130-kDa GLI1 isoform is more highly expressed in ovarian cancer, as
opposed to non-cancer ovarian tissues.
Discussion
The Hh pathway has three groups of transcriptional
regulatory proteins: GLI1, GLI2 and GLI3 (4–6,20,21).
GLI1 appears to have at least five different isoforms, GLI2 has at
least four isoforms and GLI3 has at least five isoforms (20). The transcriptional activity of each
of these depends on the specific isoform in question, the specific
tissue in question and the tissue context (4–6).
Recently, there is a growing interest in the specific functions of
the GLI1 isoforms (22,23). GLI1 has many different functions,
however, many of these functions are not ascribed to a particular
isoform. Stecca and Ruiz i Altaba investigated the 130-kDa isoform
of GLI1 in neural stem cells (18). They reported that the 130-kDa
isoform is always expressed as a doublet, which are the
phosphorylated and unphosphorylated forms of the same protein. In
an analysis of a panel of tumor cell lines, the most abundant
isoform was the 130-kDa protein and that the relative abundance of
three different GLI1 isoforms was: GLI1-130kDa > GLI1-100kDa
> GLI1 full length-160-kDa.
The interface between GLI1 and C-JUN has been
recognized by several groups (9,15,24).
Since GLI1 binds to C-JUN, this suggests that the GLI1 nexus with
C-JUN may actually be a GLI1 nexus the AP1 heterodimer and all
downstream targets of AP1 and of C-JUN. This strongly suggests that
only one of the five known isoforms of GLI1, the 130-kDa isoform,
has a role in the modulation of ERCC1 and potentially other genes
of nucleotide excision repair (9,10,12,14).
A strong link between GLI1 and the regulation of DNA
damage has been reported by Agyeman and colleagues (25), Leonard et al (26) and by our group (9). Our study has specifically focused on
platinum-based anticancer chemotherapy; and on genes in nucleotide
excision repair and base excision repair pathways. The study by
Agyeman and colleagues suggest the possibility, that the GLI1
effects on DNA repair response may in fact be more broad than these
two specific DNA repair pathways.
The 130-kDa isoform is expressed at higher levels in
ovarian cancer specimens than in non-cancer ovarian specimens,
suggesting the possibility of the importance of this isoform in the
malignancy. We have reported the role of GLI1 as a factor in
cellular resistance to cisplatin (9). The data presented here, suggest that
the 130-kDa GLI1 isoform is a determinant of cellular resistance to
cisplatin. Thus, this specific protein may possibly be a reasonable
target for reversing resistance to platinum chemotherapy drugs.
Acknowledgements
This study was supported by Intramural
Research Program of the NIH, National Institute on Minority Health
and Health Disparities. We thank Wesley Denny for his helpful
discussions and technical assistance.
References
|
1.
|
Liao X, Siu MK, Au CW, et al: Aberrant
activation of hedgehog signaling pathway in ovarian cancers: effect
on prognosis, cell invasion and differentiation. Carcinogenesis.
30:131–140. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Chen X, Horiuchi A, Kikuchi N, et al:
Hedgehog signal pathway is activated in ovarian carcinomas,
correlating with cell proliferation: it’s inhibition leads to
growth suppression and apoptosis. Cancer Sci. 98:68–76.
2007.PubMed/NCBI
|
|
3.
|
Ray A, Meng E, Reed E, Shevde LA and
Rocconi RP: Hedgehog signaling pathway regulates the growth of
ovarian cancer spheroid forming cells. Int J Oncol. 39:797–804.
2011.PubMed/NCBI
|
|
4.
|
Mas C and Ruiz i Altaba A: Small molecule
modulation of HH-GLI signaling: current leads, trials and
tribulations. Biochem Pharmacol. 80:712–723. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Stecca B and Ruiz IAA: Context-dependent
regulation of the GLI code in cancer by HEDGEHOG and non-HEDGEHOG
signals. J Mol Cell Biol. 2:84–95. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Zhu H and Lo HW: The human
glioma-associated oncogene homolog 1 (GLI1) family of transcription
factors in gene regulation and diseases. Curr Genomics. 11:238–245.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Mine T, Matsueda S, Gao H, et al: Created
Gli-1 duplex short-RNA (i-Gli-RNA) eliminates CD44 Hi progenitors
of taxol-resistant ovarian cancer cells. Oncol Rep. 23:1537–1543.
