Introduction
Hepatocellular carcinoma (HCC) is the most common
primary malignant tumor of the liver and the third most common
cause of cancer-related death worldwide (1). An increasing number of studies have
indicated that there is close correlation between HCC and
long-standing chronic inflammation (2,3).
However, the detailed molecular mechanisms linking chronic
inflammation and malignant transformation remain to be further
defined.
There is substantial evidence showing that
inflammation mediators, such as cyclooxygenase (COX)-derived
prostaglandins (PGs) may have a causally important role in
hepatocellular carcinogenesis (4,5).
Prostaglandin E2 (PGE2), which is one of the
key products of cyclooxygenase-2, could greatly promote tumor cell
proliferation, anti-apoptosis, angiogenesis and invasion. For
example, PGE2 significantly promoted hepatocellular
carcinoma cells proliferation and survival by upregulating the
expression of FUSE-binding protein 1 (6) and Survivin (7), in human glioma cells PGE2
altered the proliferative, apoptotic and migratory properties
(8), in cholangiocarcinoma cells
PGE2 was able to enhance the invasion by upregulating
the MMP2 expression through the CREB pathway (9), and in a breast cancer model it was
shown to control the tumor growth, angiogenesis, lymphangiogenesis
and metastasis to the lungs and lymph nodes through the EP4
receptor (10). However, in
hepatocellular carcinoma, the mechanisms for its ability to promote
invasiveness are not clarified.
Although surgical resection and adjuvant therapy can
cure well confined primary tumors, metastatic disease is largely
incurable because of its systemic nature and the resistance of
disseminated tumor cells to existing therapeutic agents, this also
explains why >90% of mortality from cancer is attributable to
metastases, not the primary tumors from which these malignant
lesions arise (11).
Hepatocellular carcinoma is a highly malignant disease, the
recurrence and metastasis is quite common in patients, so it is
imperative to fully understand the mechanisms underlying its
invasion and metastasis.
YB-1 (Y-box-binding protein 1) encoded by the YBX1
gene, is a member of the cold-shock protein super family, all of
which contain a highly conserved nucleic-acid-binding motif that
binds to both DNA and RNA (12).
It is a kind of nuclear-cytoplasm shuttle protein, and could
function not only as a transcription factor to regulate genes
transcription in the nuclear, but also control a subset of mRNA
translational efficiency in the cytoplasm. Plethora of publications
indicate that YB-1 can function as an oncoprotein, and is highly
correlated with cancer progression and poor prognosis. For example,
the expression level of cytoplasmic YB-1 has been shown to
correlate with progression in breast cancers (13). The importance of the correlation
between nuclear YB-1 levels with patient prognosis has been
strengthened by similar observations for non-small cell lung
carcinoma and prostate cancer (14,15).
IHC and genomic studies have shown that YB-1 protein and mRNA
levels are frequently elevated in advanced breast cancer, and are
associated with poor patient outcome (16–19).
Although YB-1 has been regarded as a useful biomarker of cancer
progression, the detailed mechanisms for its tumor-promoting
functions are not very clear, and whether the YB-1 could be viewed
as a novel therapeutic target is still controversial (20).
Given our previous results showing that E Prostanoid
1 (EP1) receptor could enhance the invasion of HCC (21,22),
and the involvement of YB-1 in the malignance of carcinoma, this
study was designed to evaluate our hypothesis that PGE2
may promote hepatocellular carcinoma cell invasive growth through
upregulation of YB-1 via the EP1 receptor and its signaling
pathway. Our data reveal that PGE2 enhances HCC cell
invasion through EP1 receptor-mediated upregulation of YB-1 and
this process involves the mTOR pathway. When binding with
PGE2 or selective agonist, EP1 receptor activates Src
and EGFR, which subsequently induces the phosphorylation of p-44/42
MAPK, another key serine and threonine protein kinase critical for
HCC cell invasion. Furthermore, mTOR is activated via the above
signal pathways upregulating the expression level of YB-1, which in
turn regulates the expression of a series of proteins associated
with the epithelial-mesenchymal transition. These findings reveal
that PGE2 could promote HCC cell invasion through
upregulating YB-1 expression level via the EP1/Src/EGFR/Erk/mTOR
pathway. This is the first study detailing the role of
PGE2-EP1 receptor signal pathway in YB-1 expression in
human HCC cell lines.
Materials and methods
Cell culture
Human HCC cell lines Hep3B and Huh7 were obtained
from American Type Culture Collection (Manassas, VA, USA). Cells
were maintained at 37°C in a humidified CO2 incubator.
Hep3B cells were cultured in minimum essential medium (MEM) and
Huh7 cells in Dulbecco’s modified Eagle’s medium (DMEM) containing
10% fetal bovine serum (FBS; Gibco).
Western blotting
At the end of each treatment, the cells were washed
twice with ice-cold phosphate-buffered saline and then sonicated on
ice in a lysis buffer (50 mM Tris-HCl, pH 8.0, containing 150 mM
NaCl, 1% Nonidet P-40, 5 mM EDTA, with the protease inhibitor
tablets from Roche, or together with 50 mM sodium fluoride, 25 mM
glycerophosphate, or 1 mM Na3VO4 for
phosphorylation assay). Cell lysates were centrifuged at 12000 × g
for 10 min at 4°C, and the supernatants were collected for western
blotting. Protein concentration was measured using a Bio-Rad
protein assay (Bio-Rad, Hercules, CA, USA). After boiling for 5 min
in the loading buffer with 10% 2-mercaptoethanol, the samples
containing 30 μg protein were separated on 10% Tris-glycine
gels; the separated proteins were transferred onto a nitro
cellulose membrane (Bio-Rad). Immunoblotting was performed using
individual antibodies (including rabbit monoclonal anti-YB-1,
phosphor-p44/42 MAPK, raptor, E-Cadherin, mouse monoclonal
anti-Snail, total-p44/42 MAPK from Cell Signaling (Boston, MA,
USA); rabbit polyclonal anti-GAPDH, Vimentin from SAB Signalway
Antibody (Nanjing, China).
Cell invasion assay
The cell invasion assay was performed in transwell
chambers (Coster Corning, USA). The Matrigel (BD Biosciences
Discovery Labware, Bedford, MA, USA) was diluted with serum-free
medium (1:5 dilutions), and then 40 μl of diluted Matrigel
was added to the upper chamber, and incubated in 37°C for 5 h to
make the gel solidified. Cells (5×104) in 100 μl
serum-free medium in the presence or absence of PGE2 or
EP1 receptor agonist 17-P-T-PGE2 (Cayman Chemical, Ann
Arbor, MI, USA) were seeded in the upper chamber. Regular medium
containing 10% FBS were added in the lower chamber as
chemoattractants. To determine the role of YB-1 protein in
PGE2 induced Huh7 and Hep3B cell invasion, the cells
transfected with the control siRNA or YB-1 siRNA (5×104)
in 100 μl of serum-free medium in the presence or absence of
PGE2 or EP1 receptor agonist were seeded in the upper
chamber; the regular medium containing 10% FBS was added in the
lower chamber. After 24 h of incubation at 37°C, the cells were
fixed with ethanol and stained with 0.1% crystal violet for 30 min.
After washing the cells with PBS, the cells on the upper surface of
the filter were mechanically removed with a cotton swab. The
invading cells on the lower surface were solubilized with 300
μl 10% acetic acid and the absorbance of which was measured
at 570 nm. These experiments were repeated three times, and three
wells were used for each treatment.
RNAi interference
The sequences of EP1 siRNA (siRNA ID: 194727),
Raptor siRNA (siRNA ID: 33215) and YB-1 siRNA (siRNA ID: s9732)
were from Ambon, Life Technology Co. HCC cells (2.3×105)
were plated in 6-well plates for 24 h, resulting in a 30–50%
confluent cell monolayer. The cells were then transfected with the
target siRNA, or a non-silencing 21-nucleotide irrelevant RNA
duplex as a negative control, using Lipofectamine™ 2000
(Invitrogen, Carlsbad, CA, USA). After 72 h, depletion of target
protein was confirmed by immunoblotting and real-time PCR and
subsequently used for further experiments.
RNA isolation and real-time PCR
Total RNA from the cultured cells was isolated using
TRIzol Reagent (Invitrogen) according to the manufacturer’s
instructions. Reverse transcription was carried out with
PrimeScript™ RT reagent kit (Takara Co., Japan) according to the
standard protocol. The sequences of the primers for EP1 receptor
are as follows: forward: 5′-AGCAGCACCTTCAACCCTCA-3′; reverse:
5′-TCCAGCAGATGCACGACAC-3′. The RNA level of EP1 receptor was
determined by the real-time PCR analysis, using the Power SYBR
Master Mix kit (ABI).
Statistical analysis
Results are expressed as the mean ± SD. Statistical
analysis was performed with Student’s t-test. p-values <0.05
were considered statistically significant.
