Introduction
After injury, cell migration and proliferation are
important mechanisms of tissue repair to restore airway and
alveolar epithelia in several respiratory diseases (1–4).
However, uncontrolled cell growth, dysregulation of cell motility
and acquisition of an invasive phenotype are some of the hallmarks
of cancer development and propagation (5,6). In
fact, cell proliferation and motility are regulated by many actors,
including growth factors and down-stream signalling effectors.
However, a growing body of evidence demonstrates that K+
channels also participate in the control of cell motility and
growth of various cell types (7–13).
Indeed, inhibiting or silencing of voltage-dependent (Kv),
calcium-activated (KCa), inward-rectifying (Kir) and 2-pore (K2P)
channels reduces the proliferation of numerous normal and cancer
cells in vitro (14–22)
and animal models (20,23,24).
The mechanisms underlying the regulation of cellular proliferation
by K+ channels may be manifold, but evidence points to
cell cycle control (10,25–27).
In addition, K+ channels could also be involved in tumor
cell invasion and metastasis propagation via regulation of cell
migration (11,14,28–31).
The role of K+ channels in lung cancer
has not been studied extensively. It has been shown that the
inhibition of Girk channels (21)
in small-cell lung cancer (SCLC) cells and Kv1.3 channels in
non-small-cell lung cancer (NSCLC) A549 cells (20) has anti-proliferative effects. We
previously reported the involvement of different types of
K+ channels, including a member of the Kv channel
subfamily, KvLQT1, in the control of normal alveolar and bronchial
epithelial cell proliferation, migration and epithelial repair
(18,19). However, the participation of this
channel in lung tumor cells has not been reported. Growing interest
in K+ channels as therapeutic targets in cancer is not
only due to their role in cell growth and motility, but also
because they have been found to be overexpressed in various tumor
tissues, including in lung cancer (16,23,25,31–36).
However, to the best of our knowledge, possible KvLQT1
overexpression in lung adenocarcinoma (AD) has never been
observed.
In the present study, we first examined the mRNA
expression of 3 Kv channels; KvLQT1, EAG1 and ERG1. The impact of
pharmacological inhibition of these channels, with various drugs,
was then evaluated in scratch assays. Because chromanol and
clofilium reduced wound-healing rates more efficiently than
astemizole and ergtoxin, we verified the involvement of KvLQT1 via
a molecular approach with specific siRNA. The impact of KvLQT1
inhibitors on A549 cell motility and proliferation mechanisms was
then assessed. KvLQT1 was also investigated in wound healing,
migration and growth of another lung cancer cell line: H460.
Finally, KvLQT1 expression levels were measured in tumor tissues
from patients with lung AD.
Materials and methods
Lung tissue samples
Our protocol on human tissues has been approved by
the Ethics Committee of our Institution (Comité d’éthique à la
recherche du CHUM). All participants provided their written
informed consent to participate in this study and all clinical
investigation has been conducted according to the principles
expressed in the Declaration of Helsinki. Tumor and matched,
adjacent, non-neoplastic lung tissues were sourced from 26 patients
with lung adenocarcinoma (AD), the most common subtype of NSCLC.
Tissues were collected immediately after surgical resection at the
CHUM Hôpital Notre-Dame (Montréal, Québec, Canada) or obtained from
the tissue bank of the Respiratory Health Network of the FRQS.
Tissues were dissected from surgical samples by a pathologist, who
confirmed the diagnosis of primary lung cancer and established
clinical and histological parameters. The different growth patterns
were quantified using a 5% increment by a thoracic pathologist, who
classified the AD tumor samples based on the latest IASLC
classification (37), into the
following sub-categories: i) AD in situ (1 patient), ii)
lepidic-predominant AD [4 patients, including formely non-mucinous
bronchioloalveolar carcinoma (BAC) class], iii) acinar-predominant
AD (8 patients), iv) papillary-predominant AD (3 patients), v)
micropapillary-predominant AD (2 patients) and vi)
solid-predominant AD (4 patients) as well as vii) invasive mucinous
AD (4 patients, formely mucinous BAC). As reported in Table I, among the 26 patients included in
our study, 46% were men and 54% were women, with a median age of 66
years (range 39–77 years); 76% had a smoking history. No patients
in our cohort had metastasis at the time of lung resection.
 | Table I.KvLQT1 expression ratio [in tumor vs
non-tumor (non-neoplastic) tissue], with individual demographic and
histopathological characteristics for the 26 patients included in
the study. |
Table I.
KvLQT1 expression ratio [in tumor vs
non-tumor (non-neoplastic) tissue], with individual demographic and
histopathological characteristics for the 26 patients included in
the study.
| Patient no. | Sex | Age | Smoking
history | AD histological
subtype | KvLQT1 ratio level
(tumor/non-tumor) |
|---|
| 1 | F | 62 | No | AD in
situ | 2.0 |
| 2 | M | 67 | No | Lepidic predominant
(70%) and papillary (30%) patterns | 5.0 |
| 3 | M | 43 | Yes | Lepidic predominant
(95%) and acinar (5%) patterns | 0.3 |
| 4 | F | 66 | Yes | Lepidic predominant
(75%) and acinar (25%) patterns | 1.1 |
| 5 | F | 71 | No | Lepidic predominant
(80%), acinar (10%) and papillary (10%) and papillary (10%)
patterns | 1.9 |
| 6 | M | 72 | No | Acinar predominant
(60%), micropapillary (30%) and lepidic (10%) patterns | 1.4 |
| 7 | F | 77 | Yes | Acinar predominant
(55%) and lepidic (45%) patterns | 1.3 |
| 8 | F | 77 | No | Acinar predominant
(40%), lepidic (20%), solid (20%) and papillary (10%) patterns | 0.7 |
| 9 | M | 73 | Yes | Acinar predominant
(90%) and micropapillary (10%) patterns | 1.7 |
| 10 | M | 59 | Yes | Acinar predominant
(75%), lepidic (20%) and solid (5%) patterns | 2.8 |
| 11 | F | 71 | Yes | Acinar predominant
(80%) and papillary (20%) patterns | 1.6 |
| 12 | F | 52 | Yes | Acinar predominant
(40%), micropapillary (30%), solid (20%) and papillary (10%)
patterns | 3.4 |
| 13 | F | 76 | Yes | Acinar predominant
(70%) and lepidic (30%) patterns | 1.1 |
| 14 | M | 40 | Yes | Papillary
predominant (60%) and lepidic (40%) patterns | 3.6 |
| 15 | M | 62 | Yes | Papillary
predominant (60%), lepidic (30%) and micropapillary (10%)
patterns | 4.5 |
| 16 | M | 39 | ? | Papillary
predominant (80%), acinar (10%) and lepidic (10%) patterns | 6.0 |
| 17 | F | 72 | Yes | Micropapillary
predominant (70%) and lepidic (30%) patterns | 3.0 |
| 18 | F | 73 | Yes | Micropapillary
predominant (100%) | 1.6 |
| 19 | M | 60 | Yes | Solid predominant
(90%) and acinar (10%) patterns and pleomorphic features | 0.9 |
| 20 | F | 44 | Yes | Solid predominant
pattern (100%) | 3.3 |
| 21 | M | 71 | Yes | Solid (70%)
predominant, micropapillary (20%) and acinar (10%) patterns | 2.9 |
| 22 | M | 50 | Yes | Solid predominant
(70%) and acinar (30%) patterns with clear cells and cribriform
(30%) features | 2.4 |
| 23 | F | 66 | No | Invasive mucinous
AD | 1.1 |
| 24 | F | 63 | Yes | Invasive mucinous
AD | 1.5 |
| 25 | F | 74 | Yes | Invasive mucinous
AD | 6.8 |
| 26 | M | 61 | Yes | Invasive mucinous
AD | 0.5 |
Cell culture
A549 and H460 lung cancer cells were purchased from
the ATCC and maintained in culture medium (A549: DMEM, Gibco,
Invitrogen, Burlington, ON, Canada; H460: RPMI, Gibco, Invitrogen)
supplemented with 10% of fetal bovine serum (FBS, Gibco,
Invitrogen), 2 mM L-glutamine (Gibco, Invitrogen) and 50 U/ml
penicillin and 50 μg/ml streptomycin (Gibco,
Invitrogen).
