Introduction
Pancreatic carcinoma is the fourth leading cause of
cancer mortality in the United States, with an incidence of 43,000
new cases per year and a very high mortality with 37,000 deaths
(1). Patients with pancreatic
carcinoma usually present with advanced stage. Despite new drugs
and therapeutic regimens with gemcitabine, the prognosis of
patients with pancreatic carcinoma has not significantly changed in
the last decades (2). Surgical
resection still remains the main therapy for pancreatic carcinoma,
but a large fraction of patients cannot undergo curative resection
(3). Moreover, molecular
mechanisms including cancer development and progression are still
unclear. Thus, innovative therapeutic targets and prognostic
markers are urgently needed for pancreatic carcinoma treatment.
MicroRNAs (miRNAs) represent a novel class of
naturally occurring small non-coding RNA molecules, which are
evolutionarily conserved. Mature miRNAs are approximately short
22-nucleotide molecules. The mature single-stranded miRNAs bind to
specific targets in mRNA 3′-untranslated regions (3′-UTRs) and
negatively regulate translation or mRNA cleavage through partial
sequence homology (4). Accumulated
evidence in cancer biology has shown frequent deregulation of
miRNAs in human malignancies (5–7).
Deregulated miRNAs play an important role in behaving as either
oncogenes or tumor suppressor genes, and some of miRNAs have been
implicated in cellular processes of proliferation, apoptosis and
chemoresistance (8–10).
Among these miRNAs, miR-301 has been reported to be
upregulated in various cancers, including pancreatic, colorectal,
oral, hepatocellular and lung cancers (11–15).
Recently, miR-301a was revealed to regulate MEOX2, which is known
to be associated with ERK/CREB pathway in lung cancer (16). Subsequently, miR-301 was implicated
in cell proliferation, clonogenicity, migration, invasion,
tamoxifen resistance, tumor growth and microvessel density.
Moreover, a study identified FOXF2, BBC3, PTEN and COL2A1 as
candidate miR-301 targets in breast cancer (17). More recently, other researchers
have shown that miR-301a is associated with NF-κB activity through
targeting NF-κB repressing factor (NKRF) in pancreatic carcinoma
(18). These results could
indicate that miR-301 may be a new class of genes involved in human
oncogenesis.
In this study, we aimed to investigate the
mechanistic role of miR-301b expression in pancreatic carcinoma and
to identify new target genes of miR-301b. We found that miR-301b
could promote cell invasion and migration in Panc-1 and BxPC-3. In
addition, we showed that the transfection of synthetic miR-301b
increased drug resistance to gemcitabine. The miR-301b was examined
for potential 3′UTR-binding sites utilizing miRNA target search
algorithms, which is TargetScan (http://genes.mit.edu/targetscan/) and microRNA.org (http://www.microrna.org/).
This result revealed that TP63 is one of the
candidates as a target of miR-301b. Inverse correlation between
miR-301b and TP63 was observed in five different pancreatic
carcinoma cell lines. The results imply that miR-301b has functions
as an oncogene and that its inhibition may have therapeutic
potential roles for treatment of pancreatic carcinoma and is a
predictive marker of response to chemotherapy in patients with
pancreatic carcinoma.
Materials and methods
Oligonucleotides
Pre-miR-301b, negative control, miR-301b inhibitor
and its negative control were purchased from Ambion (Tokyo, Japan).
TP63-shRNA and negative control were purchased from Invitrogen
(Tokyo, Japan).
Cell lines and cell culture
conditions
Pancreatic carcinoma cell lines (Panc-1 and BxPC-3)
were obtained from the American Type Culture Collection (Rockville,
MD, USA). Panc-1 was maintained in Dulbecco’s modified Eagle’s
medium (DMEM) supplemented with 10% fetal bovine serum (FBS).
BxPC-3 cells were grown in RPMI-1640 with 10% FBS. Both media
contained antibiotics (100 U/ml penicillin and 100 μg/ml
streptomycin). All cell lines were routinely passaged as monolayer
cultures at 37°C in a humidified atmosphere of 95% air and 5%
CO2.
MiR-301b or miR-301b inhibitor
transfection experiments
MiRNA precursor molecules corresponding to miR-301b
and miR-301b inhibitor were transfected using the RNAiMAX
Transfection Reagent (Invitrogen) into Panc-1 and BxPC-3, and the
effects on respective oligonucleotide were measured by quantitative
real-time PCR. Panc-1 and BxPC-3 cells were transfected with 50 nM
microRNA in a 6-well plate for RNA extraction or a 10-cm dish for
wound healing, proliferation and invasion assays following the
manufacturer’s protocol. Cells in the 6-well plate were collected
48 h after transfection to extract RNA and measured for miR-301b
expression. After 12-h transfection, transfected cells in the 10-cm
dish were seeded in 96-well plates for wound healing and
proliferation, or Matrigel coated wells for invasion assays. These
premiRNA and inhibitor transfection experiments were repeated
independently three times.
RNA preparation and real-time PCR
analysis
Total cellular RNA was extracted from cultured cells
using TRIzol (Invitrogen) according to the manufacturer’s
instructions. Cell pellets were suspended in an aliquot of 1
ml/well of TRIzol in a 6-well plate. Isolated RNA (6 μg) was
used for reverse transcription into cDNA (GE Health Care,
Buckinghamshire, UK). Random primers (6-mer) were used according to
the manufacturer’s protocol. cDNA was diluted and stored at −20°C
until use. Gene expression levels were measured with
custom-designed, TaqMan real-time polymerase-chain reaction
(Applied Biosystems, Foster City, CA, USA) containing probes to 6
genes: CDH1 (ID: Hs00156401_m1), TP63 (ID: Hs00174164_m1), IκB-α
(ID: Hs00355671_g1) and miR-301b (ID: 002392) with GAPDH (ID:
Hs99999901_s1) for mRNA or RNAU6 (ID: 001002) for miRNA as an
internal control. The relative expression levels of genes and
miR-301b, relative to GAPDH or RNAU6, were calculated using the
relative quantification ΔΔCt method. Each sample was assayed in
triplicate.
Gemcitabine sensitivity assay with
transfection of premiR-301b or miR-301b inhibitor
The drug sensitivity assay was performed essentially
as described in our previous report (19). Briefly, cells were seeded in a 6-cm
dish at 70% confluency. After 12 h, pre-miR-301b or miR-301b
inhibitor and respective controls were transfected in each dish
overnight. Then transfected cells were seeded in 96-well plates at
4,000 cells/well in triplicate. After incubating for 12 h, cell
viability was determined by treating cells with stepwise 4-fold
serial dilutions of gemcitabine (from 100 μM) and incubated
at 37°C for 96 h. To evaluate cell viability, the cells were fixed
with 25% glutaraldehyde for 30 min at room temperature and then
stained with 200 μl of 0.05% methylene blue for 20 min. The
dye was eluted with 0.33 M HCl for 20 min with agitation.