Results
YB-1 protein plays a key role in
PGE2-induced invasion of hepatocellular carcinoma
cells
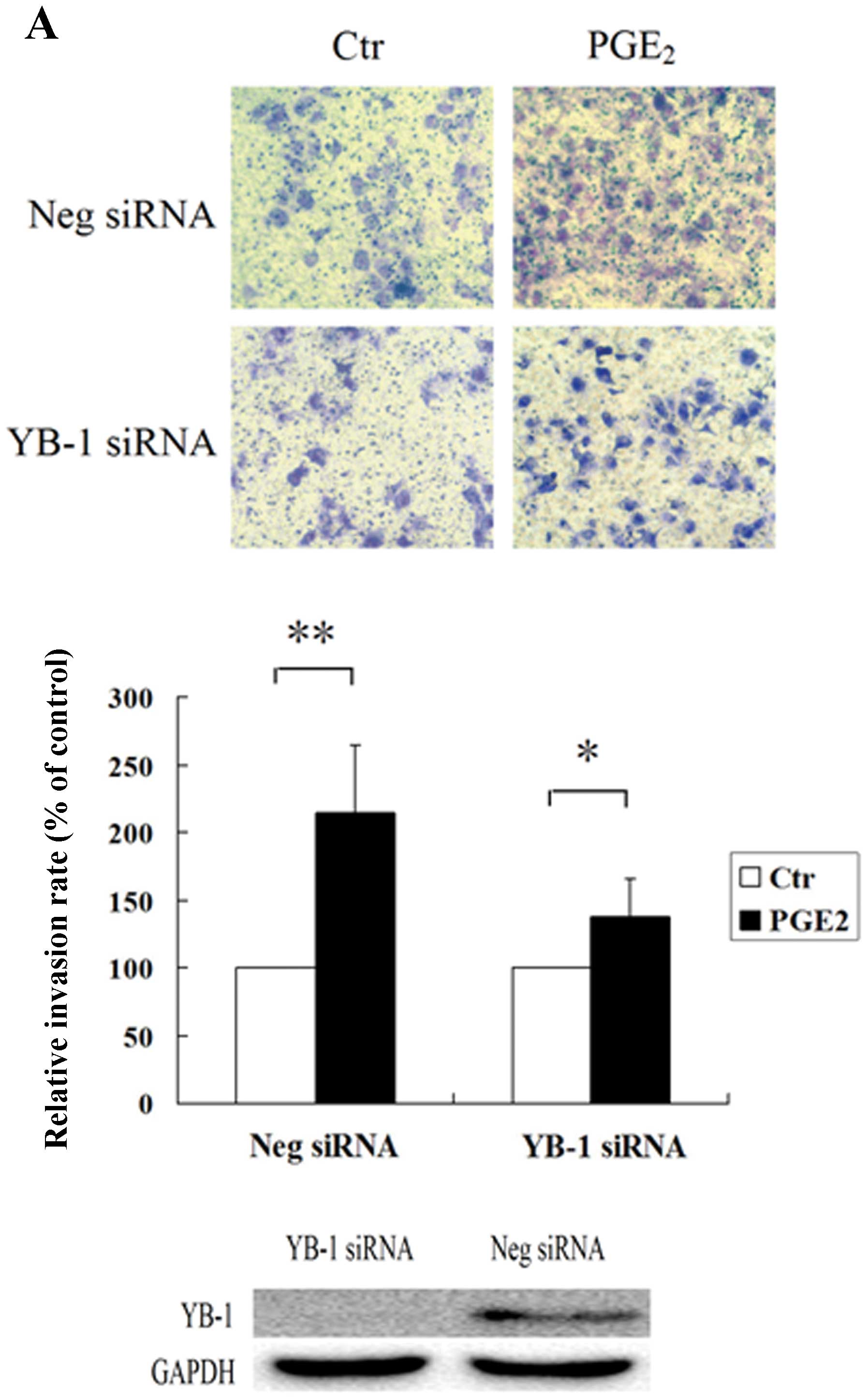
The cell invasion assay was utilized to analyze the
role of YB-1 protein in PGE2-induced invasion of HCC
cells. Huh7 and Hep3B cells were transfected with YB-1 siRNA or
negative control siRNA, and the transfected cells were then
analyzed for PGE2-induced cell invasion. As shown in
Fig. 1, PGE2 greatly
enhanced the invasion properties of Huh7 and Hep3B cells
transfected with negative control siRNA, while in the cells
transfected with YB-1 siRNA, the invasion ability induced by
PGE2 was significantly suppressed. Since YB-1 was able
to regulate the cell proliferation, in order to rule out the
suppressed invasion ability was caused by lower proliferating rate,
we performed the WST analysis and found that RNAi suppression of
YB-1 expression could not inhibit the cell proliferation rate (data
not shown). These results indicated that the YB-1 protein may play
an important role in PGE2-induced HCC cell invasion.
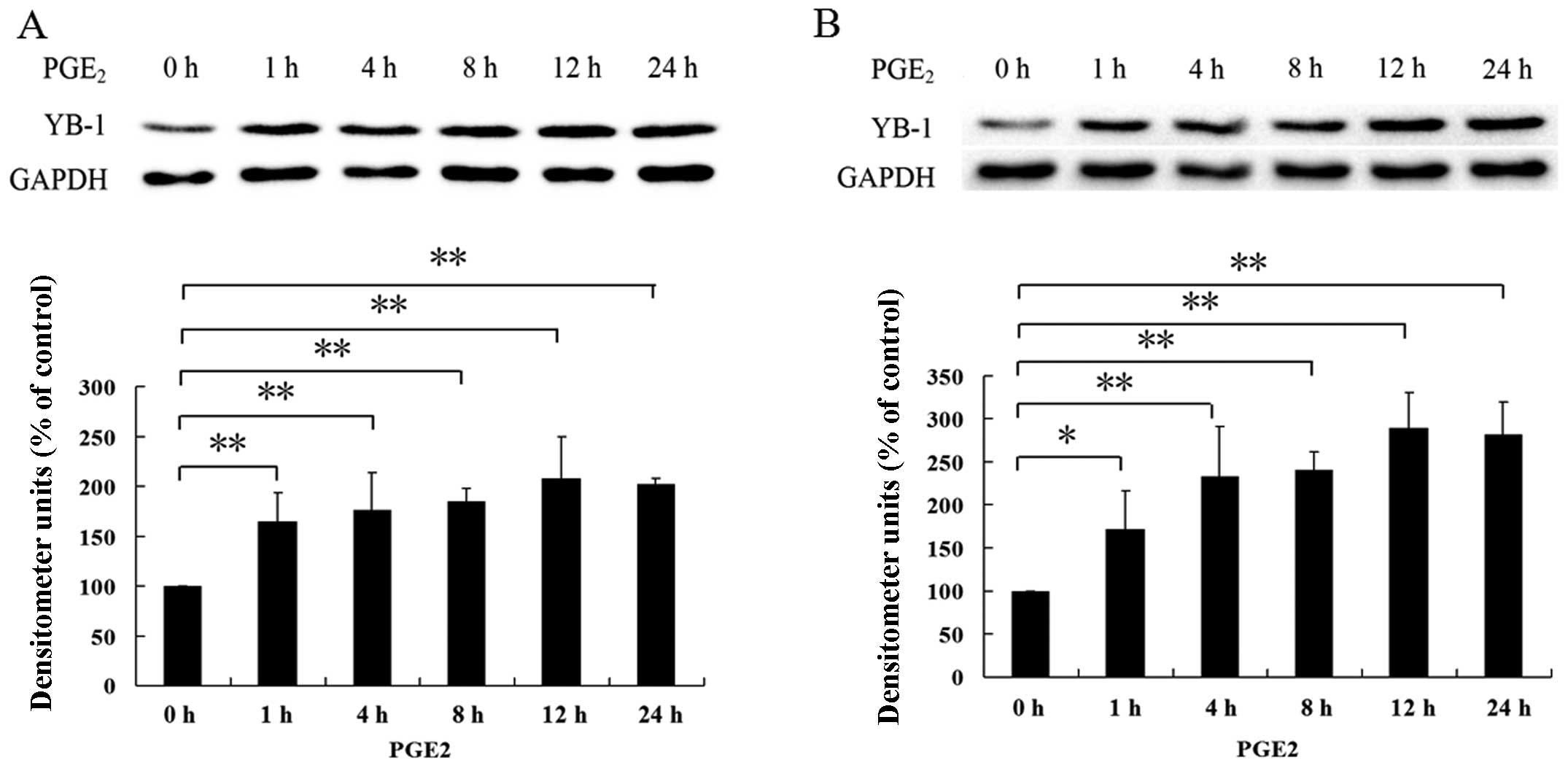
PGE2 induces YB-1 protein
expression in hepatocellular carcinoma cells
RNAi suppression of YB-1 protein significantly
blocked the PGE2-induced invasion of hepato cellular
carcinoma cells suggesting a possible interconnection between
PGE2 and YB-1. Given that PGE2 could greatly
enhance the invasion of tumor cells (23–25)
and that YB-1 has been reported to be necessary for maintaining the
malignance of cancer (12,26,27),
we postulated that PGE2 could enhance HCC invasion
through upregulation of YB-1 expression level. To evaluate this
hypothesis, we treated Huh7 and Hep3B cells with PGE2
for indicated times, as shown in Fig.