Wound-healing assays
The commonly-employed ‘wound-healing assay’, which
gives an estimation of the capacity of cells to migrate and
proliferate (Fig. 1A) after
mechanical injury, was used. Briefly, A549 and H460 cell monolayers
were injured mechanically with a pipette tip (6 wounds/Petri dish),
then washed to remove detached/injured cells, before photography
with a Nikon camera under light microscopy (×4 enlargement). Marks
on the Petri dishes allowed us to photograph the wounds exactly at
the same place at various times, with measurement of the wound
areas initially and after repair, by ImageJ software (National
Institutes of Health), as described previously (38,39).
The results, reported as the rates of wound closure in
μm2/h, are compared to the controls and in the
presence of K+ channel inhibitors, i.e., clofilium
(Sigma-Aldrich, Oakville, ON, Canada), chromanol (Tocris, Bristol,
UK), ergtoxin (Alomone Labs Ltd., Jerusalem, Israel) and astemizole
(Tocris). Wound-healing rates were also evaluated in the presence
of negative controls or KvLQT1 siRNAs (Invitrogen). For Ki67
staining during wound healing (Fig.
1A), confluent A549 cell monolayers, cultured on glass slides
in Flexiperm (Dako Denmark, Glostrup, Denmark), were fixed in cold
methanol at time 0 after injury and after 18 h of repair before
staining with anti-Ki67 antibody (Dako), then Alexa Fluor 488 goat
anti-mouse secondary antibody (proliferating cells stained in
green) and finally counterstaining with DAPI (staining of nuclei in
blue, Sigma).
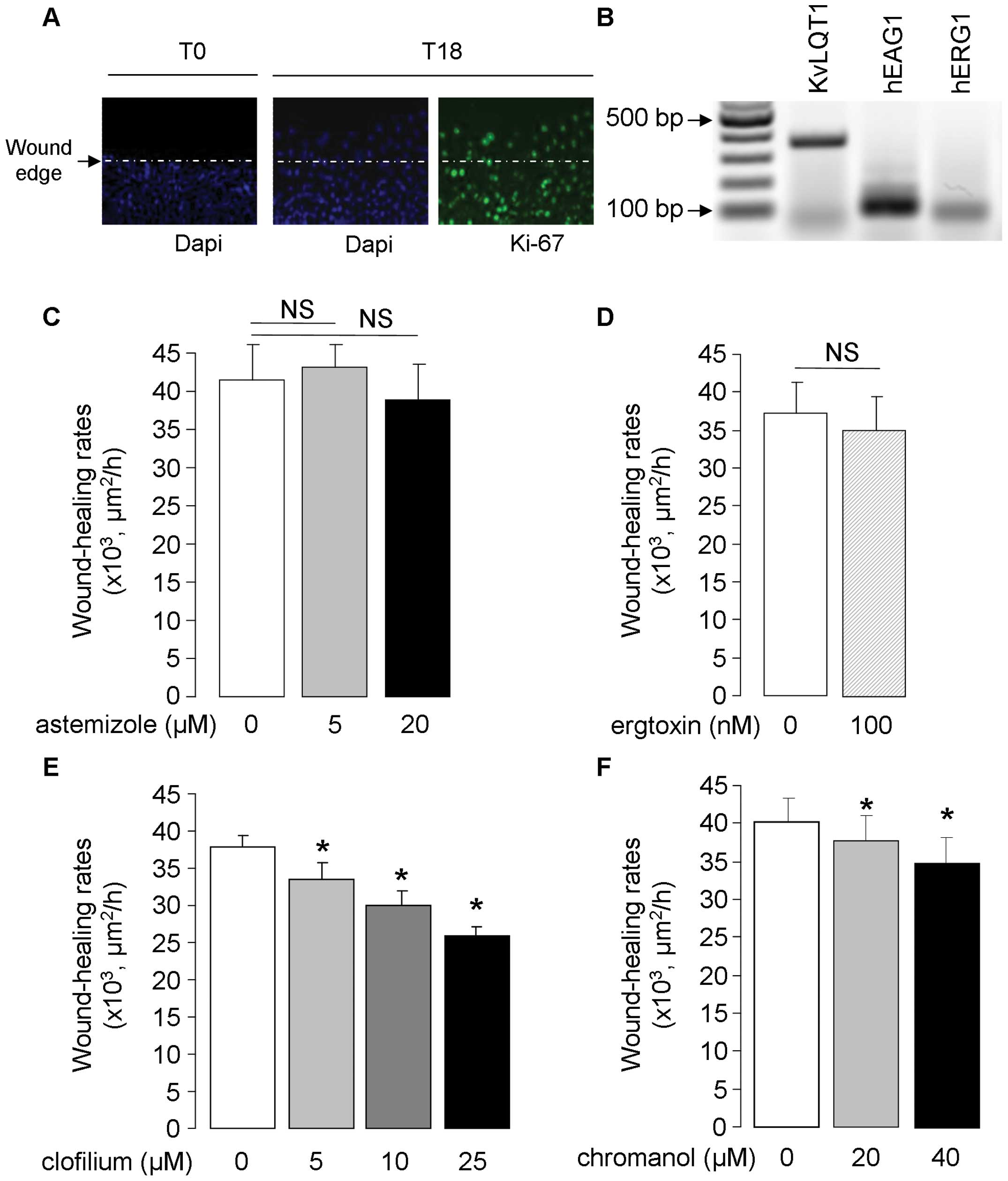 | Figure 1.Detection of Kv K+
channels and impact of their inhibition on wound healing in A549
cell monolayers. (A) Representative photograph (at ×20
magnification) showing cell migration as well as active cell
proliferation, indicated by Ki67 staining (green), at the wound
edge of a repairing A549 cell monolayers. (B) Representative
agarose gel showing PCR products amplified from A549 cell cDNA with
PCR primer pairs designed from human KvLQT1, EAG1 and ERG1
K+ channels. A549 cell monolayers were injured
mechanically and wound healing was followed over an 18-h period.
Wound-closure rates (μm2/h) were compared in the
control condition (0) and in the presence of astemizole [5 and 20
μM, n=5; (C)], ergtoxin [100 nM, n=5; (D)], clofilium [5, 10
or 25 μM, n=5; (E)] and chromanol [20 or 40 μM, n=8);
(F)]. *p<0.05. |
Video-microscopy time-lapse
experiments
The 2D cell migration rates (μm/h) of
non-confluent A549 cells were evaluated by single-cell tracking
with Axiovision software (Carl Zeiss, Toronto, ON, Canada) on
sequence images captured at 5-min intervals over an 18-h period by
digital camera connected to a Carl Zeiss microscope (Carl Zeiss,
×10 enlargement).
Cell migration in Boyden-type
chamber
The 3D migratory capacities of A549 and H460 cells
were evaluated by Boyden-type chamber migration assay (18,19).
Briefly, cells obtained after trypsinization were counted, and cell
viability was verified by trypan blue assay. The cell suspensions
were then placed in the upper compartment of 8-μm pore
filters (0.33 cm2, ThinCerts-TC inserts, Greiner
Bio-one; MJS Biolynx, Brockville, ON, Canada) coated on the lower
side with a collagen matrix. After cell migration, the filters were
washed with PBS, and the cells were fixed with
paraformaldehyde-acetone solution, then stained with hematoxylin.
Non-migrating cells of the upper compartment were scrapped-off with
cotton-tipped applicators (Fisher, Napean, ON, Canada), whereas
migrating cells that passed through the lower face of the filters
were counted in 5 different randomly-chosen fields by a
light-inverted microscope at ×20 magnification (18,19).
Cell proliferation and cell cycle
analyses
A549 and H460 cell growth was evaluated by counting
cell number over a 72-h period. Briefly, the cells were cultured at
low density (15,000/cm2) in 35-mm Petri dishes for 24 h
in the absence of inhibitor. At this time (T0), they were exposed
or not to K+ channel inhibitors for 24–72 h, then
separated with trypsin-EDTA (0.05%, Gibco, Invitrogen) at times 0,
24, 48 and 72 h, before counting by hemacytometer. The absence of
drug cytotoxicity was verified by trypan-blue exclusion assay. At
the end of growth assays, A549 cells were fixed in ethanol (70%)
for cell cycle analysis. After centrifugation, they were
re-suspended in PBS supplemented with pancreatic RNase A (Roche,
Mannheim, Germany). After 30 min, propidium iodide solution (1
mg/ml, Sigma-Aldrich) was added to each sample, and flow cytometry
performed with a flow Coulter EpicsXL cytometer (Beckman Coulter,
Inc., Mississauga, ON, Canada). Data were recorded for at least
10,000 events, and signals were analyzed by Expo32 software
(Applied Cytometry Systems, Sheffield, UK).