2010.PubMed/NCBI
|
|
8.
|
Steg AD, Bevis KS, Katre AA, et al: Stem
cell pathways contribute to clinical chemoresistance in ovarian
cancer. Clin Cancer Res. 18:869–881. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Kudo K, Gavin E, Das S, Amable L, Shevde
LA and Reed E: Inhibition of Gli1 results in altered c-Jun
activation, inhibition of cisplatin-induced upregulation of ERCC1,
XPD and XRCC1 and inhibition of platinum-DNA adduct repair.
Oncogene. 31:4718–4724. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Zhong X, Thornton K and Reed E: Computer
based analyses of the 5′-flanking regions of selected genes
involved in the nucleotide excision repair complex. Int J Oncol.
17:375–380. 2000.
|
|
11.
|
Bonovich M, Olive M, Reed E, O’Connell B
and Vinson C: Adenoviral delivery of A-FOS, an AP-1 dominant
negative, selectively inhibits drug resistance in two human cancer
cell lines. Cancer Gene Ther. 9:62–70. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Li Q, Tsang B, Bostick-Bruton F and Reed
E: Modulation of excision repair cross complementation group 1
(ERCC-1) mRNA expression by pharmacological agents in human ovarian
carcinoma cells. Biochem Pharmacol. 57:347–353. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Reed E: ERCC1 measurements in clinical
oncology. N Engl J Med. 355:1054–1055. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Reed E: DNA damage and repair in
translational oncology: an overview. Clin Cancer Res. 16:4511–4516.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Laner-Plamberger S, Kaser A, Paulischta M,
Hauser-Kronberger C, Eichberger T and Frischauf AM: Cooperation
between GLI and JUN enhances transcription of JUN and selected GLI
target genes. Oncogene. 28:1639–1651. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Li Q, Gardner K, Zhang L, Tsang B,
Bostick-Bruton F and Reed E: Cisplatin induction of ERCC-1 mRNA
expression in A2780/CP70 human ovarian cancer cells. J Biol Chem.
273:23419–23425. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Raivich G: c-Jun expression, activation
and function in neural cell death, inflammation and repair. J
Neurochem. 107:898–906. 2008.PubMed/NCBI
|
|
18.
|
Stecca B and Ruiz i Altaba A: A GLI1-p53
inhibitory loop controls neural stem cell and tumour cell numbers.
EMBO J. 28:663–676. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Cheng CK, Yeung CM, Hoo RL, Chow BK and
Leung PC: Oct-1 is involved in the transcriptional repression of
the gonadotropin-releasing hormone receptor gene. Endocrinology.
143:4693–4701. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Ruiz i Altaba A: Gli proteins encode
context-dependent positive and negative functions: implications for
development and disease. Development. 126:3205–3216.
1999.PubMed/NCBI
|
|
21.
|
Ruiz i Altaba A: Hedgehog signaling and
the Gli code in stem cells, cancer and metastases. Sci Signal.
4:pt92011.PubMed/NCBI
|
|
22.
|
Carpenter RL and Lo HW: Identification,
functional characterization and pathobiological significance of
GLI1 isoforms in human cancers. Vitam Horm. 88:115–140. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Carpenter RL and Lo HW: Hedgehog pathway
and GLI1 isoforms in human cancer. Discov Med. 13:105–113.
2012.PubMed/NCBI
|
|
24.
|
Lo HW, Zhu H, Cao X, Aldrich A and
Ali-Osman F: A novel splice variant of GLI1 that promotes
glioblastoma cell migration and invasion. Cancer Res. 69:6790–6798.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Agyeman A, Mazumdar T and Houghton JA:
Regulation of DNA damage following termination of Hedgehog (HH)
survival signaling at the level of the GLI genes in human colon
cancer. Oncotarget. 3:854–868. 2012.PubMed/NCBI
|
|
26.
|
Leonard JM, Ye H, Wetmore C and Karnitz
LM: Sonic Hedgehog signaling impairs ionizing radiation-induced
checkpoint activation and induces genomic instability. J Cell Biol.
183:385–391. 2008. View Article : Google Scholar : PubMed/NCBI
|