Absorbance was measured in a microplate reader (model 3550,
Bio-Rad, Tokyo, Japan) at 598 nm. The 50% inhibitory concentration
for cell growth (IC50) was calculated.
Cell invasion assay
Invasion assay was performed in 24-well Biocoat
Matrigel invasion chambers (Becton-Dickinson) according to the
manufacturer’s protocol. Briefly, cells were transfected with
pre-miR-301b or miR-301b inhibitor and each negative control in
10-cm dish. After 12-h transfection, cells were harvested and
plated in the Matrigel coated wells (4 or 5×104/well)
and control insert wells (4 or 5×104/well) using Panc-1
and BxPC-3 cells, respectively. After 20-h incubation, the invasive
cells through the membrane were fixed with methanol for 5 min and
stained by crystal violet for 5 min. Then under a microscope (×20
magnification), invaded cells were counted in 3 random fields. All
assays were performed in triplicate.
Wound healing assay
Transfected cells were seeded in a 6-well plate at
80% confluency, after 12 h, the monolayer of cells was scratched
with 20 μl pipette tip across the center of the well. After
scratching, each well was gently washed with medium to remove the
cell debris. Cells were then grown in appropriate medium. The cells
were allowed to close the wound for 48 h. Photographs were taken at
0, 12, 24 and 36 or 48 h at the same position of the wound in both
Panc-1 and BxPC-3 cells.
Immunofluorescence
Cells were seeded into chamber slide at 40%
confluent. After incubation overnight, pre-miR-301b or miR-301b
inhibitor or negative control (50 nM) oligonucleotides were
transfected for 48 h as described above. Cells were fixed in 4%
paraformaldehyde for 20 min. For permeabilization, 0.15% Triton
X-100 in phosphate-buffered saline (PBS) was applied for 20 min.
Consequently, cells were blocked by 5% goat serum for 1 h at room
temperature. CDH1, NF-κB and TP63 protein expression of cell lines
were detected using anti-CDH1 (Abcam, Cambridge, MA, USA),
anti-NF-κB (Cell Signaling, Tokyo, Japan) and anti-TP63 (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) antibody according to the
manufacturer’s protocol. Alexa Fluor-conjugated antibody was used
as a secondary antibody.
Target gene prediction
Target genes prediction was performed to meet the
following criteria. First, miRNA targets were analyzed using three
algorithms, including TargetScan (http://www.targetscan.org) and microRNA.org
(http://www.microrna.org/). Second, in order to
reduce the number of false positives, only putative target genes
predicted by at least two of the programs were accepted.
Transient transfections of
TP63-shRNA
To knock down endogenous TP63 in Panc-1 and BxPC-3,
the cells were both seeded at 70–80% confluence in 6-well plates.
TP63 and negative control shRNAs were purchased from Invitrogen.
Two different shRNAs were transfected at a final concentration of
80 nM per well using Lipofectamine RNAiMAX reagent (Invitrogen)
following the manufacturer’s recommendations. To validate
suppression efficiency of shRNA, cells were incubated for 48 h and
then harvested for real-time PCR analysis. Subsequently, mixed
shRNAs were transfected in both cell lines respectively. For
immunofluorescence, the cells were seeded into chamber slides.
After 48-h transfection, cells were fixed with 4% paraformaldehyde.
Details as given above.
NF-κB inhibition using IκB-α mutant
adenovirus in Panc-1 and BxPC-3 cells
To investigate the association with NF-κB and CDH1,
NF-κB activation was downregulated by IκB-α mutant adenovirus in
Panc-1 and BxPC-3 cells. Both viruses were purchased from Vector
Biolabs (Philadelphia, PA, USA). Cells were seeded in a 6-well
plate at 70–80% confluence. Twelve hours later, mutant and the
control adenovirus were infected at a MOI (multiplicity of
infection) of 100 for serial two days in the cells, respectively.
Then, the RNA was extracted for real-time PCR analysis for CDH1
expression.
Activation of NF-κB by lipopolysaccharide
(LPS) in miR-301b knock-down cells
To further examine the association with NF-κB and
CDH1, LPS (Sigma-Aldrich, St. Louis, MO, USA) was added to activate
NF-κB. Cells were seeded in a 6-well plate at 70–80% confluence.
After 12 h, miR-301b or negative control viruses were transfected
at 50 nM using RNAiMax (Invitrogen). Forty-eight hours later, LPS
was added in each well at 0, 2.5 and 5 μg/ml for 2 h. Then,
the RNA was extracted for real-time PCR analysis for CDH1
expression.
Statistical analysis
All results were performed in triplicate and carried
out on at least two times. Data are shown as the mean ± SD where
applicable. Graphpad Prism v5.0 (Graphpad Software Inc., La Jolla,
CA, USA) was used for all statistical analysis. Levels of
significance for comparison between cell lines were determined by
the Student’s t-test distribution. To assess the correlation
between miR-301b expression, TP63 expression and CDH1 expression,
Pearson correlation analysis was performed in pancreatic carcinoma
cell lines. The probability of P<0.05 was considered to be
statistically significant.
Results
MiR-301b regulated invasiveness and
promoted gemcitabine resistance in Panc-1 and BxPC-3 cells
To evaluate functional role of miR-301b in
pancreatic carcinoma, pre miR-301b or miR-301b inhibitor were
transfected in Panc-1 and BxPC-3 cells using lipofectamine.