KvLQT1 channel silencing with siRNAs
As described previously (40), A549 cells (40–50% confluence) were
exposed to control siRNAs (Stealth siRNA-negative control,
12935-400, 100 pmol, Invitrogen) or siRNA directed against KvLQT1
(Stealth™ siRNA duplex oligonucleotides; Kcnq1-HSS142716,
Invitrogen, 100 pmol), mixed with Lipofectamine™ RNAiMax
transfection reagent in OptiMEM (Invitrogen). Fresh, complete DMEM
was added onto cells after 5-h exposure to siRNAs, and then
replaced every 24 h for 72–96 h. Transfection efficiency >90%
was observed under these experimental conditions (40).
Reverse transcription-polymerase chain
reaction (RT-PCR) amplification
Total RNA purified from A549 cells with TRIzol
reagent (Invitrogen) was reverse-transcribed with MMLV reverse
transcriptase (Invitrogen) in the presence of oligodT primers.
cDNAs were amplified with Taq polymerase (Invitrogen), and specific
primers designed from sequences of human KvLQT1 [Kv7.1 (41), NM_000218, 5′-taaggaagagccc
aacactg-3′, 5′-cgatccttgctcttttctga-3′, 1 μM, 355-bp
product], human ether-a-go-go 1 (hEAG1, Kv10.1, NM_002238.3, 5′-gag
aacgtggatgagggcat-3′, 5′-gtagtggtccagcttacggg-3′, 1 μM,
96-bp product), human ether-a-go-go-related gene (hERG1, Kv11.1,
NM_000238.3, NM_172056.2, 5′-ggccagagccgtaagttcat-3′,
5′-aagccgtcgttgcagtagat-3′, 1 μM, 74-bp product) and β-actin
(hβ-actin, 5′-agagctacgagctgcctgac-3′, 5′-aaagccatgccaatctcatc-3′,
0.25 μM, 499-bp product), for normalization. Primer pairs
were designed in distinct exons to avoid genomic DNA amplification.
KvLQT1, EAG and ERG products were amplified for 30 cycles, while
β-actin amplification was stopped after 18 cycles. RT-PCR products
were finally separated on 1% agarose gel and stained with
SYBR® Safe (Invitrogen). Signals were detected by
Typhoon Gel Imager and analyzed by ImageQuant software (Molecular
Dynamics, Baie d’Urfe, QC, Canada). KvLQT1 silencing with siRNA was
also verified by RT-PCR (40).
Immunoblotting
Immunoblotting was undertaken according to a
well-established laboratory protocol (18,40).
Total A549 proteins were solubilized in lysis buffer [150 mM NaCl,
50 mM Tris-HCl, pH 7.6, 1% Triton X-100, 0.1% SDS, protease
inhibitor cocktail (Complete Mini EDTA-free protease inhibitor
cocktail, Roche)] on ice for 30 min. Proteins from lung tissues
were extracted by homogenization, with a glass putter for 2 min in
lysis buffer (150 mM NaCl, 25 mM Tris-HCl, 1 mM EDTA, leupeptin,
aprotinin, PMSF protease and orthovana-date phosphatase
inhibitors). Cell lysates were centrifuged, supernatants collected,
and protein concentration measured by Bradford assay. Proteins were
separated by SDS-PAGE and transferred onto nitrocellulose
membranes. After blocking, the membranes were incubated with
anti-KvLQT1 (sc-10645, Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA) and anti-β-actin antibodies (CLT9001, Cedarlane Laboratory
Ltd., Burlington, ON, Canada) on the same blot, to ensure
equivalent loading. KvLQT1 antibody specificity was verified with
its blocking peptide [sc10645P, Santa Cruz, (18)]. After washing, the membranes were
incubated with donkey anti-goat (for KvLQT1, Santa Cruz
Biotechnology, Inc.) and horse anti-mouse (for β-actin; Cell
Signaling Technology, Boston, MA, USA) IgG linked to horseradish
peroxidase for 1 h. The intensity of each specific band was
quantified with ChemiDoc XRS+ Molecular Imager and Image Lab
software (Bio-Rad, Mississauga, ON, Canada), then normalized to the
β-actin signal.
Statistical analyses
The data are presented as mean ± standard error of
the mean (SEM) and were compared with Statview software (SAS
Institute, Cary, NC, USA). Statistical analyses were made using
paired Student’s t-test, Wilcoxon signed rank test and one sample
sign test. For comparison between more than two means, we used
Friedman analysis followed by Dunn’s test. Differences were
considered significant when p<0.05.
Results
Detection of Kv channels and impact of
their inhibition on wound healing in A549 cell monolayers
The presence of human KvLQT1, EAG1 and ERG1 mRNA was
first detected by RT-PCR in lung AD A549 cells (Fig. 1B). To evaluate the contribution of
these channels, A549 cells were exposed to various pharmacological
Kv channel inhibitors in scratch-assays, which engages cell
migration and proliferation processes (Fig. 1A). Neither astemizole (EAG and ERG
channel inhibitor, 5 and 20 μM), nor ergtoxin (specifically
blocking ERG channels, 100 nM) significantly affected A549
wound-healing rates (Fig. 1C and
D). On the contrary, clofilium, which is frequently used to
study KvLQT1 function in respiratory epithelia (42–44),
dose-dependently decreased wound-healing rates of A549 cell
monolayers (11, 21 and 31% inhibition in the presence of 5, 10 and
25 μM clofilium, respectively, Fig. 1E). Similarly, the KvLQT1 blocker
chromanol reduced A549 wound healing (Fig. 1F).
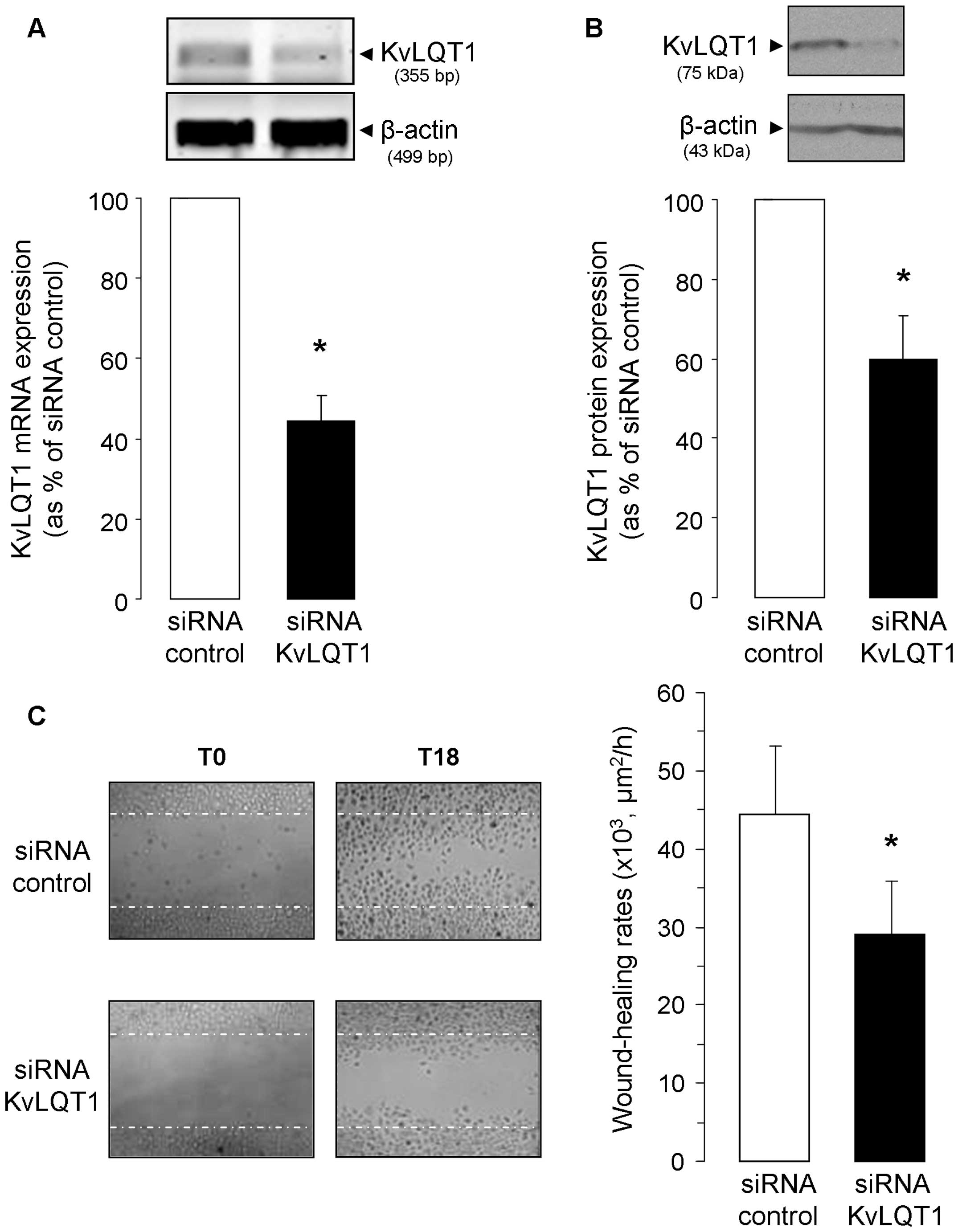
To confirm the role of KvLQT1, we adopted a
complementary approach with stealth KvLQT1 siRNAs to test if its
specific silencing would subsequently affect wound-healing rates.