2, treatment of Huh7 and Hep3B cells with PGE2
greatly increased the expression level of YB-1. These findings
indicate that PGE2 was able to upregulate YB-1
expression in hepatocellular carcinoma.
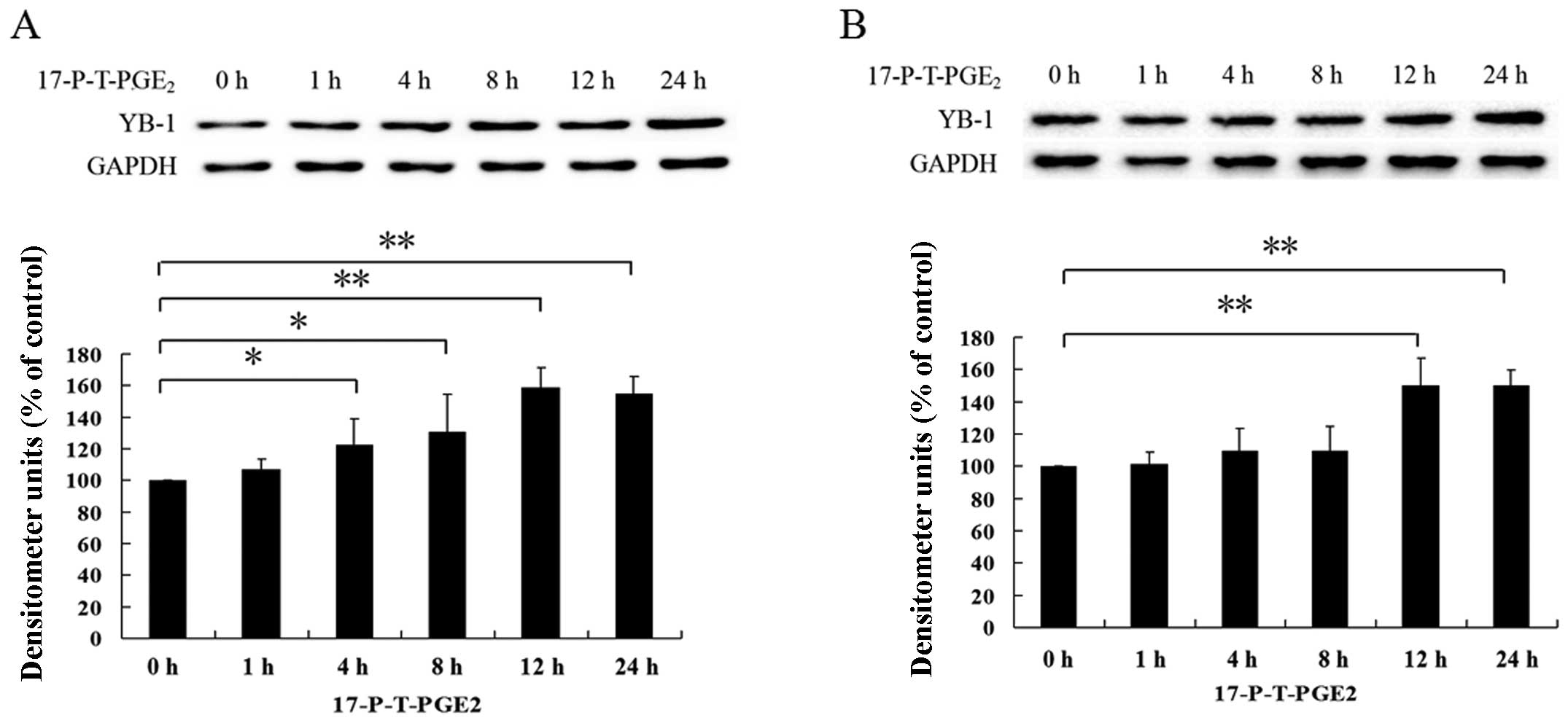
EP1 receptor-mediated upregulation of
YB-1 enhances HCC cell invasion
The EP1 receptor plays an important role in
promoting HCC cell invasion, although the detailed mechanisms are
not very clear (28). RNAi
suppression of YB-1 protein also blocked PGE2-induced
cell invasion, which suggests that YB-1 is closely associated with
HCC cell invasion. Therefore, we investigated whether EP1 receptor
is involved in PGE2-induced YB-1 expression. As shown in
Fig. 3A and B, we found that
treatment of HCC cells with 17-PT-PGE2 significantly
increased the expression level of YB-1. In order to further confirm
this result, we downregulated the EP1 receptor expression level, as
shown in Fig. 3C and D, we found
that when the EP1 receptor expression was suppressed,
PGE2-induced YB-1 expression was almost completely
inhibited. These observations indicate that EP1 receptor plays an
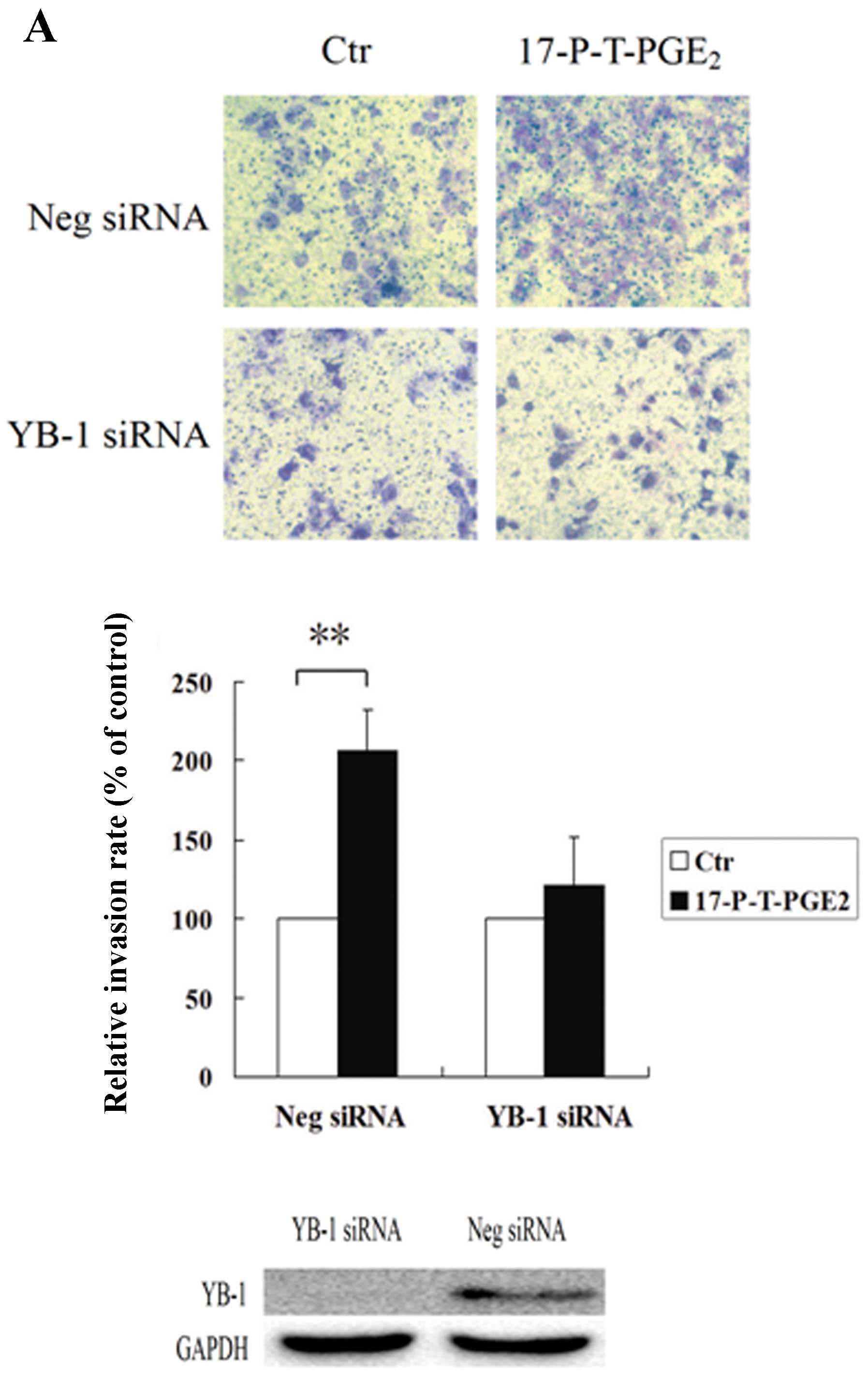
important role in PGE2-induced YB-1 expression. Since
YB-1 is critical to PGE2-induced HCC cell invasion,
further experiment was performed to investigate the role of YB-1 in
17-P-T-PGE2-induced HCC cell invasion. As shown in
Fig. 4, knockdown of YB-1
expression dramatically suppressed the HCC cell invasion induced by
17-P-T-PGE2, whereas, the HCC cells transfected with
negative control siRNA, when treated with EP1 receptor agonist,
still exhibited greatly enhanced invasion ability.