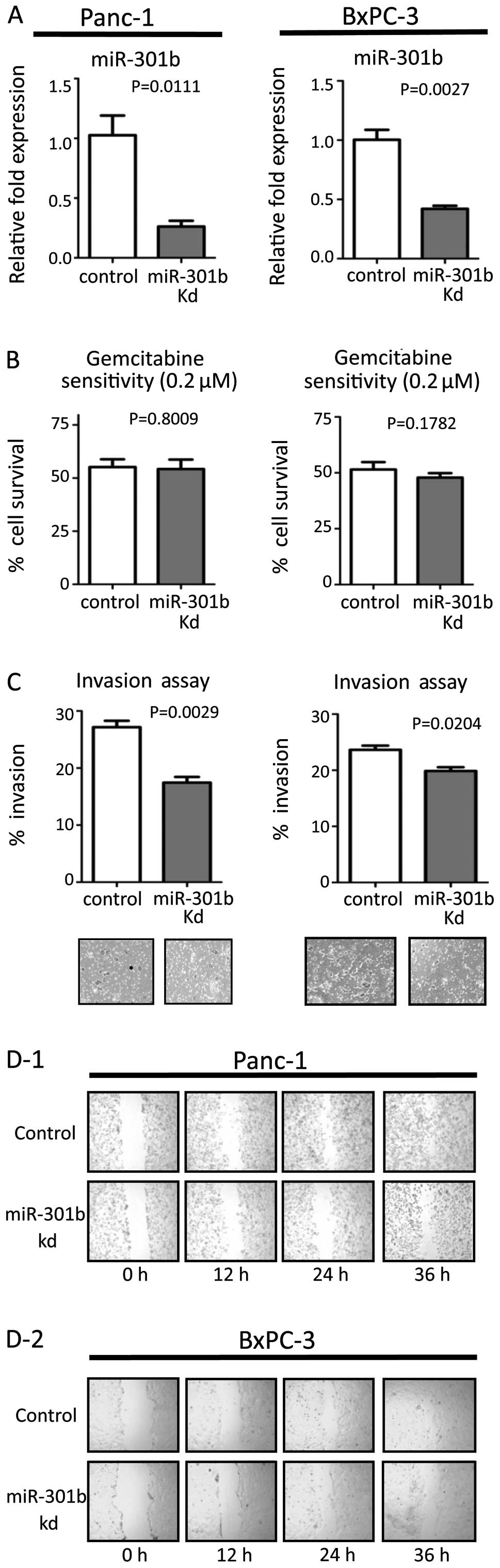
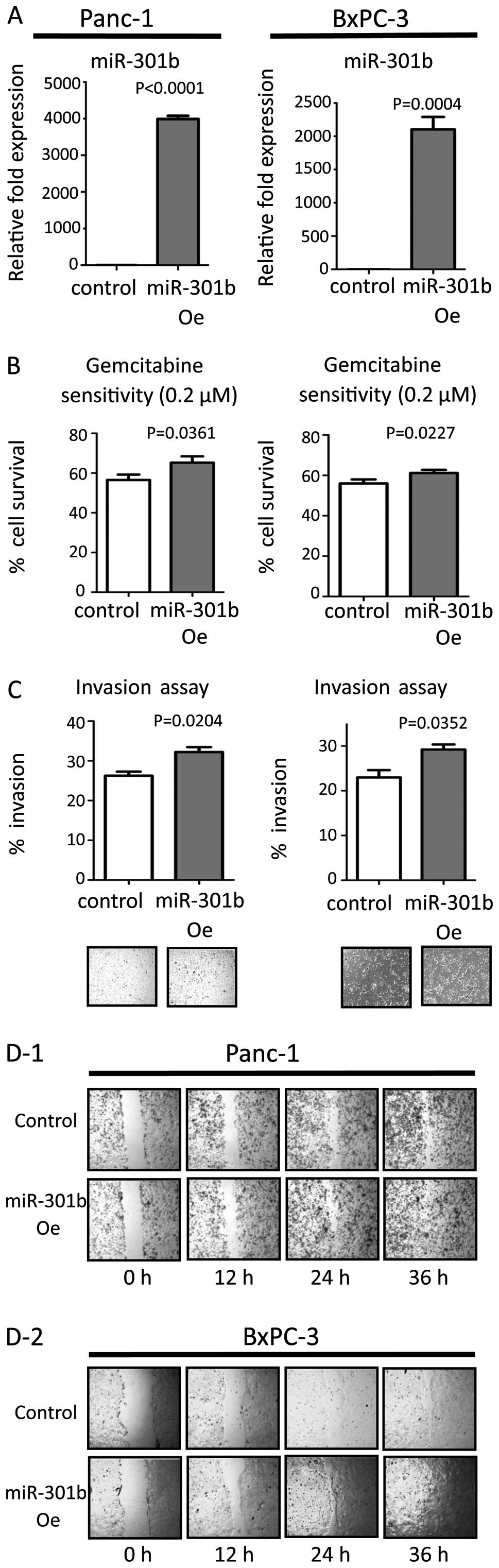
Efficacy of transfection was confirmed by real-time PCR (Figs. 1A and 2A). MiR-301b inhibitor transfection
suppressed their invasiveness, while inhibition of miR-301b did not
affect sensitivity to gemcitabine (Fig. 1B and C). On the other hand,
miR-301b precursor molecules enhanced invasiveness in both cell
line types. Moreover, miR-301b transfection increased gemcitabine
resistance (Fig. 2B and C). Next,
migration ability was evaluated by the wound healing assay. We
transiently inhibited or forced miR-301b expression in both cells.
Inhibition of miR-301b reduced migration (Fig. 1D). On the other hand, forced
miR-301b promoted the migration ability compared to control
transfected cells (Fig. 2D). These
results were consistent in both cell lines. Transfected cells did
not show any change by the proliferation assay in either cell line
(data not shown).
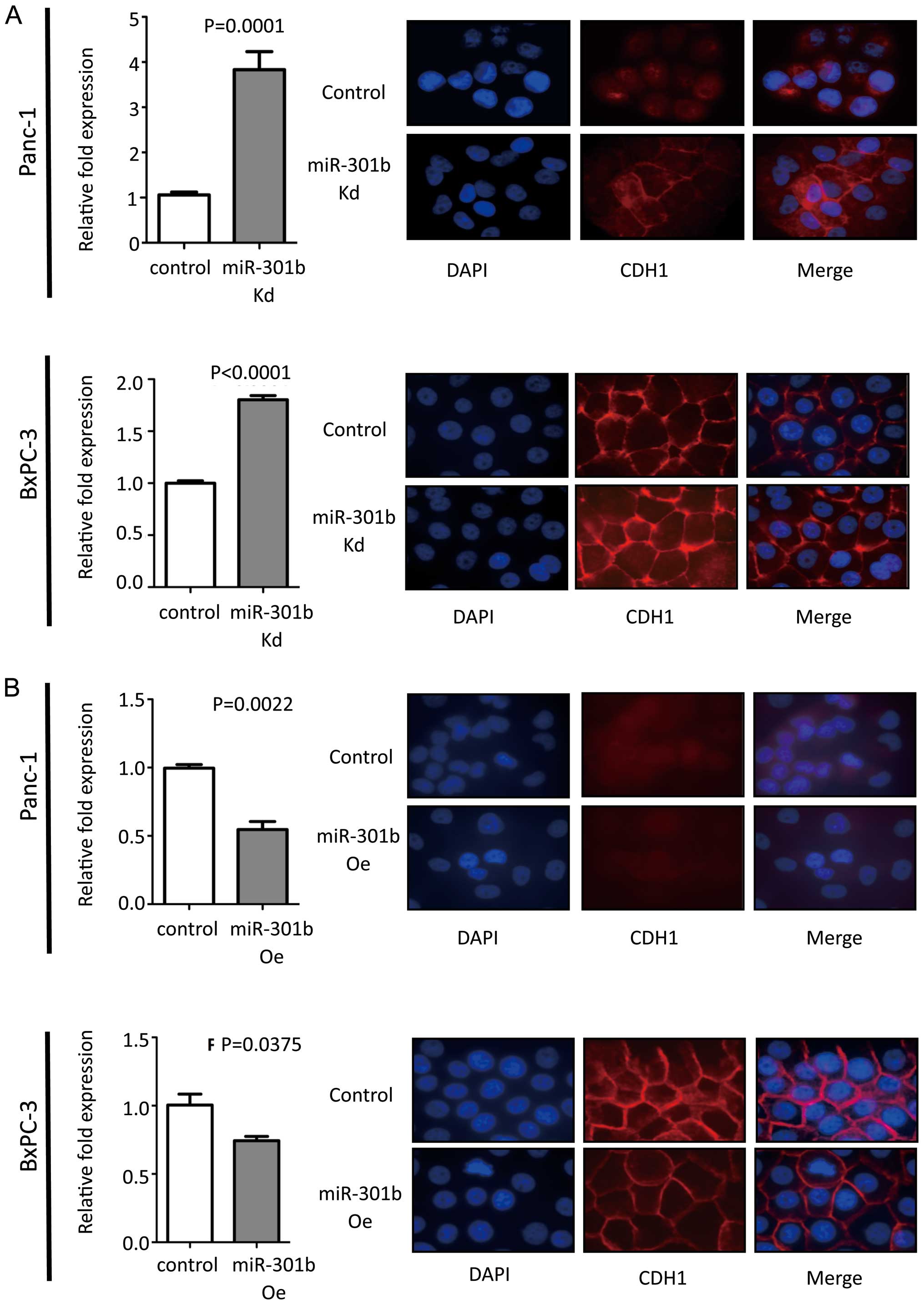