First, the efficiency of KvLQT1 siRNA in downregulating KvLQT1 mRNA
(Fig. 2A) and protein (Fig. 2B) expression was verified. We then
found that partial KvLQT1 silencing elicited a significant decrease
in wound-healing (28.9±6.9×103 μm2/h,
Fig. 2C), compared to A549 cells
exposed to negative control siRNA (44.3±8.9×103
μm2/h, i.e., 35% inhibition). Data from both
pharmacological and molecular approaches thus point toward KvLQT1
involvement in the regulation of A549 wound healing. We then
undertook complementary assays to dissect its implication in both
cell migration and proliferation.
Impact of clofilium on A549 cell
motility
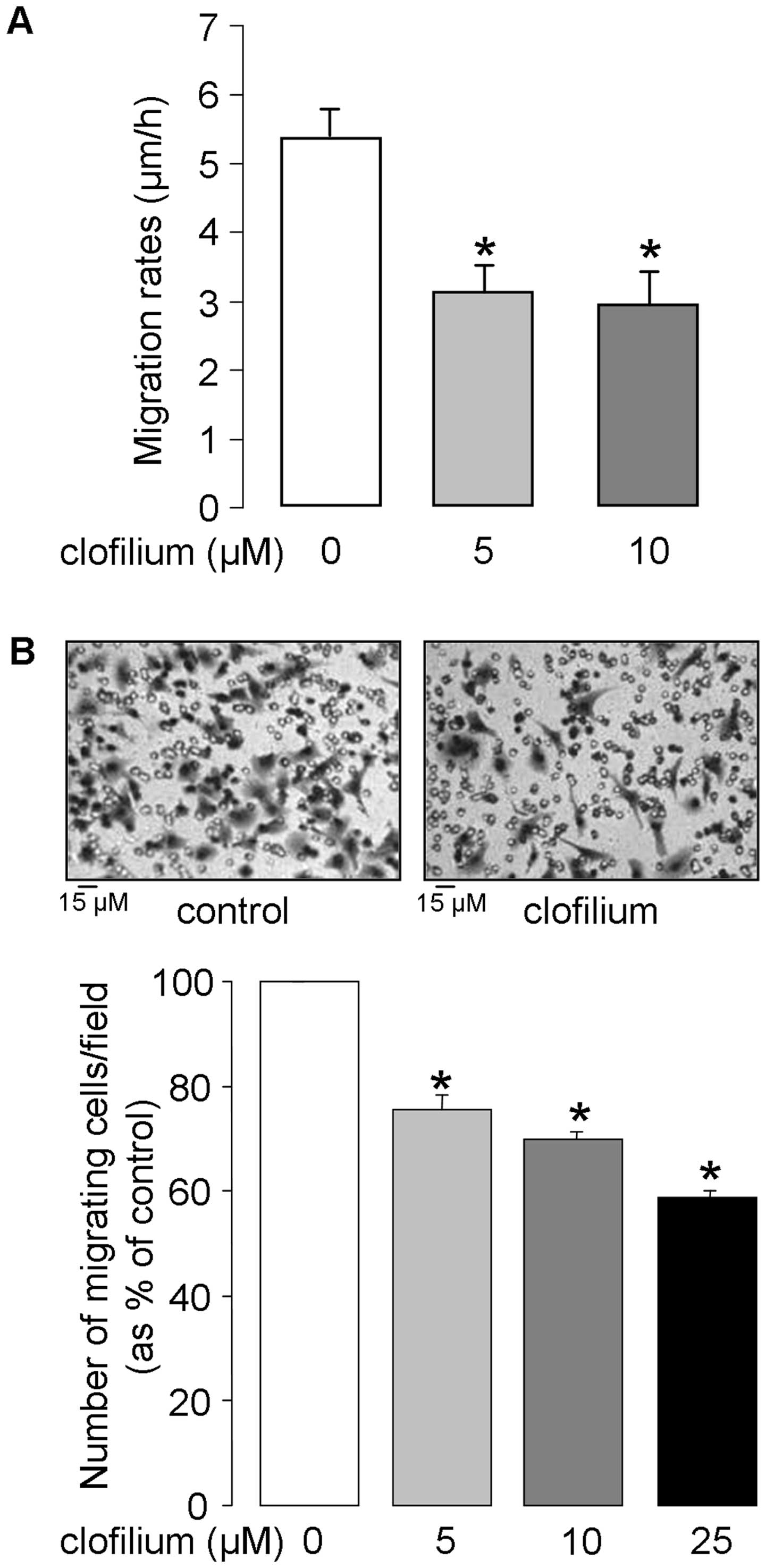
The effect of clofilium on 2D cell migration
dynamics was assessed by single-cell tracking of sub-confluent A549
cells in time-lapse video-microscopy experiments. As illustrated in
Fig. 3A, cell migration rates were
reduced by 42% in the presence of 5 μM clofilium; no further
inhibition (45%) was elicited by higher (10 μM) clofilium
concentration. 3D cell motility, an important determinant of cancer
propagation, was then estimated by Boyden-type chamber assay. It
was observed that the number of cells migrating through the
chambers was significantly reduced by clofilium (25, 31 and 42%
inhibition at 5, 10 and 25 μM, respectively, Fig. 3B). Altogether, these data indicate
that clofilium could depress 2D and 3D A549 cell motility.
Anti-proliferative effect of KvLQT1
inhibition through A549 cell cycle control
Because cell proliferation is a crucial component of
tumor growth, we then evaluated KvLQT1’s contribution to cell
proliferation, by counting the number of A549 cells after a 24- to
72-h period in the presence of KvLQT1 inhibitors. We noted that 20
μM chromanol significantly reduced cell growth, compared to
untreated cells (Fig. 4A).
Similarly, increasing clofilium concentrations (from 5 to 25
μM) decreased A549 cell proliferation in a dose- and
time-dependent manner (Fig. 4B).
At the 72-h post-treatment time-point, we estimated that <5
μM of clofilium was required to inhibit 50% of cell
growth.