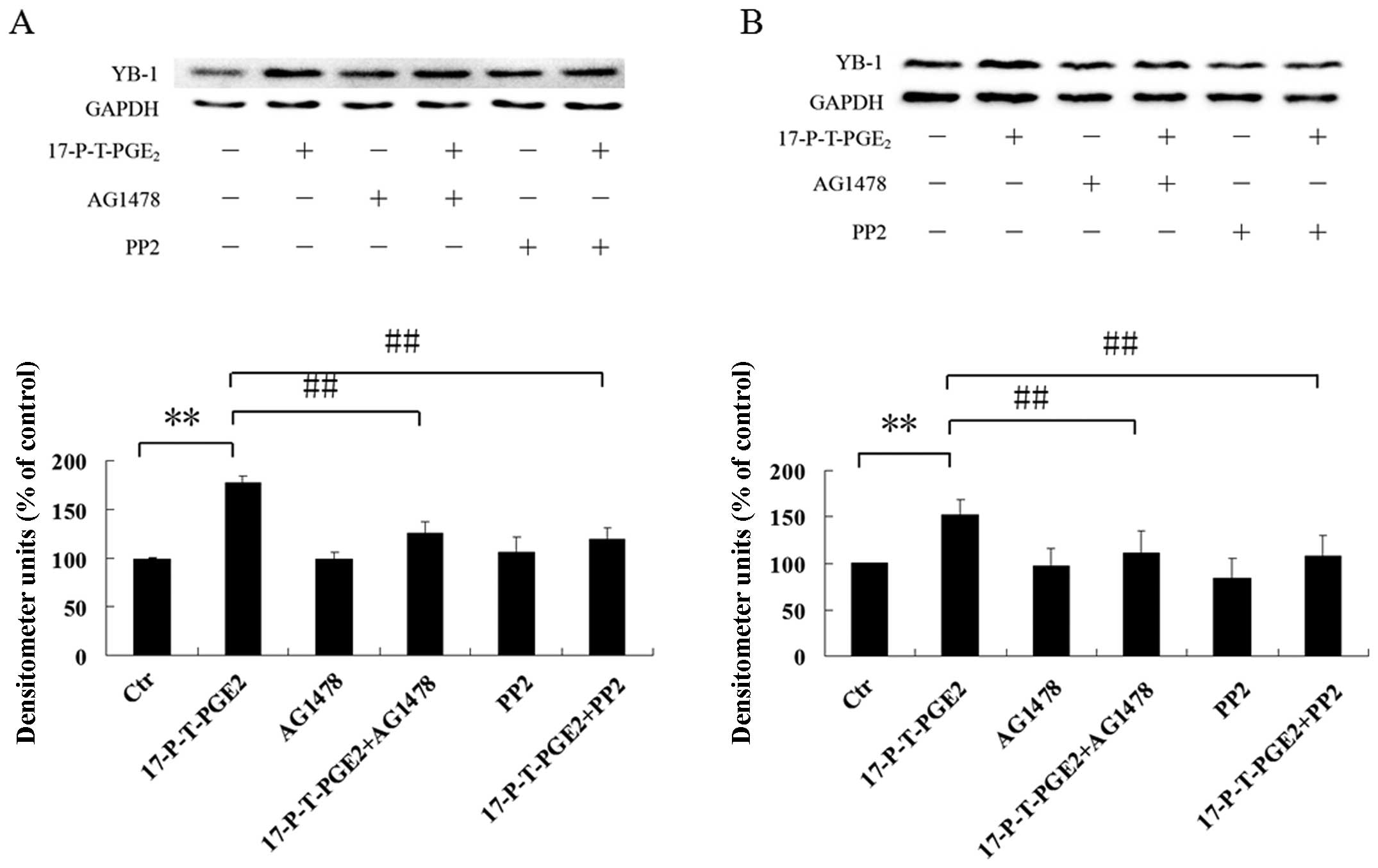
Involvement of EGFR and Src in
PGE2-induced YB-1 expression
EGFR and Src have been reported to be downstream of
the EP1 receptor, and are both important for cancer cell invasion
(28,29). We further investigated whether EGFR
and Src are also involved in the YB-1 expression induced by
17-P-T-PGE2. As shown in Fig. 5, pretreatment of Huh7 and Hep3B
cells with EGFR inhibitor AG1478 or Src inhibitor PP2 significantly
suppressed the YB-1 expression induced by 17-P-T-PGE2.
These observations indicate that EGFR and Src both play important
roles in YB-1 expression induced by 17-P-T-PGE2.
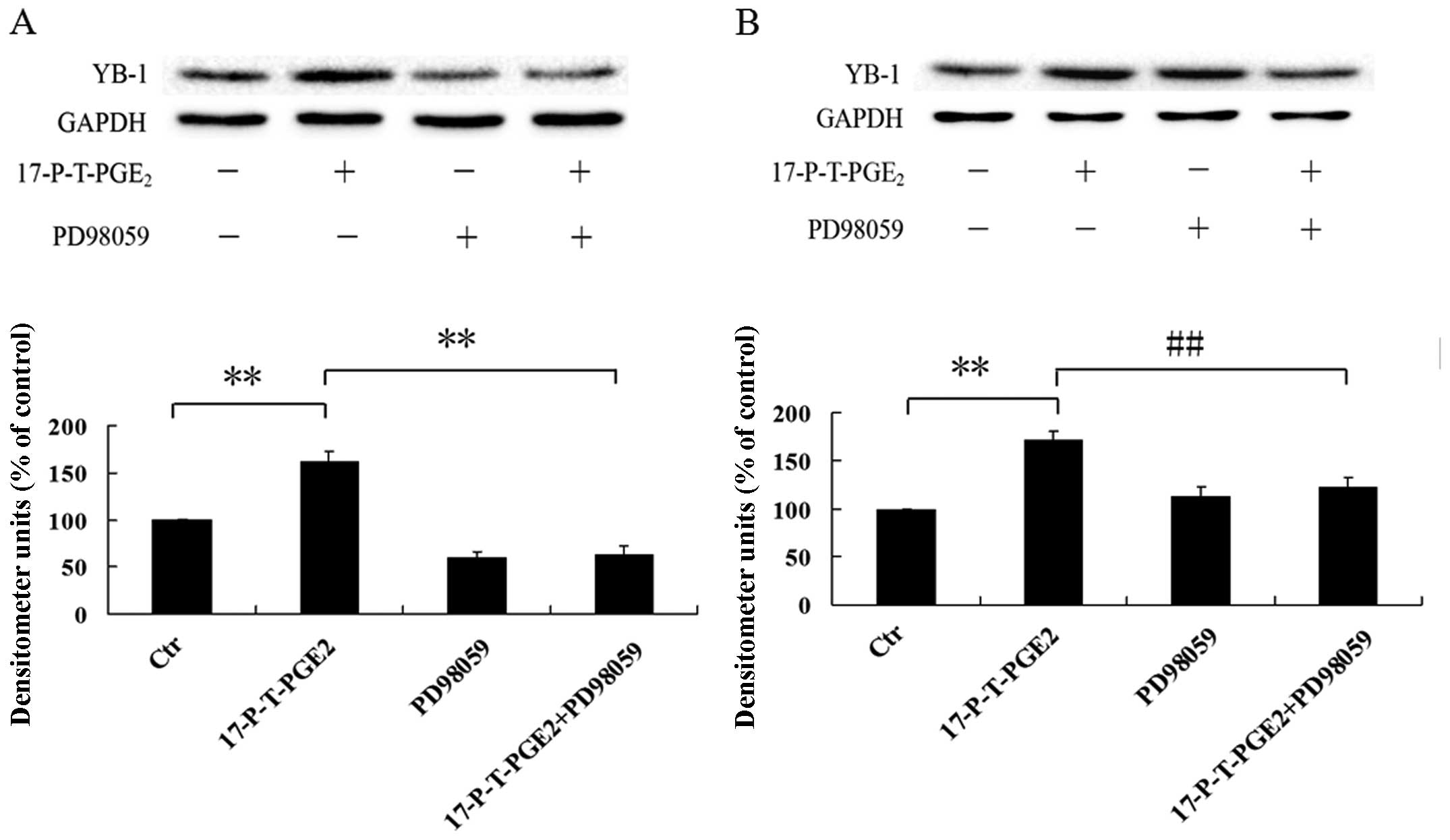
Activation of p44/42 MAPK via EP1
receptor increases YB-1 expression
p44/42 MAPK is a kind of serine/threonine protein
kinase, which is believed to be a critical factor associated with
cancer cell invasion (30–32). Many studies pointed out that
PGE2 could activate p44/42 MAPK via EP1 receptor, so the
potential involvement of p44/42 MAPK in upregulating YB-1
expression via EP1 receptor was determined by using the MEK
inhibitor PD98059. As shown in Fig. 6A
and B, pretreatment of Huh7 and Hep3B cells with PD98059 could
greatly inhibited the YB-1 expression induced by
17-P-T-PGE2. Furthermore, we also found that the p44/42
MAPK could be activated by 17-P-T-PGE2 in a
time-dependent manner (Fig. 6C and
D). These findings indicate that PGE2-induced YB-1
expression via EP1 receptor is mediated through activation of
p44/42 MAPK.