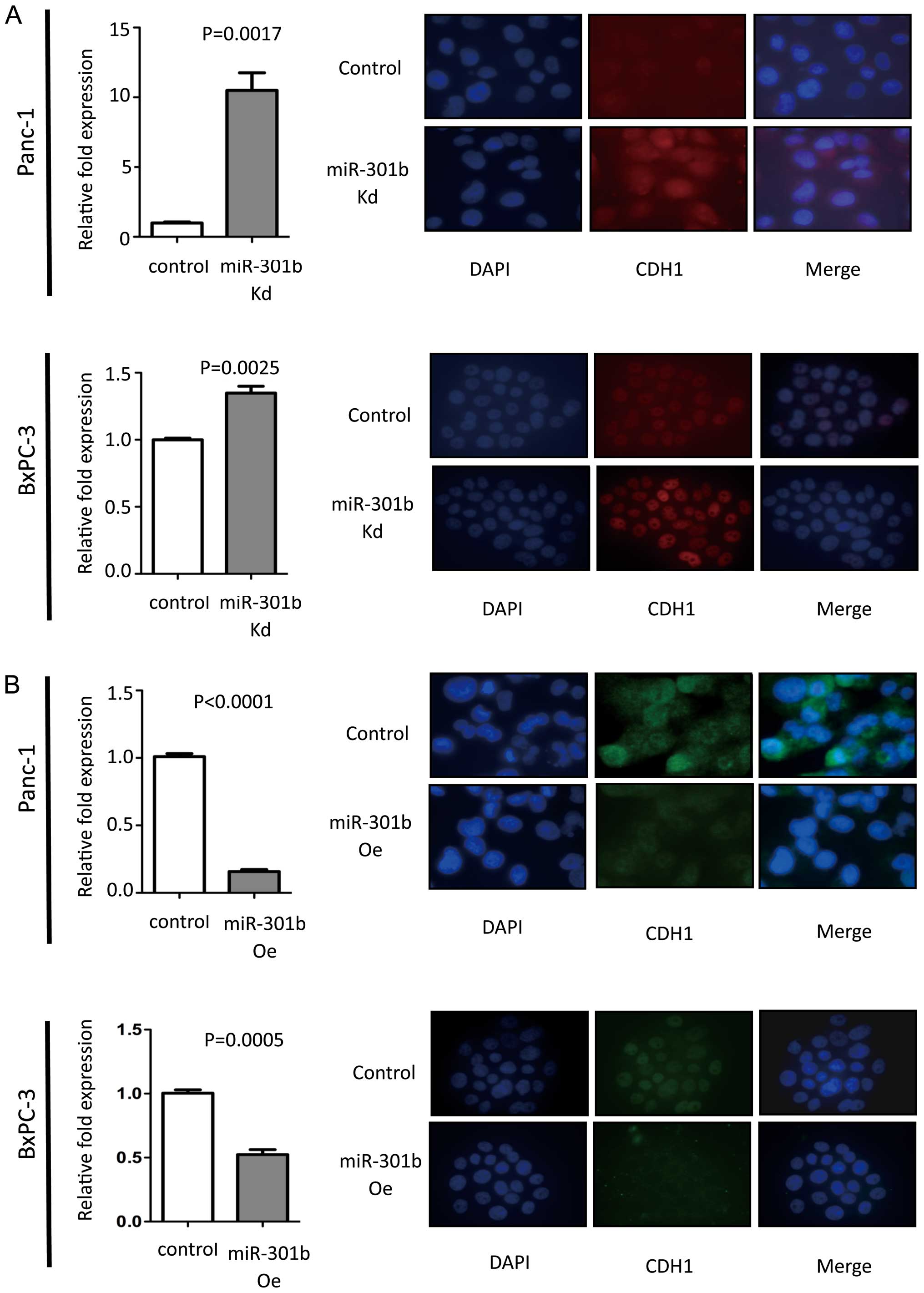
MiR-301b expression level was inversely
correlated with CDH1 expression
MiR-301b inhibition exhibited increased CDH1
expression at mRNA level compared to control in Panc-1 cells. Then
immunofluorescence was performed for CDH1. MiR-301b knock-down
cells showed enhanced CDH1 protein in Panc-1 cells. Inversely,
overexpressed miR-301b cells showed reduced CDH1 expression levels.
To validate this phenomenon, real-time PCR and immunofluorescence
experiments were carried out in BxPC-3 cells. We obtained the same
results with Panc-1 cells (Fig.
3). However, morphology remained unchanged in miR-301b
overexpression and knock-down cells (data not shown). We also
measured epithelial to mesenchymal transition (EMT)-related genes,
such as Vimentin, Snail, Slug, Twist, Zeb1 and Zeb2 by real-time
PCR. Unfortunately, the results did not exhibit consistent pattern
of such gene expression to explain how miR-301b affected CDH1
expression in the cell lines (data not shown).