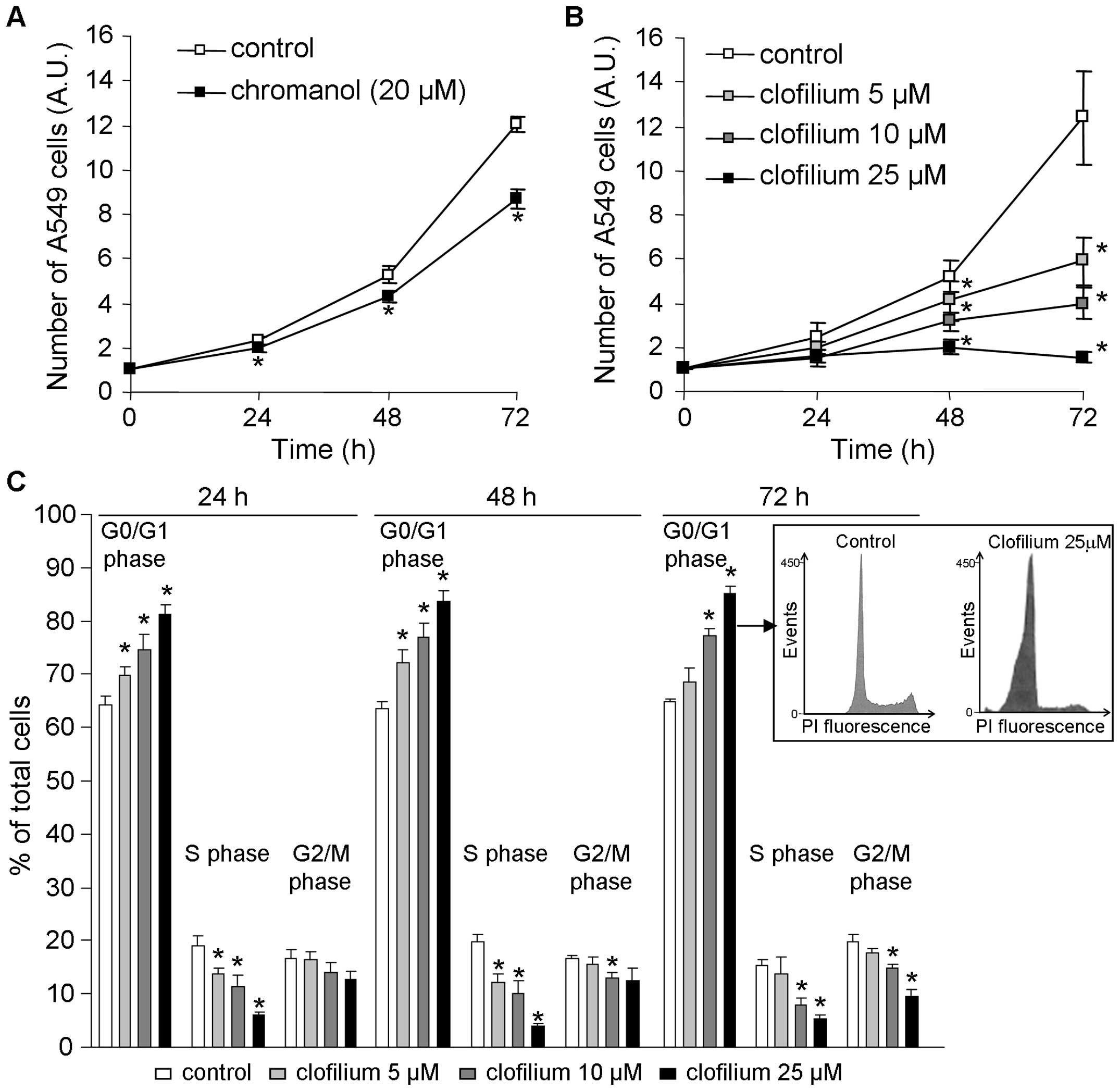 | Figure 4.Impact of KvLQT1 inhibition on
proliferation and cell cycle distribution. Sub-confluent A549 cells
were incubated with chromanol [20 μM, n=6; (A)] and
increasing clofilium concentrations [5, 10 or 25 μM, n=7;
(B)] for 24, 48 or 72 h. Cell growth was then estimated at each
time-point by counting the number of A549 cells after cell
separation by trypsinization. Values are reported in arbitrary
units, normalized to the number of cells at time 0.
*p<0.05. (C) Cell cycle distribution of A549 cells,
after 24-, 48- or 72-h exposure to 5, 10 or 25 μM clofilium
was assessed by analyzing isolated nuclei stained with propidium
iodide by flow cytometry. Bar diagrams show the percentages of
cells in the G0/G1, S and G2/M phases. The results are expressed as
mean percentage of total cells ± SEM of 5–7 experiments.
*p<0.05. Insert, flow cytometry histograms of A549
cells in the control condition and after 72-h treatment with 25
μM clofilium. |
It has been proposed that cell cycle control by
K+ channels may be a possible mechanism whereby they
could regulate cell proliferation (10,25–27,45).
We thus decided to examine the effect of 24-, 48- and 72-h
clofilium application on A549 cell cycle distribution, by flow
cytometry. First, it has to be mentioned that clofilium, even at 25
μM concentration for 72 h, did not elicit cytotoxicity as
indicated by the absence of significant accumulation of sub-G0
apoptotic cells (Fig. 4C, insert).
Moreover, we discerned a dose-dependent effect of clofilium on the
cell cycle with an increasing number of cells in the G0/G1 phase
and subsequent reduction of cells in the S and G2/M phases
(Fig. 4C). These results showed
that KvLQT1 inhibition in A549 cells significantly curbed cell
proliferation and impacted cell cycle progression.
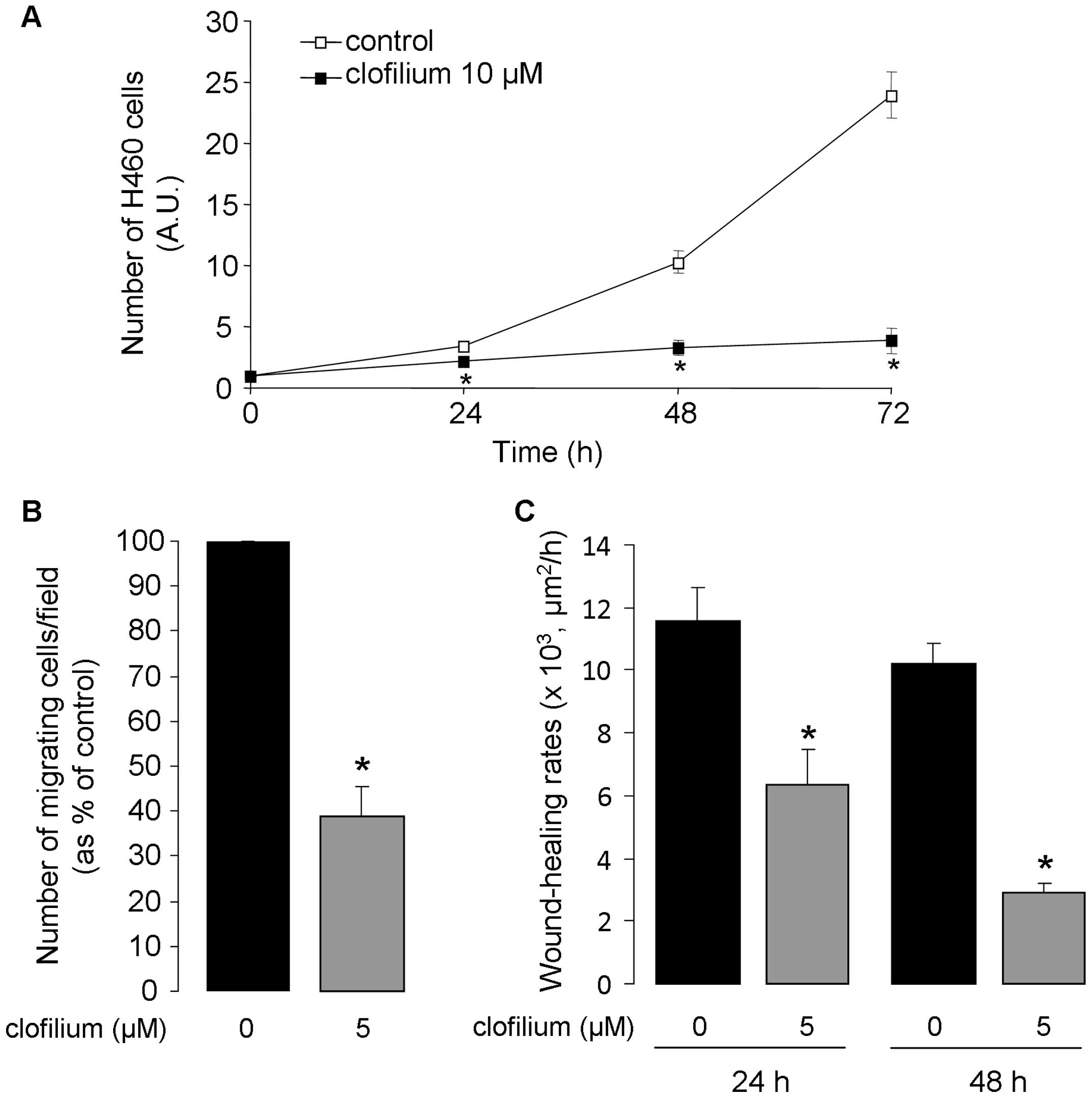
Reduction of H460 cell proliferation,
migration and wound healing by clofilium
The effect of clofilium was also assessed in a
second lung cancer cell model, the H460 cell line. As depicted in
Fig. 5, KvLQT1 inhibition severely
reduced H460 cell growth (Fig. 5A)
and migration in the Boyden-type chamber (Fig. 5B). Accordingly, healing rates,
measured at 24 and 48 h after wounding, were affected by clofilium
(Fig. 5C).
