Introduction
DNA topoisomerases are essential nuclear enzymes
that function to resolve topological problems in DNA, which
normally occur during replication, transcription, and other
DNA-associated processes. The family of topoisomerases has two
major members - type I (topo I) and type II (topo II), and their
catalytic activity involves the formation of transient covalent
bridges of enzyme-DNA complexes (1–5). The
involvement of these enzymes in essential cellular processes tagged
topoisomerases as important targets for anticancer treatments and
for the development of potent, more effective, anticancer
drugs.
Topoisomerase I interacts with camptothecin (CPT)
and several of its analogs (i.e. topotecan, irinotecan) at the
interface of the enzyme-DNA complex to induce cell-death. Topo I is
the only target of these alkaloid compounds (6,7).
Clinical activity includes phase I–III studies for the indicated
FDA approved agents for ovarian, colon, small- and non-small cell
lung cancers. Other topoisomerase I inhibitors are being tested in
the clinic, in the treatment of pancreatic, breast and hematologic
malignancies (6).
Preclinical studies on cell lines and tumor
xenografts demonstrated the antitumor activity of
EGFR-TK-inhibitors, as single agent or in combination with other
drugs, including topoisomerase interacting agents (8–11).
The role of epidermal growth factor receptor (EGFR)
has been identified in various tumors (12,13).
The EGFR signaling pathway is one of the most important pathways
that regulate growth, survival, proliferation, and differentiation
in cells.
The EGFR is overexpressed, dysregulated or mutated
in many epithelial malignancies, and its activity is important in
tumor growth, progression and metastatic ability. This has made
these receptors the target of development of anticancer treatment
approaches (13,14).
The most predominant clinical approach of EGFR
inhibition includes monoclonal antibodies that target the
extracellular domain of the receptor, and small molecule tyrosine
kinase inhibitors (TKIs) that inhibit the receptor’s catalytic
activity (15). Erlotinib
(Tarceva) and gefitinib (Iressa, ZD-1839) are small-molecule
tyrosine kinase inhibitors directed against the epidermal growth
factor receptor. These molecules block the intracellular
autophosphorylation of the receptor, and affects EGFR-mediated cell
proliferation (16).
It was previously demonstrated in our lab, and
others, that certain small synthetic tyrosine kinase antagonist
molecules, tyrphostins, inhibit the catalytic activity of purified
topo I enzyme (17), as well as
the cellular topo I in drug-treated cells (18).
In recent years, it has been shown in vitro
and in vivo, that the combined treatment of human colorectal
cancer cells, with anti-EGFR drugs and with topoisomerase I
inhibitors increased the antitumor activity of either agent alone
(8,9,19)
and with no increased toxicity (10,19).
Phase II studies have demonstrated that combined therapy of triple
negative breast cancer (TNBC) with cetuximab (an anti-EGFR
antibody) and cisplatin dramatically increased patients’ response
rate, compared to cisplatin treatment alone and doubled their
progression-free survival duration (20).
Gefitinib was found to modulate SN-38 (the active
metabolite of CPT-11) ability to inhibit topo I in colorectal tumor
cell lines, to accumulate cells in S-phase, and to induce apoptosis
(21). Erlotinib was found to
inhibit tumor growth and metastasis in a TNBC xenograft model
(20).
These findings indicate that EGFR-TKIs can enhance
the antitumor activity of other anticancer agents, as well as
topoisomerase inhibitors, without enhanced toxicity, and will be
effective against tumor cells.
There is an immense value in finding drugs that can
modulate several cellular targets. Therefore, understanding the
full potential of these drugs and their mode of action can ensure
effective treatment protocols.
As tyrphostins are not clinically approved, we
sought to investigate the effect of erlotinib and gefitinib on topo
I. In this study we show for the first time that topo I is an
additional target of erlotinib and gefitinib in drug-treated tumor
cells. Their mechanism differs from camptothecins, known inhibitors
of topo I. While erlotinib inhibits topo I by affecting the ability
of the enzyme to bind DNA, gefitinib probably affects the enzyme
through regulation of the EGFR signaling pathway. Furthermore,
combined treatments based on low doses of erlotinib or gefitinib
and CPT enhanced the inhibitory effect of CPT in MCF7 cells.
Materials and methods
Cells
MCF7 and PC3 cell lines were cultured as a monolayer
in DMEM or RPMI-1640 medium (Biological Industries Beith Haemek,
Israel), respectively, supplemented with 10% fetal bovine serum,
100 U/ml penicillin, 100 μg/ml streptomycin and L-glutamine.
Cell lines were grown in a humidified incubator supplemented with
5% CO2, at 37°C.