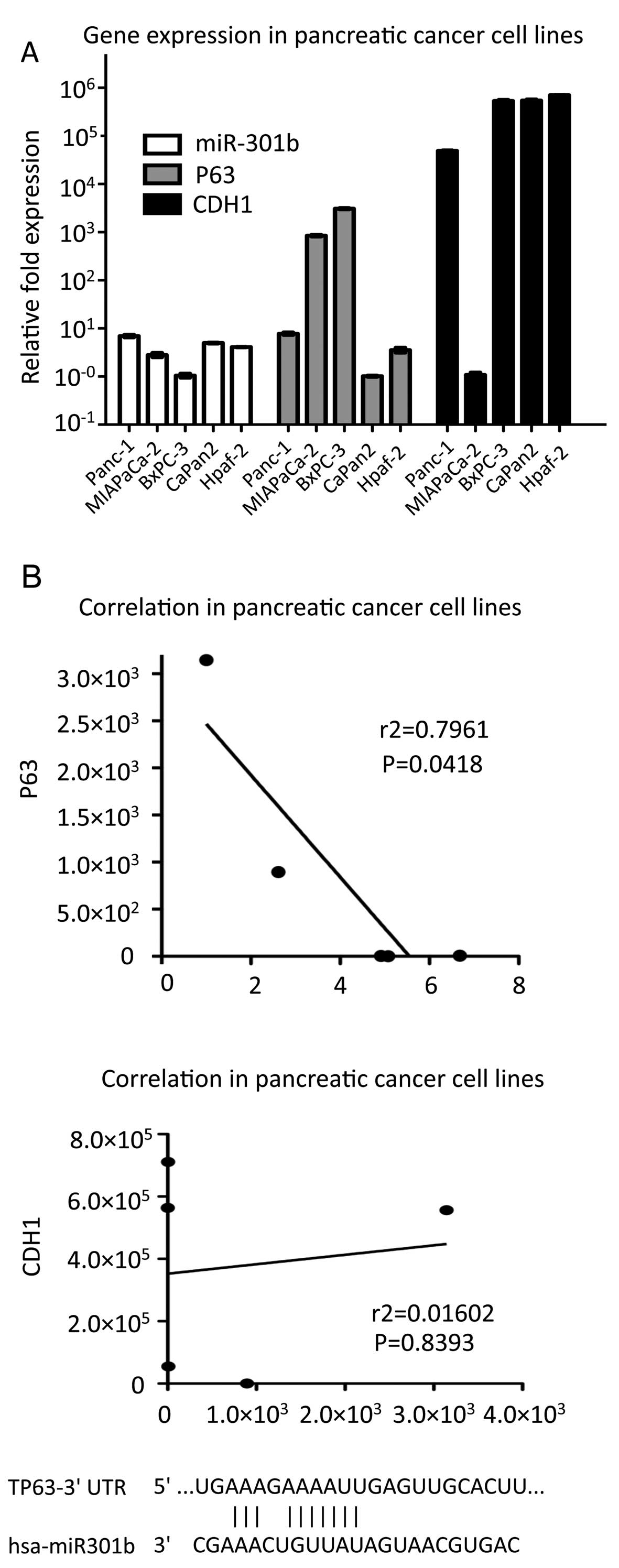
TP63 is suggested as a potent target for
miR-301b
We explored candidates of miR-301b target using
miRNA target search algorithms which were available at TargetScan
and microRNA.org. There were 2,136 and 7,903 potential
targets respectively. Among the putative considerable targets, we
focused on specific genes which were associated with CDH1
expression. We chose PTEN and TP63 as potential targets of
miR-301b. Initially, to identify pathways which are involved in
miR-301b, we investigated the miR-301b, TP63, PTEN and CDH1
expression using five different cell lines (Panc-1, MIAPaCa-2,
BxPC-3, Capan-2 and Hpaf-2) by real-time PCR method. Interestingly,
the result showed a clear inverse correlation between miR-301b and
TP63 expression in Pearson data (r2= 0.7961) (Fig. 4A). However, there was no
correlation in miR-301b-PTEN, PTEN-CDH1 and TP63-CDH1 relation,
respectively (data not shown). These data indicated that TP63 was
one of the putative targets for miR-301b. Therefore, we
hypothesized a possible pathway to demonstrate how miR-301b
regulates CDH1 expression (Fig.
4B).
MiR-301b regulates TP63 as one of several
miR-301b target genes in pancreatic carcinoma
Further study revealed that miR-301b transfection
attenuated endogenous TP63 expression relative to control cells in
Panc-1 and BxPC-3 cells (Fig. 5B).
In contrast, downregulated miR-301b demonstrated increased TP63
expression in both cell lines as well (Fig. 5A). These results suggested that
TP63 was the most likely candidate to have interaction with
miR-301b.