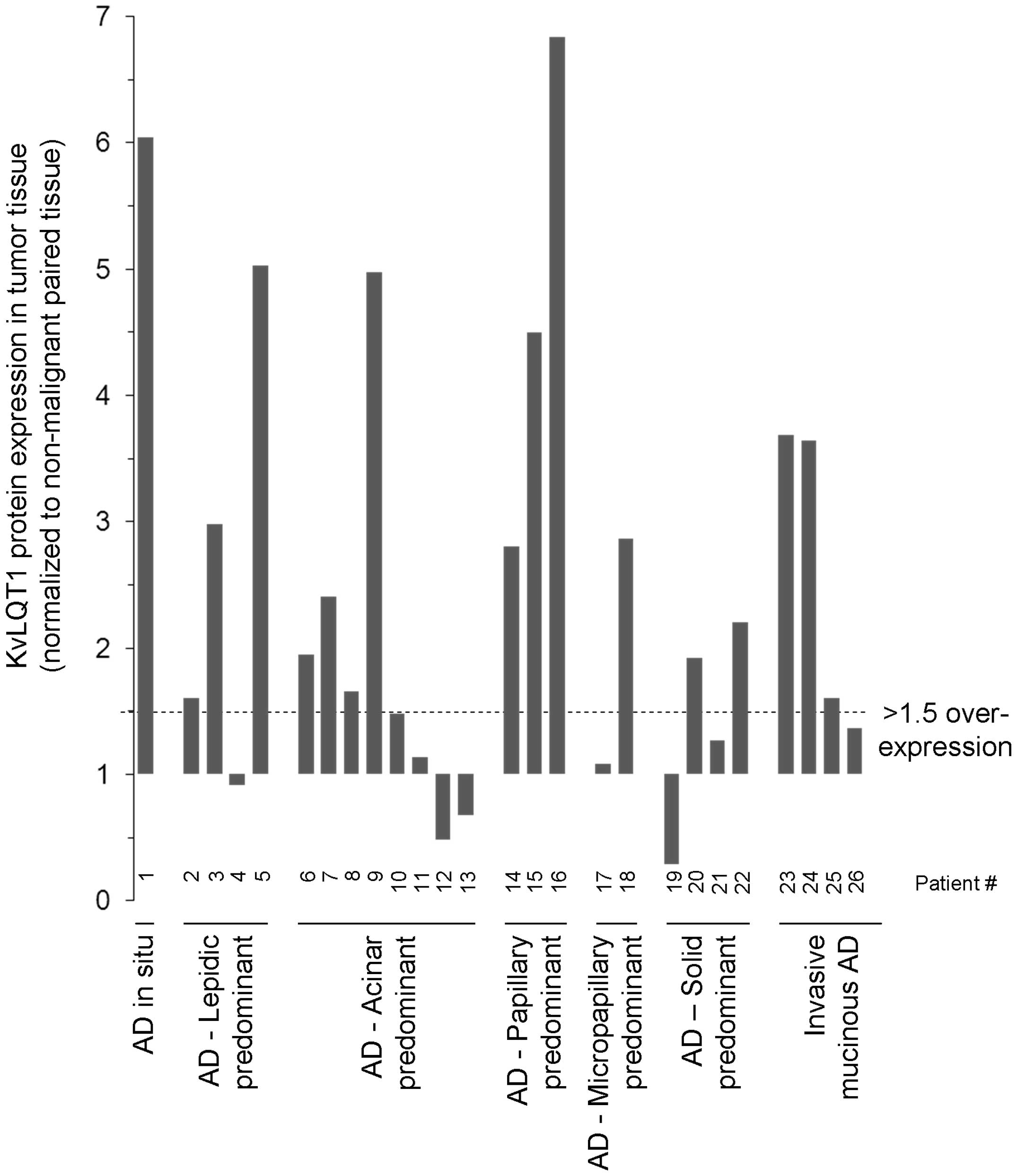
KvLQT1 protein overexpression in tumor
tissues from patients with lung AD
KvLQT1 expression levels were then examined by
immunoblotting of human lung AD tumor tissues, compared to matched,
adjacent, non-neoplastic lung parenchyma, from 26 patients
(Fig. 6 and Table I). An elevation (1.5- to 7-fold) of
KvLQT1 protein was apparent in AD tumor tissues from 17 among 26
patients studied (i.e., 65%). More precisely, KvLQT1 overexpression
(>1.5-fold) was found in 1/1 patient with AD in situ, 3/4
patients with lepidic-predominant AD, 4/8 patients with
acinar-predominant AD, 3/3 patients with papillary-predominant AD,
1/2 patients with micropapillary-predominant AD, 2/4 patient with
solid-predominant AD and 3/4 patients with invasive mucinous AD.
KvLQT1 overexpression was detected in both males and females, with
or without smoking history (Table
I).
Discussion
Our data first disclosed that clofilium and
chromanol as well as KvLQT1 silencing with siRNA significantly
reduced the wound-healing rates of A549 monolayers. Moreover, A549
cell migration, proliferation and cell cycle were affected after
KvLQT1 inhibition. The role of this channel was also verified in
the lung cancer cell line H460. Finally, KvLQT1 overexpression was
detected in 17 of 26 lung AD human samples.
KvLQT1, EAG1 and ERG1 mRNA was noted in A549 cells.
However, application of ergtoxin failed to decrease A549
wound-healing rates. Similarly, astemizole, which blocks EAG and
ERG channels, did not significantly affect wound healing, whereas
clofilium, used frequently to study KvLQT1 channel function in
respiratory epithelia (42–44),
dose-dependently reduced wound healing. We are aware that this
compound may have off-target effects, e.g., on TASK (46), EAG (47) and ERG (48) K+ channels. However,
higher clofilium concentrations (∼100 μM) are necessary for
TASK inhibition (46,49). Moreover, the absence of inhibitory
effects of ergtoxin and astemizole suggests that the suppressive
action of clofilium in our assays is mainly occurring via KvLQT1
inhibition. This hypothesis is reinforced by the observed
inhibition of wound healing by chromanol and confirmed by data
obtained after KvLQT1 silencing, which caused a marked decline of
wound-healing rates.
The decrease in A549 and H460 cell growth with
clofilium and/or chromanol that we report here is the first
evidence of such a role for KvLQT1 in NSCLC cells. These results
are in agreement with other studies showing that K+
channel function modulates cell proliferation both in vitro
and in vivo (9,10,15,16,23).
In A549 cells, an anti-proliferative effect after Kv1.3 inhibition
or silencing has already been cited (20). In SCLC cells, the involvement of
other K+ channels in proliferation, i.e., Girk (21) and 4-AP sensitive K+
channels (22), has been
demonstrated.
Our results indicate that KvLQT1 may regulate cell
growth by controlling cell cycle progression from the G0/G1 to the
S phase. Indeed, clofilium blockade affected cell cycle
distribution, with a dose-dependent increase of A549 cells in the
G0/G1 phase (and a decrease in G2/M and S). A similar result was
observed after Kv1.3 silencing in the same cell model (20). The regulation of cell proliferation
by K+ channels, through cell cycle control, has also
been postulated in many other cancer types (10,45,50).
Moreover, it has been hypothesized that transient
hyperpolarization, after K+ channel activation, could be
essential for G1 phase progression (10,50,51).
In fact, other mechanisms, including calcium signaling, changes in
cell volume and/or activation of signaling pathways, e.g., may also
be involved (10,15,20,27,50).
Our experiments provide proof that clofilium not
only decreases cell proliferation but also 2D and 3D cell
migration, as shown by time-lapse video-microscopy and Boyden-type
chamber assays. This is the first evidence of KvLQT1 involvement in
lung cancer cell motility/proliferation, although we previously
described its role in non-cancer alveolar and bronchial cells
(18,19). Our present study is in agreement
with previous reports that the migration of other cancer cells is
reduced by inhibiting or silencing many K+ channel types
(11,14,29–31).
Several hypotheses have been proposed to explain how these channels
regulate cell migration, e.g., through the modulation of membrane
potential, cell volume, intracellular calcium and signaling
pathways (11). However, it has
also been proposed that K+ channels may regulate cell
motility through their coupling with migratory machinery proteins,
such as integrins (52).
At present, it is not clear if KvLQT1 protein per
se or its K+ conducting function is responsible for
the observed effect of KvLQT1 modulation on cell
migration/proliferation. However, the inhibitory effect of
clofilium and/or chromanol indicates that K+ conductance
via KvLQT1 may be involved. Similarly, K+ permeation is
the central mechanism by which TASK3 exerts its oncogenic potential
in vitro and in vivo, as supported by experiments on
point mutations at the channel pore (53). In contrast, another study showed
that ERG protein downregulation after silencing reduced SCLC cell
proliferation, whereas its blockade had no impact, suggesting that
its ion-conducting property was unnecessary (54).