Enzymes, antibodies and compounds
Erlotinib (Tarceva®) was kindly provided
by Roche Diagnostics GmbH Pharma Research, Penzberg, Germany.
Gefitinib (Iressa™) was kindly provided by AstraZeneca
Pharmaceuticals (Cheshire, UK). Stock solutions of erlotinib and
camptothecin (Sigma, Israel), at 20 mM (dissolved in 100% DMSO),
and of gefitinib, at 50 mM, were stored in aliquots at −70°C and
diluted in DMSO before being added to the reaction mixture or to
the cell culture medium. Stock solution of etoposide (Teva,
Israel), at 34 mM, was stored at room temperature.
Purified calf thymus topoisomerase I was purchased
from Takara Bio Inc. Supercoiled DNA plasmid pUC19 and E.
coli (cells with a cloned pseT gene of bacteriophage T4)
T4 polynucleotide kinase were purchased from Fermentas (Hanover,
MD, USA).
The primary antisera were as follows: monoclonal
mouse anti-β-actin antibody (MP Biomedicals, LLC); goat polyclonal
IgG (C-15) anti-topo I (Santa Cruz Biotechnology Inc., CA,
USA).
Appropriate horseradish peroxidase secondary
antibodies were purchased from Santa Cruz Biotechnology Inc.
Enhanced chemiluminescence (ECL) reagents were purchased from
Biological Industries Beith Haemek, Israel.
Topo I DNA-binding sequence oligonucleotides were
obtained from Sigma-Aldrich, Israel. [γ-32P]ATP was
purchased from Saifan Precision Instruments Ltd., Israel.
Cell proliferation assay
Cells were plated as triplicate in 96-well plates at
a density of 5,000 cells/well in 100 μl of DMEM medium and
incubated overnight. Various concentrations of the drugs were added
for different time intervals. Control cultures received medium
containing the highest concentration of the vehicle (DMSO) present
in any treatment group. Plates were incubated at 37°C. Cell
cytotoxicity was measured by the Neutral Red assay (22). The CC50 value for each
drug was calculated.
Nuclear protein extracts preparation
Nuclear extracts for topoisomerase assays and
western blot analysis from MCF7 and PC3 cells was prepared as
described (23,24) except that a mixture of protease
inhibitors (final concentrations: 2 μg/ml aprotinin, 2
μg/ml leupeptin, 1 μg/ml pepstatin A, 2 μg/ml
antipain, 100 μg/ml PMSF) was added to the extraction
buffers. Total protein concentration was determined using the
Bio-Rad protein assay kit (Bio-Rad Lab, CA, USA).
Topoisomerase I assay
Purified calf thymus topo I (1 U) or 25 ng of total
nuclear proteins from drug-treated and untreated cell lines was
added to a topo I specific reaction mixture containing, at a final
volume of 25 μl: 20 mM Tris-HCl (pH 8.0), 1 mM
dithiothreitol, 20 mM KCl, 10 mM MgCl2, 1 mM EDTA, 30
μg/ml bovine serum albumin and 225 ng pUC19 supercoiled DNA
plasmid. Erlotinib, at various concentrations, was added to the
reaction mixture prior to the addition of the enzyme. Following
incubation at 37°C for 30 min, the reaction was terminated by
adding 5 μl of stopping buffer [final concentration; 1%
sodium dodecyl sulfate (SDS), 15% glycerol, 0.5% bromophenol blue
and 50 mM EDTA pH 8.0]. The reaction products were analyzed by
electrophoresis on 1% agarose gel using a TBE buffer (89 mM
Tris-HCl, 89 mM boric acid, and 62 mM EDTA) at 1 V/cm, stained by
ethidium bromide (1 μg/ml), and photographed using a short
wavelength UV lamp (Chemilmager™ 5500 equipment, Alpha Inotech
Corp., CA, USA). Densitometric analysis of the results was
performed using the EZQuant-Gel software (EZQuant, Rehovot,
Israel), and the percentage of topo I inhibition was calculated
(17,18).
Determination of the level of topo I
protein by western blot analysis
Equal amounts of nuclear proteins derived from MCF7
cells that were subjected to different treatments, were analyzed by
western blot analysis as previously described (24,25)
using either anti topo I antibodiy (Santa Cruz Biotechnology Inc.),
or anti-β-actin antibodies (MP Biomedicals, LLC). The
immunocomplexes were detected by enhanced chemiluminescence
(ECL).