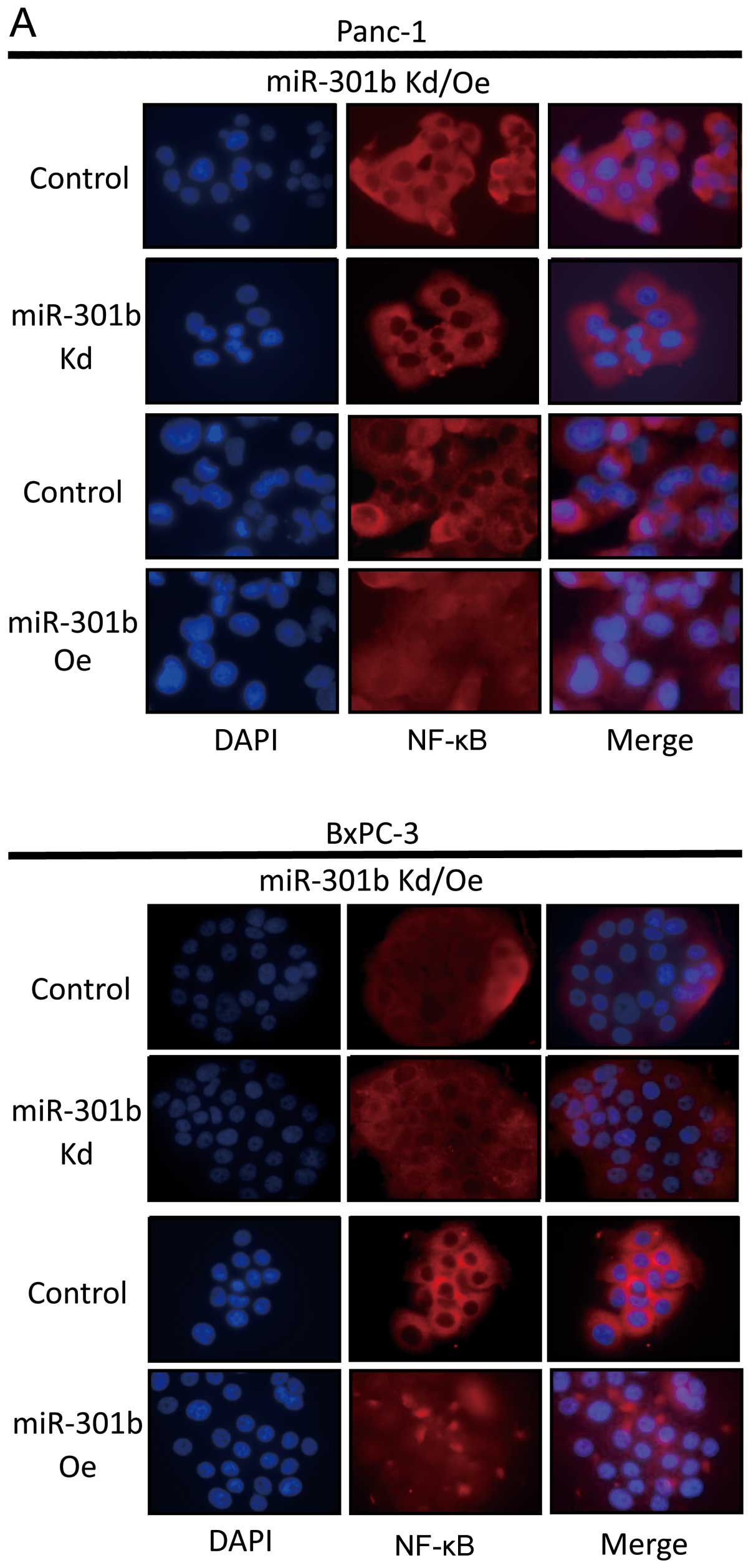
MiR-301b expression level is associated with NF-κB
activity. In miR-301b-inhibited cells, we found that NF-κB was
downregulated in Panc-1 and BxPC-3. Moreover, when pre-miR-301b was
transfected, NF-κB moved into nucleus from cytoplasm in both cell
lines (Fig. 6A). To further study
miRNA-mediated NF-κB activation in pancreatic carcinoma cells,
endogenous TP63 was repressed by TP63 shRNA. In both cell lines,
inhibition efficiency showed ≥60% inhibition at each shRNA. TP63
knock-down cells exhibited activated NF-κB by immunofluorescence
analysis and downregulated CDH1 by real-time PCR and
immunofluorescence analysis (Fig.
6B).
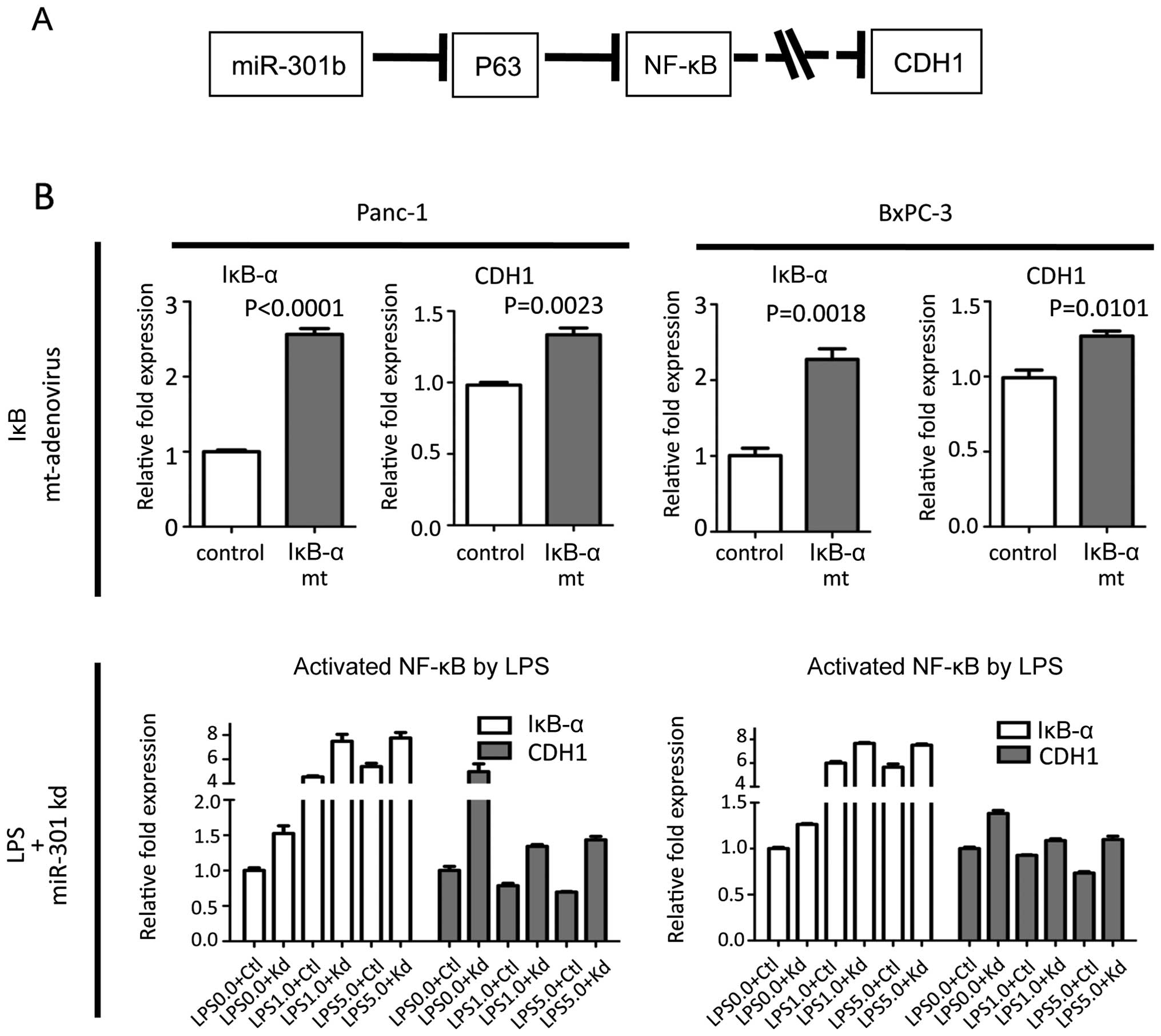
NF-κB regulated CDH1 expression
To examine our putative pathway (Fig. 7A), NF-κB was suppressed using IκB-α
mutant adenovirus. When Panc-1 and BxPC-3 cells were infected with
adenovirus, CDH1 was slightly upregulated. In contrast, when NF-κB
was activated by lipopolysaccharide (LPS) in miR-301 knock-down
cells, NF-κB activation by LPS significantly negated the beneficial
effect on upregulation of CDH1 by miR-301b inhibition (Fig. 7B). These data suggested that the
NF-κB regulates CDH1 expression.
Discussion
Deregulated expression of miRNAs is responsible for
cancer initiation and development in many types of cancer. Recent
reports have revealed that aberrant expression of miRNAs is implied
in cell proliferation, cell invasion and chemo-sensitivity. Among
these miRNAs, there is accumulated evidence suggesting that miR-301
is upregulated in several malignant tumors (11,14,15).