Our western blot analysis of tissues from patients
with lung AD revealed, for the first time, KvLQT1 overexpression
ranging from 1.5- to 7-fold, in 17/26 of them. To the best of our
knowledge, our study provides the first evidence of KvLQT1
overexpression in lung cancer tissues, although higher KvLQT1
protein levels have already been reported in testicular seminoma
tumor samples (35) and in
gastrointestinal cancers (36).
Other K+ channels (Girk, TASK3) have also been shown to
be overexpressed in SCLC tissues (21,32).
In fact, higher expression levels of many K+ channels
have been detected in several cancer tissue types (16,23,31–36),
and it has been demonstrated that their overexpression highly
increased cell proliferation/migration (14,15).
It is not yet clear why K+ channels are modulated in
cancer tissues but it has been postulated that they may be
regulated during cell dedifferentiation or by high cytokine levels
found in tumors (25).
Elevated KvLQT1 levels were detected in our study in
all AD tested sub-categories, including AD in situ,
lepidic-, acinar-, papillary-, micropapillary-, solid-predominant
AD as well as invasive mucinous AD. However, a larger cohort would
be necessary to define if the frequency and level of KvLQT1
overexpression change among these different classes. Moreover,
patient number was not sufficient to establish a possible
correlation between KvLQT1 expression level and cancer severity
(e.g., tumor grade or the presence of lymph node metastasis).
Nevertheless, correlation between clinical parameters and prognosis
outcomes with K+ channel expression has already been
reported in other studies (reviewed in ref. 12).
In conclusion, our data demonstrate for the first
time, efficient reduction of A549 and H460 cell growth/motility
after KvLQT1 inhibition, with clofilium and/or chromanol. Our study
highlights increased expression of this channel in human AD
samples. It would thus be interesting to further evaluate KvLQT1’s
oncogenic potential promise and the potential effect of KvLQT1
inhibitors on lung cancer development in vivo.
Acknowledgements
We thank Dr P. Romeo, pathologist at
the CHUM and the tissue bank of the FRQS Respiratory Health
Network, for providing human tissue samples. We acknowledge the
logistical assistance received from the Research Support Office,
CRCHUM. This study was funded by the Canadian Institutes of Health
Research (CIHR, MOP-111054), the CRCHUM and Université de Montréal
(scholarship to E.B.), the CIHR training program of the Respiratory
Health Network and the Fonds de Recherche du Québec - Santé
(fellowships to A.G.).
References
|
1.
|
Berthiaume Y, Lesur O and Dagenais A:
Treatment of adult respiratory distress syndrome: plea for rescue
therapy of the alveolar epithelium. Thorax. 54:150–160. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Sacco O, Silvestri M, Sabatini F, Sale R,
Defilippi AC and Rossi GA: Epithelial cells and fibroblasts:
structural repair and remodelling in the airways. Paediatr Respir
Rev. 5(Suppl A): S35–S40. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Zahm JM, Kaplan H, Herard AL, Doriot F,
Pierrot D, Somelette P and Puchelle E: Cell migration and
proliferation during the in vitro wound repair of the respiratory
epithelium. Cell Motil Cytoskeleton. 37:33–43. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Crosby LM and Waters CM: Epithelial repair
mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol.
298:L715–L731. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Mareel M and Leroy A: Clinical, cellular,
and molecular aspects of cancer invasion. Physiol Rev. 83:337–376.
2003.
|
|
7.
|
Prevarskaya N, Skryma R and Shuba Y: Ion
channels and the hallmarks of cancer. Trends Mol Med. 16:107–121.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
O’Grady SM and Lee SY: Molecular diversity
and function of voltage-gated (Kv) potassium channels in epithelial
cells. Int J Biochem Cell Biol. 37:1578–1594. 2005.PubMed/NCBI
|
|
9.
|
Wonderlin WF and Strobl JS: Potassium
channels, proliferation and G1 progression. J Membr Biol.
154:91–107. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Villalonga N, Ferreres JC, Argiles JM,
Condom E and Felipe A: Potassium channels are a new target field in
anticancer drug design. Recent Pat Anticancer Drug Discov.
2:212–223. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Schwab A, Hanley P, Fabian A and Stock C:
Potassium channels keep mobile cells on the go. Physiology.
23:212–220. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Arcangeli A, Crociani O, Lastraioli E,
Masi A, Pillozzi S and Becchetti A: Targeting ion channels in
cancer: a novel frontier in antineoplastic therapy. Curr Med Chem.
16:66–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Girault A, Haelters JP, Potier-Cartereau
M, Chantome A, Jaffres PA, Bougnoux P, Joulin V and Vandier C:
Targeting SKCa channels in cancer: potential new therapeutic
approaches. Curr Med Chem. 19:697–713. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Afrasiabi E, Hietamaki M, Viitanen T,
Sukumaran P, Bergelin N and Tornquist K: Expression and
significance of HERG (KCNH2) potassium channels in the regulation
of MDA-MB-435S melanoma cell proliferation and migration. Cell
Signal. 22:57–64. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Huang L, Li B, Li W, Guo H and Zou F:
ATP-sensitive potassium channels control glioma cells proliferation
by regulating ERK activity. Carcinogenesis. 30:737–744. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Lallet-Daher H, Roudbaraki M, Bavencoffe
A, Mariot P, Gackiere F, Bidaux G, Urbain R, Gosset P, Delcourt P,
Fleurisse L, Slomianny C, Dewailly E, Mauroy B, Bonnal JL, Skryma R
and Prevarskaya N: Intermediate-conductance
Ca2+-activated K+ channels (IKCa1) regulate
human prostate cancer cell proliferation through a close control of
calcium entry. Oncogene. 28:1792–1806. 2009.
|
|
17.
|
Alvarez-Baron CP, Jonsson P, Thomas C,
Dryer SE and Williams C: The two-pore domain potassium channel
KCNK5: induction by estrogen receptor alpha and role in
proliferation of breast cancer cells. Mol Endocrinol. 25:1326–1336.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Trinh NT, Prive A, Kheir L, Bourret JC,
Hijazi T, Amraei MG, Noel J and Brochiero E: Involvement of KATP
and KvLQT1 K+ channels in EGF-stimulated alveolar
epithelial cell repair processes. Am J Physiol Lung Cell Mol
Physiol. 293:L870–L882. 2007.PubMed/NCBI
|
|
19.
|
Trinh NT, Prive A, Maille E, Noel J and
Brochiero E: EGF and K+ channel activity control normal
and cystic fibrosis bronchial epithelia repair. Am J Physiol Lung
Cell Mol Physiol. 295:L866–L880. 2008.PubMed/NCBI
|
|
20.
|
Jang SH, Choi SY, Ryu PD and Lee SY:
Anti-proliferative effect of Kv1.3 blockers in A549 human lung
adenocarcinoma in vitro and in vivo. Eur J Pharmacol. 651:26–32.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Plummer HK III, Dhar MS, Cekanova M and
Schuller HM: Expression of G-protein inwardly rectifying potassium
channels (GIRKs) in lung cancer cell lines. BMC Cancer. 5:1042005.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Pancrazio JJ, Tabbara IA and Kim YI:
Voltage-activated K+ conductance and cell proliferation
in small-cell lung cancer. Anticancer Res. 13:1231–1234. 1993.
|
|
23.
|
Wang ZH, Shen B, Yao HL, Jia YC, Ren J,
Feng YJ and Wang YZ: Blockage of intermediate-conductance-Ca(2+)
-activated K(+) channels inhibits progression of human endometrial
cancer. Oncogene. 26:5107–5114. 2007.PubMed/NCBI
|
|
24.
|
Gomez-Varela D, Zwick-Wallasch E, Knotgen
H, Sanchez A, Hettmann T, Ossipov D, Weseloh R, Contreras-Jurado C,
Rothe M, Stuhmer W and Pardo LA: Monoclonal antibody blockade of
the human Eag1 potassium channel function exerts antitumor
activity. Cancer Res. 67:7343–7349. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Felipe A, Vicente R, Villalonga N,
Roura-Ferrer M, Martinez-Marmol R, Sole L, Ferreres JC and Condom
E: Potassium channels: new targets in cancer therapy. Cancer Detect
Prev. 30:375–385. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Woodfork KA, Wonderlin WF, Peterson VA and
Strobl JS: Inhibition of ATP-sensitive potassium channels causes
reversible cell-cycle arrest of human breast cancer cells in tissue
culture. J Cell Physiol. 162:163–171. 1995. View Article : Google Scholar
|
|
27.
|
Zhanping W, Xiaoyu P, Na C, Shenglan W and
Bo W: Voltage-gated K+ channels are associated with cell
proliferation and cell cycle of ovarian cancer cell. Gynecol Oncol.