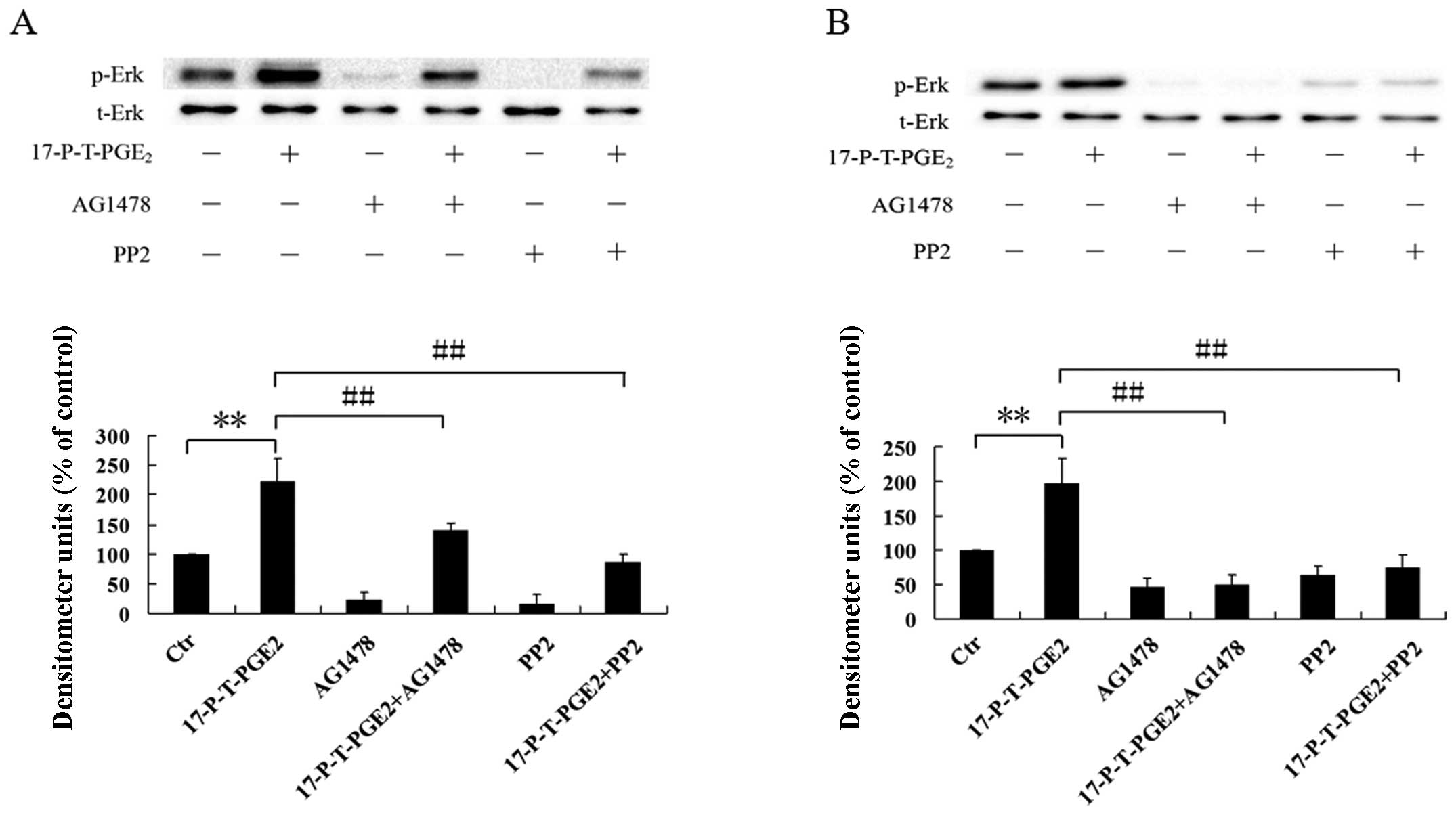
EGFR and Src are involved in the p44/42
MAPK activation induced by PGE2
EGFR and Src are both involved in YB-1 expression
induced by 17-P-T-PGE2, while p44/42 MAPK, activated by
PGE2 via EP1 receptor, also plays a key role in
regulating YB-1 expression. Therefore, we postulated that p44/42
MAPK activation induced by 17-P-T-PGE2 is mediated, at
least in part, through EGFR and Src. To evaluate this hypothesis,
we pretreated Huh7 and Hep3B cells with EGFR inhibitor AG1478 or
Src inhibitor PP2 for 1 h, then treated the cells with
17-P-T-PGE2 for indicated times. As shown in Fig. 7, pretreatment of Huh7 cells with
AG1478 or PP2 significantly suppressed the phosphorylation level of
p44/42 MAPK induced by 17-P-T-PGE2. In Hep3B cells,
AG1478 or PP2 almost completely inhibited the phosphorylation of
p44/42 MAPK. These results clearly indicate that EGFR and Src are
both involved in p44/42 MAPK activation induced by
17-P-T-PGE2.
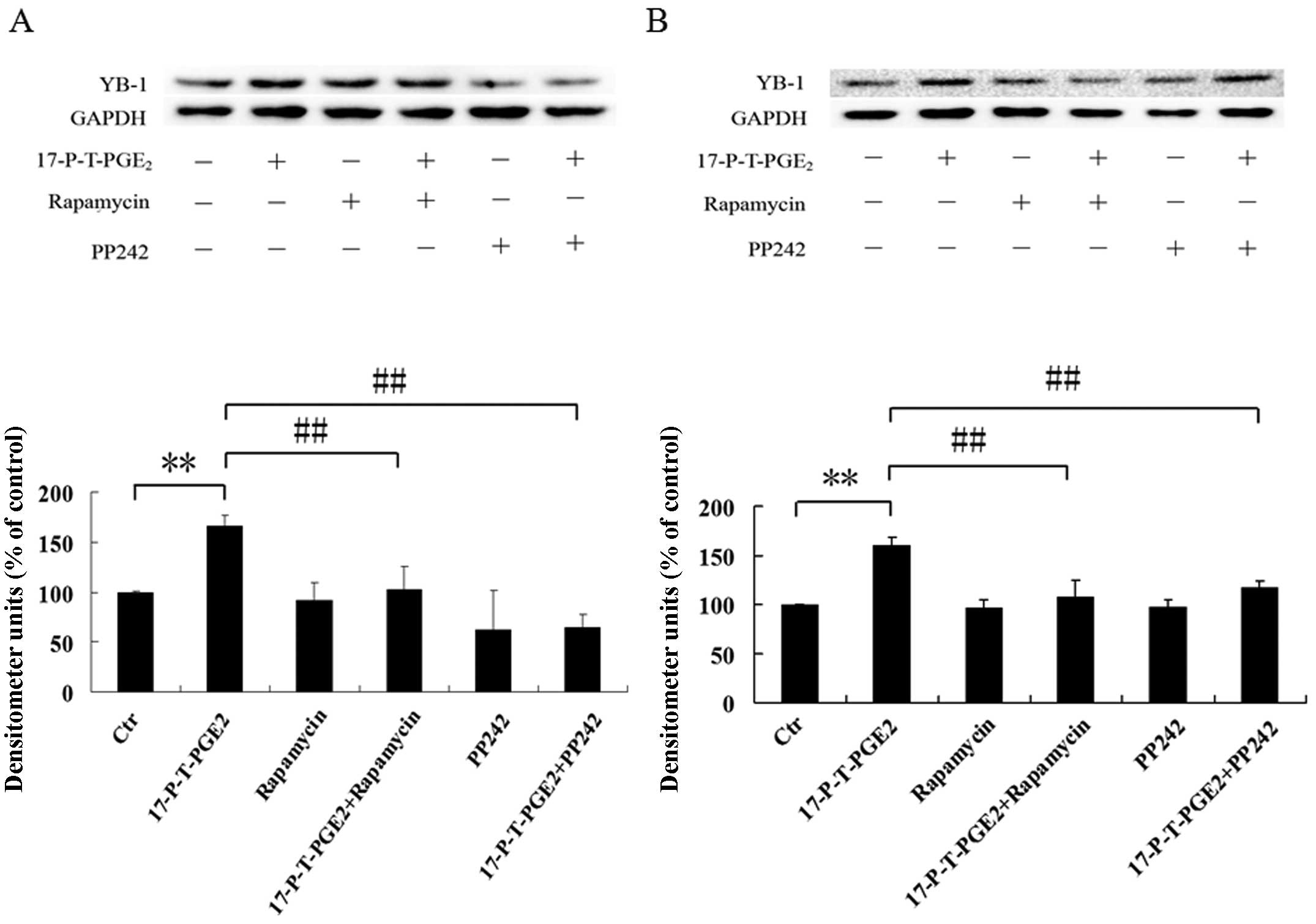
mTOR regulates the YB-1 expression
induced by PGE2
The mechanistic target of rapamycin (mTOR) signaling
pathway senses and integrates a variety of environmental cues to
regulate organismal growth and homeostasis. This pathway regulates
many major cellular processes and is implicated in an increasing
number of pathological conditions, including cancer and others
disease (33). Dysregulated mTOR
pathway was able to influence many aspects of tumor formation, such
as proliferation, anti-apoptosis, cell cycle activation,
angiogenesis and metastasis, and p44/42 MAPK could activate mTOR
pathway by inhibiting the activity of TSC1/ TSC2. We have shown
that PGE2 activated p44/42 MAPK via EP1 receptor, thus,
we speculated that mTOR pathway is involved in YB-1 expression
induced by 17-P-T-PGE2. Pretreatment of Huh7 and Hep3B
cells with rapamycin or PP2, as shown in Fig. 8A and B, significantly suppressed
the YB-1 expression level induced by 17-P-T-PGE2. Since
mTOR forms two complexes to show its functions, we investigated
through which complex the 17-P-T-PGE2 upregulated YB-1
expression. As shown in Fig. 8C and
D, RNAi suppression of raptor expression, existing only in mTOR
complex 1, functions as scaffold for assembling the complex and for
binding substrates and regulators, almost completely blocked the
YB-1 expression induced by 17-P-T-PGE2. These
observations suggest that 17-P-T-PGE2 increased the
expression level of YB-1 through the mTOR complex 1 pathway.