Furthermore, functional role of miR-301 has been reported (16,17).
Shi et al demonstrated that miR-301 overexpression is
implicated as a negative prognostic factor in lymph node-negative
invasive ductal breast cancer. They also showed that miR-301
attenuation decreased cell proliferation, clonogenicity, migration,
invasion, tamoxifen resistance, tumor growth, and microvessel
density through targeting FOXF2, BBC3, PTEN, and COL2A1 (17). In addition, Cao et al showed
that miR-301 targeted MEOX2 to affect the ERK/CREB pathway in a
lung carcinoma cell line (16). A
recent study in pancreatic carcinoma demonstrated that miR-301a
contributed to activation of NF-κB by repressing NKrf which
interacted with specific negative regulatory elements to mediate
transcriptional repression of NF-κB (18). These findings indicate that miR-301
may function as an oncogene by inhibiting tumor suppressor
genes.
In this study we focused on investigating how
miR-301b regulated invasiveness of pancreatic carcinoma, since
miR-301b attenuation showed reduced invasiveness. Moreover,
exogenous miR-301b enhanced invasiveness in both Panc-1 and BxPC-3
cell lines. Consistent with previous report in breast cancer
(17), our data showed similar
results even in pancreatic carcinoma cell lines. As a factor of
enhanced invasiveness, we found that CDH1 might be a candidate to
explain the phenomenon among the epithelial to mesenchymal
transition (EMT)-related gene expression, such as CDH1, Vimentin,
Zeb1, Zeb2, Twist, Snail and Slug, which were measured using
real-time PCR when miR-301b was forced or inhibited in both Panc-1
and BxPC-3 cells. However, miR-301b expression level did not affect
their morphology in spite of alteration of CDH1 expression (data
not shown). To investigate the mechanism how miR-301b could
influence CDH1 expression using TargetScan and microRNAs.org prediction tools for possible mRNA
targets, we selected two genes, PTEN and TP63 on the condition
that: i) the target has to overlap with both algorithms, ii) the
target has to be a tumor suppressor gene, and iii) the target has
to be associated with CDH1. PTEN was already published as a target
of miR-301 in breast cancer (17).
However, in our data, PTEN expression was not associated with
miR-301b expression level in pancreatic carcinoma cells (data not
shown). Therefore, we examined TP63 as a novel putative target of
miR-301b. i) Statistical analysis demonstrated strong correlation
between CDH1 and miR-301b (r2= 0.7961, P= 0.0418) in
five pancreatic carcinoma cell lines. ii) Forced
expression/knock-down miR-301b reduced/induced TP63 expression.
According to these findings, we concluded that TP63 is a new
potential target for miR-301b.
TP63, which is a member of the P53 tumor suppressor
gene family, is critical for the development of stratified
epithelial tissues, such as epidermis, breast (20,21)
and for cell viability (22). TP63
gene is transcribed from two different promoters, generating two
types of isoforms which either contain or lack an amino-terminal
transactivation domain referred to as TA and ΔN isoforms,
respectively. TAp63 has been implicated in regulation of cell
proliferation, apoptosis and differentiation. Elevated TAp63 has
several functions inducing p53-responsive genes, inhibiting cell
proliferation and promoting apoptosis (23). Inhibition of TAp63 induced
chemoresistance (24). TAp63
isoforms acted as tumor suppressors by regulating senescence
through p53-independent pathways (25), whereas ΔNp63 isoforms enhance
proliferation and inhibit apoptosis (26,27).
A recent report showed that TAp63 regulates NF-κB transcription and
protein stability, subsequently leading to the cell death phenotype
(28). Another report showed TAp63
is a transcriptional target of NF-κB, which may play a role in cell
proliferation, differentiation and survival upon NF-κB activation
(29,30). In contrast, NF-κB repressed CDH1
expression through enhancing Zeb1 expression (31). Based on this evidence, we
hypothesized that miR-301b regulates CDH1 expression through TP63
and NF-κB (Fig. 7A). Overexpressed
miR-301b cells exhibited more activated NF-κB than control cells in
both pancreatic carcinoma cell lines. Furthermore, TP63 inhibition
by shRNA showed activated NF-κB and reduced CDH1 expression. On the
other hand, NF-κB inhibition using IκB-α mutant adenovirus revealed
elevated CDH1. In addition, NF-κB activation by lipopolysaccharide
(LPS) induced downregulated CDH1 expression in both cell lines.
Effect of LPS on NF-κB compensated for upregulated CDH1 expression
by miR-301b inhibition. Considering these data, TP63 might control
CDH1 expression through NF-κB activation. However, unlike previous
published data (31), our data did
not show an association between NF-κB and Zeb1/CDH1 axis. Contrary
to our expectations, activation/inactivation of NF-κB did not show
any major change in CDH1 expression in either cell line, and TP63
did not have any significant correlation with CDH1 in five
different cell line data. These data suggested that NF-κB does not
regulate CDH1 expression exclusively through Zeb1, but also through
other transfactors. Our results indicated that miR-301b expression
contributed to NF-κB activation in pancreatic carcinoma, which was
the same as previous reports, however, the target to regulate NF-κB
was different (18). Consistent
with some published findings, our results supported the hypothesis
that miR-301b might suppress CDH1 through the TP63/NF-κB pathway as
an oncogene in pancreatic carcinoma. Hence, with increased amount
of evidence toward an explanation for pancreatic carcinoma,
treatment tailored to each individual’s gene profiling will be
extremely desired (32,33). The limitations of our study include
the unclear mechanism how TP63 regulates CDH1 expression through
NF-κB, and lack of a mouse study.
In conclusion, our data indicated that TP63 could be
targeted by miR-301b in pancreatic carcinoma. Our data suggested
that miR-301b might be useful as a biomarker of malignant
potential. We also demonstrated that miR-301b promotes cell
invasion through inhibition of CDH1 by targeting the tumor
suppressor gene TP63 in pancreatic carcinoma. Finally, exogenous
miR-301b was involved in gemcitabine resistance. These data
indicated that tailored treatments according to gene expression in
each patient will be employed for treatment of pancreatic carcinoma
in the near future.
Acknowledgements
The authors would like to thank Dr
Mitsuhiro Yoneda for helpful discussions throughout this study.
References
|
1.
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J, et
al: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249. 2009.
View Article : Google Scholar
|
|
2.
|
Eltawil KM, Renfrew PD and Molinari M:
Meta-analysis of phase III randomized trials of molecular targeted
therapies for advanced pancreatic cancer. HPB (Oxford). 14:260–268.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Li D, Xie K, Wolff R and Abbruzzese JL:
Pancreatic cancer. Lancet. 363:1049–1057. 2004. View Article : Google Scholar
|
|
4.
|
Bartel D: MicroRNAs: genomics, biogenesis,
mechanism, and function. Cell. 116:281–297. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Piepoli A, Tavano F, Copetti M, Mazza T,
Palumbo O, et al: MiRNA expression profiles identify drivers in
colorectal and pancreatic cancers. PLoS One. 7:e336632012.