104:455–460. 2007.
|
|
28.
|
Restrepo-Angulo I, Sanchez-Torres C and
Camacho J: Human EAG1 potassium channels in the
epithelial-to-mesenchymal transition in lung cancer cells.
Anticancer Res. 31:1265–1270. 2011.PubMed/NCBI
|
|
29.
|
Hammadi M, Chopin V, Matifat F,
Dhennin-Duthille I, Chasseraud M, Sevestre H and Ouadid-Ahidouch H:
Human ether a-gogo K(+) channel 1 (hEag1) regulates MDA-MB-231
breast cancer cell migration through Orai1-dependent calcium entry.
J Cell Physiol. 227:3837–3846. 2012.
|
|
30.
|
Yasukagawa T, Niwa Y, Simizu S and Umezawa
K: Suppression of cellular invasion by glybenclamide through
inhibited secretion of platelet-derived growth factor in ovarian
clear cell carcinoma ES-2 cells. FEBS Lett. 586:1504–1509. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Potier M, Joulin V, Roger S, Besson P,
Jourdan ML, Leguennec JY, Bougnoux P and Vandier C: Identification
of SK3 channel as a new mediator of breast cancer cell migration.
Mol Cancer Ther. 5:2946–2953. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Mu D, Chen L, Zhang X, See LH, Koch CM,
Yen C, Tong JJ, Spiegel L, Nguyen KC, Servoss A, Peng Y, Pei L,
Marks JR, Lowe S, Hoey T, Jan LY, McCombie WR, Wigler MH and Powers
S: Genomic amplification and oncogenic properties of the KCNK9
potassium channel gene. Cancer Cell. 3:297–302. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Ohya S, Kimura K, Niwa S, Ohno A, Kojima
Y, Sasaki S, Kohri K and Imaizumi Y: Malignancy grade-dependent
expression of k(+)-channel subtypes in human prostate cancer. J
Pharmacol Sci. 109:148–151. 2009.PubMed/NCBI
|
|
34.
|
Voloshyna I, Besana A, Castillo M, Matos
T, Weinstein IB, Mansukhani M, Robinson RB, Cordon-Cardo C and
Feinmark SJ: TREK-1 is a novel molecular target in prostate cancer.
Cancer Res. 68:1197–1203. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Tsevi I, Vicente R, Grande M,
Lopez-Iglesias C, Figueras A, Capella G, Condom E and Felipe A:
KCNQ1/KCNE1 channels during germ-cell differentiation in the rat:
expression associated with testis pathologies. J Cell Physiol.
202:400–410. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Than BL, Goos JA, Sarver AL, O’Sullivan
MG, Rod A, Starr TK, Fijneman RJ, Meijer GA, Zhao L, Zhang Y,
Largaespada DA, Scott PM and Cormier RT: The role of KCNQ1 in mouse
and human gastrointestinal cancers. Oncogene. Aug 26–2013.(Epub
ahead of print). View Article : Google Scholar
|
|
37.
|
Travis WD, Brambilla E, Noguchi M,
Nicholson AG, Geisinger KR, Yatabe Y, Beer DG, Powell CA, Riely GJ,
Van Schil PE, Garg K, Austin JH, Asamura H, Rusch VW, Hirsch FR,
Scagliotti G, Mitsudomi T, Huber RM, Ishikawa Y, Jett J,
Sanchez-Cespedes M, Sculier JP, Takahashi T, Tsuboi M,
Vansteenkiste J, Wistuba I, Yang PC, Aberle D, Brambilla C, Flieder
D, Franklin W, Gazdar A, Gould M, Hasleton P, Henderson D, Johnson
B, Johnson D, Kerr K, Kuriyama K, Lee JS, Miller VA, Petersen I,
Roggli V, Rosell R, Saijo N, Thunnissen E, Tsao M and Yankelewitz
D: International association for the study of lung cancer/american
thoracic society/european respiratory society international
multidisciplinary classification of lung adenocarcinoma. J Thorac
Oncol. 6:244–285. 2011. View Article : Google Scholar
|
|
38.
|
Maille E, Trinh NT, Prive A, Bilodeau C,
Bissonnette E, Grandvaux N and Brochiero E: Regulation of normal
and cystic fibrosis airway epithelial repair processes by TNF-alpha
after injury. Am J Physiol Lung Cell Mol Physiol. 301:L945–L955.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Trinh NT, Bardou O, Prive A, Maille E,
Adam D, Lingée S, Ferraro P, Desrosiers M, Coraux C and Brochiero
E: Improvement of defective cystic fibrosis airway epithelial wound
repair after CFTR rescue. Eur Respir J. 40:1390–1400. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Bardou O, Prive A, Migneault F,
Roy-Camille K, Dagenais A, Berthiaume Y and Brochiero E: K(+)
channels regulate ENaC expression via changes in promoter activity
and control fluid clearance in alveolar epithelial cells. Biochim
Biophys Acta. 1818:1682–1690. 2012.
|
|
41.
|
Wang Q, Curran ME, Splawski I, Burn TC,
Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de
Jager T, Schwartz PJ, Toubin JA, Moss AJ, Atkinson DL, Landes GM,
Connors TD and Keating MT: Positional cloning of a novel potassium
channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat
Genet. 12:17–23. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Cowley EA and Linsdell P: Characterization
of basolateral K+ channels underlying anion secretion in
the human airway cell line Calu-3. J Physiol. 538:747–757.
2002.PubMed/NCBI
|
|
43.
|
Devor DC, Bridges RJ and Pilewski JM:
Pharmacological modulation of ion transport across wild-type and
DeltaF508 CFTR-expressing human bronchial epithelia. Am J Physiol
Cell Physiol. 279:C461–C479. 2000.PubMed/NCBI
|
|
44.
|
MacVinish LJ, Hickman ME, Mufti DA,
Durrington HJ and Cuthbert AW: Importance of basolateral
K+ conductance in maintaining Cl- secretion in murine
nasal and colonic epithelia. J Physiol. 510:237–247. 1998.
|
|
45.
|
Ouadid-Ahidouch H and Ahidouch A: K(+)
channels and cell cycle progression in tumor cells. Front Physiol.
4:2202013.
|
|
46.
|
Inglis SK, Brown SG, Constable MJ,
McTavish N, Olver RE and Wilson SM: A Ba2+-resistant,
acid-sensitive K+ conductance in
Na+-absorbing H441 human airway epithelial cells. Am J
Physiol Lung Cell Mol Physiol. 292:L1304–L1312. 2007.PubMed/NCBI
|
|
47.
|
Asher V, Warren A, Shaw R, Sowter H, Bali
A and Khan R: The role of Eag and HERG channels in cell
proliferation and apoptotic cell death in SK-OV-3 ovarian cancer
cell line. Cancer Cell Int. 11:62011. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Perry M, Stansfeld PJ, Leaney J, Wood C,
de Groot MJ, Leishman D, Sutcliffe MJ and Mitcheson JS: Drug
binding interactions in the inner cavity of HERG channels:
molecular insights from structure-activity relationships of
clofilium and ibutilide analogs. Mol Pharmacol. 69:509–519. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Niemeyer MI, Cid LP, Barros LF and
Sepulveda FV: Modulation of the two-pore domain acid-sensitive
K+ channel TASK-2 (KCNK5) by changes in cell volume. J
Biol Chem. 276:43166–43174. 2001.PubMed/NCBI
|
|
50.
|
Pardo LA: Voltage-gated potassium channels
in cell proliferation. Physiology. 19:285–292. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Klimatcheva E and Wonderlin WF: An
ATP-sensitive K(+) current that regulates progression through early
G1 phase of the cell cycle in MCF-7 human breast cancer cells. J
Membr Biol. 171:35–46. 1999.
|
|
52.
|
Arcangeli A and Becchetti A: Complex
functional interaction between integrin receptors and ion channels.
Trends Cell Biol. 16:631–639. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Pei L, Wiser O, Slavin A, Mu D, Powers S,
Jan LY and Hoey T: Oncogenic potential of TASK3 (Kcnk9) depends on
K+ channel function. Proc Natl Acad Sci USA.
100:7803–7807. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Glassmeier G, Hempel K, Wulfsen I, Bauer
CK, Schumacher U and Schwarz JR: Inhibition of HERG1 K+
channel protein expression decreases cell proliferation of human
small cell lung cancer cells. Pflugers Arch. 463:365–376. 2012.
|