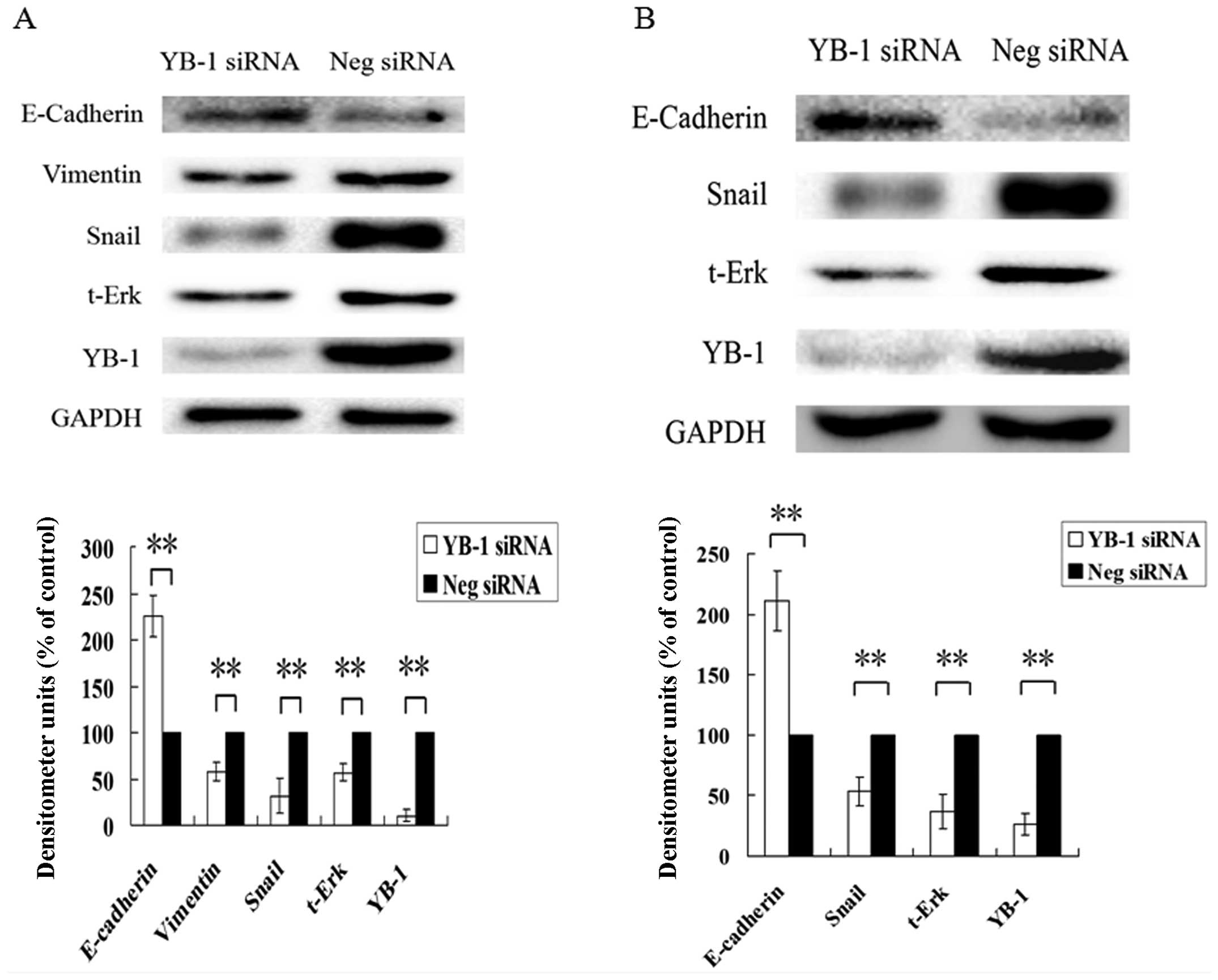
YB-1 regulates EMT-associated gene
expression
Based on our results, we known that YB-1 is critical
for hepatocellular carcinoma cell invasion induced by
PGE2. A recent study showed that in breast cancer cells,
YB-1 is associated with the EMT process, and could regulate a
series of EMT-associated gene expression (20). Thus, we further investigated
whether in HCC cells, YB-1 could regulate EMT-associated gene
expression. As shown in Fig. 9A,
RNAi suppression of YB-1 in Huh7 significantly increased the
expression level of E-Cadherin, an epithelial marker protein, and
downregulated the expression level of vimentin, a mesenchymal
marker protein. In addition, the expression level of Snail protein,
which is one of the key transcription factors controlling the EMT
process, was also greatly suppressed. The expression level of
total-p44/42 MAPK, which, as we discussed above, is a
Serine/Threonine protein kinase associated with cancer cell
invasion, was also dramatically decreased. Similar results were
also observed in Hep3B cells (Fig.
9B), except vimentin expression, which we could not detect in
Hep3B cells. These results indicate that YB-1 was able to regulate
a series of EMT-associated genes expression in HCC cells, and have
the potential to control EMT progression.
Discussion
PGE2, an inflammation mediator, exerts
its biological functions mainly through four G-protein-coupled
receptors (GPCR) on the cell surface membrane, designated as EP1,
EP2, EP3 and EP4, respectively. The EP1 receptor is coupled with
Gαq protein and thus signals through phospholipase C and
intracellular Ca2+. The EP2 and EP4 receptors are
coupled with Gαs protein, signaling through elevation of
intracellular cAMP level and activation of protein kinase A (PKA),
while the EP3 receptor is more complex, according to our previous
results, it is believed to have multiple isoforms generated through
alternative mRNA splicing in the carboxyl tail of the EP3 receptor
(6). The different EP3 receptor
splice variants, coupled with a different G protein, may have
multiple signal transduction pathways in different tissues. The
interaction between the four EP receptors subtypes and
PGE2 depends on the different expression level of an
individual receptor on the cell membrane, the binding affinity to
PGE2, and the differential threshold value for
activation. On one hand, each subtype of EP receptor could transmit
different signals to the downstream pathway, and regulate the
different aspects of cellular functions; on the other hand, the
different signals from the four subtypes could be integrated as a
whole to control the physiological or the pathological phenotypes
of the cells.
The EP1 receptor has been reported to be closely
associated with cancer cell migration and invasion. EP1 receptor
enhanced the phosphorylation of FAK promoting the hepatocellular
carcinoma cell migration and invasion (21,22).
In cholangiocarcinoma cells, EP1 receptor has been shown to
upregulate the MMP-2 expression through the CREB pathway. α2β1
integrin is associated with cell migration, and EP1 receptor
enhanced the cell migration through upregulating the expression of
α2β1 integrin (34). Some studies
have indicated that EP1 receptor was able to transactivate the EGFR
receptor and subsequently activate the Akt kinase, but the detailed
mechanisms how the activated Akt kinase would promote cancer cell
invasion are not clear (28,29).