View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Nikitina EG, Urazova LN and Stegny VN:
MicroRNAs and human cancer. Exp Oncol. 34:2–8. 2012.
|
|
7.
|
Iorio MV and Croce CM: MicroRNA
dysregulation in cancer: diagnostics, monitoring and therapeutics.
A comprehensive review. EMBO Mol Med. 4:143–159. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Li C, Hashimi SM, Good DA, Cao S, Duam W,
et al: Apoptosis and microRNA aberrations in cancer. Clin Exp
Pharmacol Physiol. 39:739–746. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Garofalo M, Romano G, Di Leva G, Nuovo G,
Jeon YJ, et al: EGFR and MET receptor tyrosine kinase-altered
microRNA expression induces tumorigenesis and gefitinib resistance
in lung cancers. Nat Med. 18:74–82. 2011.PubMed/NCBI
|
|
10.
|
Wu Y, Xiao Y, Ding X, Zhuo Y, Ren P, et
al: A miR-200b/200c/429-binding site polymorphism in the 3′
untranslated region of the AP-2α gene is associated with cisplatin
resistance. PLoS One. 6:e290432011.PubMed/NCBI
|
|
11.
|
Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner
MR, et al: Expression profiling identifies microRNA signature in
pancreatic cancer. Int J Cancer. 120:1046–1054. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Jiang J, Gusev Y, Aderca I, Mettler TA,
Nagorney DM, et al: Association of MicroRNA expression in
hepatocellular carcinomas with hepatitis infection, cirrhosis, and
patient survival. Clin Cancer Res. 14:419–427. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Miko E, Czimmerer Z, Csánky E, Boros G,
Buslig J, et al: Differentially expressed microRNAs in small cell
lung cancer. Exp Lung Res. 35:646–664. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Lu YC, Chen YJ, Wang HM, Tsai CY, Chen WH,
et al: Oncogenic function and early detection potential of
miRNA-10b in oral cancer as identified by microRNA profiling.
Cancer Prev Res. 5:665–674. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Wang YX, Zhang XY, Zhang BF, Yang CQ, Chen
XM and Gao HJ: Initial study of microRNA expression profiles of
colonic cancer without lymph node metastasis. J Dig Dis. 11:50–54.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Cao G, Huang B, Liu Z, Zhang J, Xu H, et
al: Intronic miR-301 feedback regulates its host gene, ska2, in
A549 cells by targeting MEOX2 to affect ERK/CREB pathways. Biochem
Biophys Res Commun. 396:978–982. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Shi W, Gerster K, Alajez NM, Tsang J,
Waldron L, et al: MicroRNA-301 mediates proliferation and invasion
in human breast cancer. Cancer Res. 71:2926–2937. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Lu Z, Li Y, Takwi A, Li B, Zhang J, et al:
miR-301a as an NF-κB activator in pancreatic cancer cells. EMBO J.
30:57–67. 2011.
|
|
19.
|
Funamizu N, Okamoto A, Kamata Y, Misawa T,
Uwagawa T, et al: Is the resistance of gemcitabine for pancreatic
cancer settled only by overexpression of deoxycytidine kinase?
Oncol Rep. 23:471–475. 2010.PubMed/NCBI
|
|
20.
|
Yang A, Kaghad M, Wang Y, Gillett E,
Fleming MD, et al: p63, a p53 homolog at 3q27-29, encodes multiple
products with trans-activating, death-inducing, and
dominant-negative activities. Mol Cell. 2:305–316. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Yang A, Schweitzer R, Sun D, Kaghad M,
Walker N, et al: p63 is essential for regenerative proliferation in
limb, craniofacial and epithelial development. Nature. 398:714–718.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Yuan M, Luong P, Hudson C, Gudmundsdottir
K and Basu S: c-Abl phosphorylation of ΔNp63α is critical for cell
viability. Cell Death Dis. 1:e162010.
|
|
23.
|
Helton ES, Zhang J and Chen X: The
proline-rich domain in p63 is necessary for the transcriptional and
apoptosis-inducing activities of TAp63. Oncogene. 27:2843–2850.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Gressner O, Schilling T, Lorenz K, Schulze
Schleithoff E, Koch A, et al: TAp63alpha induces apoptosis by
activating signaling via death receptors and mitochondria. EMBO J.
24:2458–2471. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Guo X, Keyes WM, Papazoglu C, Zuber J, Li
W, et al: TAp63 induces senescence and suppresses tumorigenesis in
vivo. Nat Cell Biol. 11:1451–1457. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Chiang CT, Chu WK, Chow SE and Chen JK:
Overexpression of delta Np63 in a human nasopharyngeal carcinoma
cell line downregulates CKIs and enhances cell proliferation. J
Cell Physiol. 219:117–122. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Schavolt KL and Pietenpol JA: p53 and
Delta Np63 alpha differentially bind and regulate target genes
involved in cell cycle arrest, DNA repair and apoptosis. Oncogene.
26:6125–6132. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Sen T, Sen N, Huang Y, Sinha D, Luo ZG, et
al: Tumor protein p63/nuclear factor κB feedback loop in regulation
of cell death. J Biol Chem. 286:43204–43213. 2011.PubMed/NCBI
|
|
29.
|
Wu J, Bergholz J, Lu J, Sonenshein GE and
Xiao ZX: TAp63 is a transcriptional target of NF-kappaB. J Cell
Biochem. 109:702–710. 2010.PubMed/NCBI
|
|
30.
|
Hayden MS and Ghosh S: Shared principles
in NF-kappaB signaling. Cell. 132:344–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Chua HL, Bhat-Nakshatri P, Clare SE,
Morimiya A, Badve S and Nakshatri H: NF-kappaB represses E-cadherin
expression and enhances epithelial to mesenchymal transition of
mammary epithelial cells: potential involvement of ZEB-1 and ZEB-2.
Oncogene. 26:711–724. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Funamizu N, Kamata Y, Misawa T, Uwagawa T,
Lacy CR, et al: Hydroxyurea decreases gemcitabine resistance in
pancreatic carcinoma cells with highly expressed ribonucleotide
reductase. Pancreas. 41:107–113. 2010. View Article : Google Scholar
|
|
33.
|
Funamizu N, Lacy CR, Fujita K, Furukawa K,
Takeyuki M, et al: Tetrahydrouridine inhibits cell proliferation
through cell cycle regulation regardless of cytidine deaminase
expression levels. PLoS One. 7:e374242012. View Article : Google Scholar : PubMed/NCBI
|