Metastasis is responsible for as much as 90% of
cancer-associated mortality, yet it remains the most pooly
understood component of cancer pathogenesis. During metastatic
dissemination, a cancer cell from a primary tumor executes the
following sequence of steps: it locally invades the surrounding
tissue, enters the microvasculature of the lymph and blood systems
(intravasation), survives and translocates largely through the
bloodstream to microvessels of distant tissues, exits from the
bloodstream (extravasation), survives in the microenvironment of
distant tissues, and finally adapts to the foreign microenvironment
of these tissues in ways that facilitate cell proliferation and the
formation of a macroscopic secondary tumor (35).
Some researchers pointed out that the complex
metastatic cascade can be conceptually organized and simplified
into two major phases: i) physical translocation of a cancer cell
from the primary tumor to the microenvironment of a distant tissue
and ii) colonization (36). EMT
(epithelial mesenchymal transition) has been implicated as a
critical process that drives the epithelial derived tumor to gain
the malignant properties. During the EMT process, the epithelial
cells would lose the expression profile of epithelial cell marker
proteins, and begin to express the mesenchymal cell marker
proteins. This expression profile switch makes the epithelial cell
to appear as mesenchymal phenotype. Thus, the EMT confers on
epithelial cells precisely the set of traits that would empower
them to disseminate from primary tumors and seed metastases
(37). Long standing chronic
inflammation is required to initiate the EMT process, and which is
tightly controlled by EMT-inducing transcription factors (EMT-TFs).
However, whether PGE2, as a kind of inflammation
mediator, could promote the tumor EMT process is still unclear and
an associated study is undergoing in our laboratory.
Y box binding protein 1 (YB-1) belongs to the family
of the cold-shock containing proteins which could not only function
as a transcription factor in nuclear (38,39),
but also control subsets of mRNA translational efficiency in the
cytoplasm (40). YB-1 has been
regarded as an oncoprotein and prognostic marker (12), as it can promote the cancer
development and progression. In gastric cancer cells, RNAi
knockdown YB-1 expression greatly inhibited cell migration
(41). While in breast cancer,
YB-1 was able to promote cancer cell invasion and metastasis by
altering MT1-MMP trafficking (27). Recent work showed that YB-1 could
regulate the expression of some EMT associated genes, such as Snail
(20), and have an important role
in controlling the EMT (epithelial-mesenchymal transition) and MET
(mesenchymal-epithelial transition) processes. Therefore, we
believe that YB-1 is a critical regulator to promote cancer cell
invasion.
Since PGE2 and YB-1 are both involved in
cancer cell invasion, and have the potential to regulate the EMT
process, the internal relationship between PGE2 and YB-1
is of particular interest to us. In our study, we found that
PGE2 could significantly increase the invasion ability
of HCC cells, and this effect is primarily mediated via EP1
receptor, which is consistent with previous results of others
(28). In order to determine the
role of YB-1 in PGE2-induced HCC cell invasion, we
downregulated the YB-1 expression level, and observed that
knockdown YB-1 expression greatly suppressed the HCC cell invasion
ability induced by PGE2. All these results firmly
confirm that YB-1 is a critical regulator in
PGE2-induced HCC cell invasion.
YB-1 could regulate Snail protein expression and
promote EMT process in breast cancer (20), so we investigated whether YB-1
would influence the expression of some EMT-associated genes in our
HCC cells. According to our results, we found that knockdown of
YB-1 expression dramatically suppressed Snail and vimentin
expression, while increased E-Cadherin expression. These
observations further confirm that YB-1 could regulate the cell
invasion ability.
Since YB-1 is involved in PGE2-induced
HCC cell invasion, we next analyzed whether PGE2 could
directly regulate YB-1 expression. Our results show that
PGE2 could significantly increase YB-1 expression, and
EP1 receptor is the primary receptor responsible for it. These
results are also consistent with our previous cell invasion assay,
indicating that EP1 receptor plays an important role in cell
invasion induced by PGE2.
An increasing number of evidence indicates that GPCR
could transactivate the receptor-tyrosine kinase on the cell
membrane (42). It has been
reported that EP1 receptor, as a kind of GPCR located mainly on the
cell surface membrane, could transactivate the EGFR by forming the
complex with EGFR and Src (28,29).
Therefore, EGFR and Src may be downstream proteins of EP1 receptor.
We hypothesized that Src and EGFR are involved in YB-1 expression
induced by PGE2. The chemical inhibitor analysis
confirmed our hypothesis. Src and EGFR are located upstream in the
EP1 receptor mediated signal pathway, which would activate other
effector proteins to exert their functions. Akt and p44/42 MAPK are
kinases downstream of EGFR. p44/42 MAPK regulates many cellular
functions. Recently, it has been reported that p44/42 MAPK is
mainly responsible for regulating cancer cell invasion (30,43,44).
In our study, chemical inhibitor analysis showed that when the
activity of p44/42 MAPK was inhibited, the PGE2 induced
YB-1 expression was suppressed. Further experiments confirmed that
the phosphorylation level of p44/42 MAPK was increased when EP1
receptor was activated, and when the activities of EGFR or Src were
inhibited, the phosphorylation level of p44/42 MAPK was also
suppressed. These results altogether indicate that p44/42 MAPK is
involved in YB-1 expression induced by PGE2, and the
activity of which is regulated, at least in part, through EGFR and
Src kinase.
mTOR is an atypical serine/threonine protein kinase
that belongs to the phosphoinositide 3-kinase (PI3K)-related kinase
family and interacts with several proteins to form two distinct
complexes the mTOR complex 1 (mTORC 1) and 2 (mTORC 2) (33). The mTOR pathway regulates many
major cellular functions, such as cell growth, proliferation,
metabolism and autophagy. Thus, dysregulated mTOR pathway has
significant promoting effect on tumor progression. mTOR pathway is
activated mainly by Akt and p44/42 MAPK. It is known that
PGE2 could activate p44/42 MAPK via EP1 receptor, so we
hypothesized that mTOR participated in YB-1 expression induced by
PGE2. In our results, we found that two kinds of mTOR
inhibitors could greatly suppress the YB-1 expression induced by
17-P-T-PGE2, which suggests mTOR is involved in this
process. However, mTOR exerts its functions mainly through two mTOR
complexes, so further experiments were performed to analyze which
complex is responsible for this regulation. RNAi suppression of
raptor expression, which exists only in mTORC 1, almost completely
blocked YB-1 expression induced by 17-P-T-PGE2, which
indicates that mTORC 1 is the primary complex involved in this
regulation. All these results verify our hypothesis that
PGE2 increases YB-1 expression level through activating
the mTOR pathway and the mTORC 1 plays the major role in this
process.
It has been reported that YB-1 was able to promote
cancer cell proliferation and RNAi suppression of YB-1 expression
might inhibit cell proliferation (45). However, we found RNAi suppression
of YB-1 expression did not significantly affect the cell
proliferation rate in 17-P-T-PGE2 treatment group
compared with negative siRNA transfected HCC cells by WST analysis
(data not shown). This finding suggests that
PGE2-induced YB-1 upregulation mainly involved cell
invasion but not proliferation in HCC cells.
In summary, our study revealed a signal transduction
pathway through which PGE2 regulates YB-1 expression.
PGE2 increased the YB-1 expression through EP1 receptor;
EGFR, Src and p44/42 MAPK are all involved in this process; p44/42
MAPK activated the mTOR pathway, which in turn upregulate the YB-1
expression, while YB-1 has significant role in promoting cancer
cell invasion through regulating EMT-associated gene expression. To
our knowledge, this is the first study detailing the role of
PGE2-EP1 receptor signal pathway in YB-1 expression in
human hepatocellular carcinoma cells. Our findings reveal the new
mechanisms through which PGE2 enhances hepatocellular
carcinoma cell invasion and may be helpful in finding a new
therapeutic strategy to prevent and cure malignant diseases.
Acknowledgements
This study was supported by National
Natural Science Foundation of China (30871015, 81172003) and a
project funded by the Priority Academic Program Development of
Jiangsu Higher Education Institutions (PAPD).
References
|
1.
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Berasain C, Castillo J, Perugorria MJ,
Latasa MU, Prieto J and Avila MA: Inflammation and liver cancer:
new molecular links. Ann NY Acad Sci. 1155:206–221. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Stauffer JK, Scarzello AJ, Jiang Q and
Wiltrout RH: Chronic inflammation, immune escape and oncogenesis in
the liver: a unique neighborhood for novel intersections.
Hepatology. 56:1567–1574. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Hu KQ: Cyclooxygenase 2 (COX2)-prostanoid
pathway and liver diseases. Prostaglandins Leukot Essent Fatty
Acids. 69:329–337. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Wu T: Cyclooxygenase-2 in hepatocellular
carcinoma. Cancer Treat Rev. 32:28–44. 2006. View Article : Google Scholar
|
|
6.
|
Ma J, Chen M, Xia SK, et al: Prostaglandin
E2 promotes liver cancer cell growth by the upregulation
of FUSE-binding protein 1 expression. Int J Oncol. 42:1093–1104.
2013.
|
|
7.
|
Bai XM, Jiang H, Ding JX, et al:
Prostaglandin E2 upregulates survivin expression via the
EP1 receptor in hepatocellular carcinoma cells. Life Sci.
86:214–223. 2009.
|
|
8.
|
Gomes RN and Colquhoun A: E series
prostaglandins alter the proliferative, apoptotic and migratory
properties of T98G human glioma cells in vitro. Lipids Health Dis.
11:1712012. View Article : Google Scholar
|
|
9.
|
Sun B, Rong R, Jiang H, et al:
Prostaglandin E2 receptor EP1 phosphorylate CREB and
mediates MMP2 expression in human cholangiocarcinoma cells. Mol
Cell Biochem. 378:195–203. 2013.
|
|
10.
|
Xin X, Majumder M, Girish GV, Mohindra V,
Maruyama T and Lala PK: Targeting COX-2 and EP4 to control tumor
growth, angiogenesis, lymphangiogenesis and metastasis to the lungs
and lymph nodes in a breast cancer model. Lab Invest. 92:1115–1128.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Valastyan S and Weinberg RA: Tumor
metastasis: molecular insights and evolving paradigms. Cell.
147:275–292. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Lasham A, Print CG, Woolley AG, Dunn SE
and Braithwaite AW: YB-1: oncoprotein, prognostic marker and
therapeutic target? Biochem J. 449:11–23. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Bargou RC, Jurchott K, Wagener C, et al:
Nuclear localization and increased levels of transcription factor
YB-1 in primary human breast cancers are associated with intrinsic
MDR1 gene expression. Nat Med. 3:447–450. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Shibahara K, Sugio K, Osaki T, et al:
Nuclear expression of the Y-box binding protein, YB-1, as a novel
marker of disease progression in non-small cell lung cancer. Clin
Cancer Res. 7:3151–3155. 2001.PubMed/NCBI
|
|
15.
|
Gimenez-Bonafe P, Fedoruk MN, Whitmore TG,
et al: YB-1 is upregulated during prostate cancer tumor progression
and increases P-glycoprotein activity. Prostate. 59:337–349. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Lasham A, Samuel W, Cao H, et al: YB-1,
the E2F pathway and regulation of tumor cell growth. J Natl Cancer
Inst. 104:133–146. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Dahl E, En-Nia A, Wiesmann F, et al:
Nuclear detection of Y-box protein-1 (YB-1) closely associates with
progesterone receptor negativity and is a strong adverse survival
factor in human breast cancer. BMC Cancer. 9:4102009. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Habibi G, Leung S, Law JH, et al:
Redefining prognostic factors for breast cancer: YB-1 is a stronger
predictor of relapse and disease-specific survival than estrogen
receptor or HER-2 across all tumor subtypes. Breast Cancer Res.
10:R862008. View Article : Google Scholar
|
|
19.
|
Huang J, Tan PH, Li KB, Matsumoto K,
Tsujimoto M and Bay BH: Y-box binding protein, YB-1, as a marker of
tumor aggressiveness and response to adjuvant chemotherapy in
breast cancer. Int J Oncol. 26:607–613. 2005.PubMed/NCBI
|
|
20.
|
Evdokimova V, Tognon C, Ng T, et al:
Translational activation of snail1 and other developmentally
regulated transcription factors by YB-1 promotes an
epithelial-mesenchymal transition. Cancer Cell. 15:402–415. 2009.
View Article : Google Scholar
|
|
21.
|
Bai X, Wang J, Zhang L, et al:
Prostaglandin E2 receptor EP1-mediated phosphorylation
of focal adhesion kinase enhances cell adhesion and migration in
hepatocellular carcinoma cells. Int J Oncol. 42:1833–1841.
2013.
|
|
22.
|
Bai XM, Zhang W, Liu NB, et al: Focal
adhesion kinase: important to prostaglandin E2-mediated
adhesion, migration and invasion in hepatocellular carcinoma cells.
Oncol Rep. 21:129–136. 2009.PubMed/NCBI
|
|
23.
|
Ho MY, Liang SM, Hung SW and Liang CM:
MIG-7 controls COX-2/PGE2-mediated lung cancer
metastasis. Cancer Res. 73:439–449. 2012.PubMed/NCBI
|
|
24.
|
Pai R, Nakamura T, Moon WS and Tarnawski
AS: Prostaglandins promote colon cancer cell invasion; signaling by
cross-talk between two distinct growth factor receptors. FASEB J.
17:1640–1647. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Dohadwala M, Batra RK, Luo J, et al:
Autocrine/paracrine prostaglandin E2 production by
non-small cell lung cancer cells regulates matrix
metalloproteinase-2 and CD44 in cyclooxygenase-2-dependent
invasion. J Biol Chem. 277:50828–50833. 2002.PubMed/NCBI
|
|
26.
|
Wu Y, Yamada S, Izumi H, et al: Strong
YB-1 expression is associated with liver metastasis progression and
predicts shorter disease-free survival in advanced gastric cancer.
J Surg Oncol. 105:724–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Lovett DH, Cheng S, Cape L, Pollock AS and
Mertens PR: YB-1 alters MT1-MMP trafficking and stimulates MCF-7
breast tumor invasion and metastasis. Biochem Biophys Res Commun.
398:482–488. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Han C, Michalopoulos GK and Wu T:
Prostaglandin E2 receptor EP1 transactivates EGFR/MET
receptor tyrosine kinases and enhances invasiveness in human
hepatocellular carcinoma cells. J Cell Physiol. 207:261–270.
2006.
|
|
29.
|
Han C and Wu T: Cyclooxygenase-2-derived
prostaglandin E2 promotes human cholangiocarcinoma cell
growth and invasion through EP1 receptor-mediated activation of the
epidermal growth factor receptor and Akt. J Biol Chem.
280:24053–24063. 2005.PubMed/NCBI
|
|
30.
|
Caramel J, Papadogeorgakis E, Hill L, et
al: A switch in the expression of embryonic EMT-inducers drives the
development of malignant melanoma. Cancer Cell. 24:466–480. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Caslavsky J, Klimova Z and Vomastek T: ERK
and RSK regulate distinct steps of a cellular program that induces
transition from multicellular epithelium to single cell phenotype.
Cell Signal. 25:2743–2751. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Weiss MB, Abel EV, Mayberry MM, Basile KJ,
Berger AC and Aplin AE: TWIST1 is an ERK1/2 effector that promotes
invasion and regulates MMP-1 expression in human melanoma cells.
Cancer Res. 72:6382–6392. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Liu JF, Fong YC, Chang CS, et al:
Cyclooxygenase-2 enhances alpha2beta1 integrin expression and cell
migration via EP1 dependent signaling pathway in human
chondrosarcoma cells. Mol Cancer. 9:432010. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Fidler IJ: The pathogenesis of cancer
metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev
Cancer. 3:453–458. 2003.
|
|
36.
|
Chaffer CL and Weinberg RA: A perspective
on cancer cell metastasis. Science. 331:1559–1564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelialmesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Matsumoto K and Wolffe AP: Gene regulation
by Y-box proteins: coupling control of transcription and
translation. Trends Cell Biol. 8:318–323. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Kuwano M, Uchiumi T, Hayakawa H, et al:
The basic and clinical implications of ABC transporters,
Y-box-binding protein-1 (YB-1) and angiogenesis-related factors in
human malignancies. Cancer Sci. 94:9–14. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Evdokimova V, Ovchinnikov LP and Sorensen
PH: Y-box binding protein 1: providing a new angle on translational
regulation. Cell Cycle. 5:1143–1147. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Guo TT, Yu YN, Cheong Yip GW, Matsumoto K
and Bay BH: Silencing the YB-1 gene inhibits cell migration in
gastric cancer in vitro. Anat Rec. 296:891–898. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Delcourt N, Bockaert J and Marin P:
GPCR-jacking: from a new route in RTK signalling to a new concept
in GPCR activation. Trends Pharmacol Sci. 28:602–607. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Santamaria PG and Nebreda AR:
Deconstructing ERK signaling in tumorigenesis. Mol Cell. 38:3–5.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Ellenrieder V, Hendler SF, Boeck W, et al:
Transforming growth factor beta1 treatment leads to an
epithelial-mesenchymal trans-differentiation of pancreatic cancer
cells requiring extracellular signal-regulated kinase 2 activation.
Cancer Res. 61:4222–4228. 2001.
|
|
45.
|
Fujiwara-Okada Y, Matsumoto Y, Fukushi J,
et al: Y-box binding protein-1 regulates cell proliferation and is
associated with clinical outcomes of osteosarcoma. Br J Cancer.
108:836–847. 2013. View Article : Google Scholar : PubMed/NCBI
|