Electromobility shift assays
In vitro topo I DNA-binding activity was
assayed by incubating in a total volume of 25 μl for 5 min
at 37°C. A [γ-32P]-labeled double-stranded
oligonucleotide (31-bp) containing the consensus sequence for topo
I was added to the reaction mixture. Erlotinib (100 pM) or CPT (120
μM) were added to each sample. The reaction products were
electrophoresed on a 6% native polyacrylamide gel that had been
pre-electrophoresed for 1 h. Gel was then dried for 30 min at 80°C
and authoradiography was performed.
Oligonucleotides
Oligonucleotides sequence: sense 5′-CATG
AAAAAAGACTTAGAAAAATTTTTAAAA-3′; antisense
5′-TTTTAAAAATTTTTCTAAGTCTTTTTTCATG-3′ (26). Annealing was performed as followed:
equal molar concentrations of oligonucleotides were incubated in
STE buffer (10 mM Tris pH 8.0, 50 mM NaCl, 1 mM EDTA) in a PCR
cycle, at 95°C for 2 min, afterwards it was cooled to 25°C for 45
min. Products were stored at 4°C.
Labeling 5′-protruding termini of DNA by
exchange reaction
T4 polynucleotide kinase (10 U) was added to a
specific reaction mixture containing, at a final volume of 20
μl: 20 pmol specific oligonucleotides, 0.1 M imidazole-HCl
(pH 6.4 at 25°C), 18 mM MgCl2, 5 mM DTT, 0.1 mM
spermidine, 0.1 mM EDTA, 0.1 mM ADP, 40 pmol [γ-32P]ATP
and 4.8% (w/v) polyethylene glycol 6000. Following incubation at
37°C for 30 min, the reaction was terminated by adding 1 μl
0.5 M EDTA (pH 8.0). Labeled DNA was separated using a HiYield
Gel/PCR DNA fragment extraction mini-prep kit (RBC Bioscience).
Band depletion assay
The band depletion assay was performed essentially
as previously described (10).
MCF7 cells (3×106 cells/flask) were preincubated with
erlotinib for 3 h prior to the treatment with CPT for 1 h. The
cells were removed from the flask by scraping without removing the
medium (to prevent reversal of the cleavable complex), followed by
centrifugation (1,000 rpm, 5 min, 4°C). Denaturing buffer (2% SDS,
62.5 mM Tris-HCl pH 6.8, and 1 mM EDTA) was immediately added to
the cell pellet, and the samples were mixed by vortexing until the
turbidity disappeared. Samples were sonicated to diminish viscosity
(40 bursts of 2 min each at two-thirds of the maximum output of
microtip). Equal volumes of protein samples were analyzed on 7.5%
SDS-PAGE followed by western blot analysis with anti-topo I
antibodies.
Results
Combined treatment of erlotinib or
gefitinib with CPT increases the anticancer effect of CPT in
MCF7
In order to investigate the mechanism of the
combined treatment with topoisom-erase- and tyrosine kinase
inhibitors, we examined this effect in MCF7 breast and PC3 prostate
cancer cell lines. First, cells were exposed to various
concentrations (0–20 μM) of erlotinib or gefitinb for 24 h.
While cells exhibit higher sensitivity to gefitinib treatment
(IC50 concentrations are 9±1 μM for MCF7 and
16±1.4 μM for PC3), MCF7 and PC3 cells were quite resistant
to erlotinib treatment (data not shown), suggesting that erlotinib
alone is not efficient as a cytotoxic drug for these cells.
Therefore, an acceptable dose of erlotinib was chosen for the
evaluation of the combined treatment. Low doses of CPT were also
selected for the investigation of the combined treatment on the
viability of MCF7 and PC3 cells. CPT (0.1 or 0.02 μM) and
either erlotinib (0.5 μM) or gefitinib (5 μM) were
administered as single agents, or in combination, and cell
viability was examined for ≤72 h. While PC3 cells displayed no
beneficial cytotoxic effect with the combination of CPT and
erlotinib, a trend of increased cytotoxicity with gefitinib was
observed, however, it was not statistically significant (data not
shown). On the contrary, the combination of either erlotinib or
gefitinib with CPT increased the cytotoxic effect of CPT in MCF7
cells, compared to each treatment administered alone, as depicted
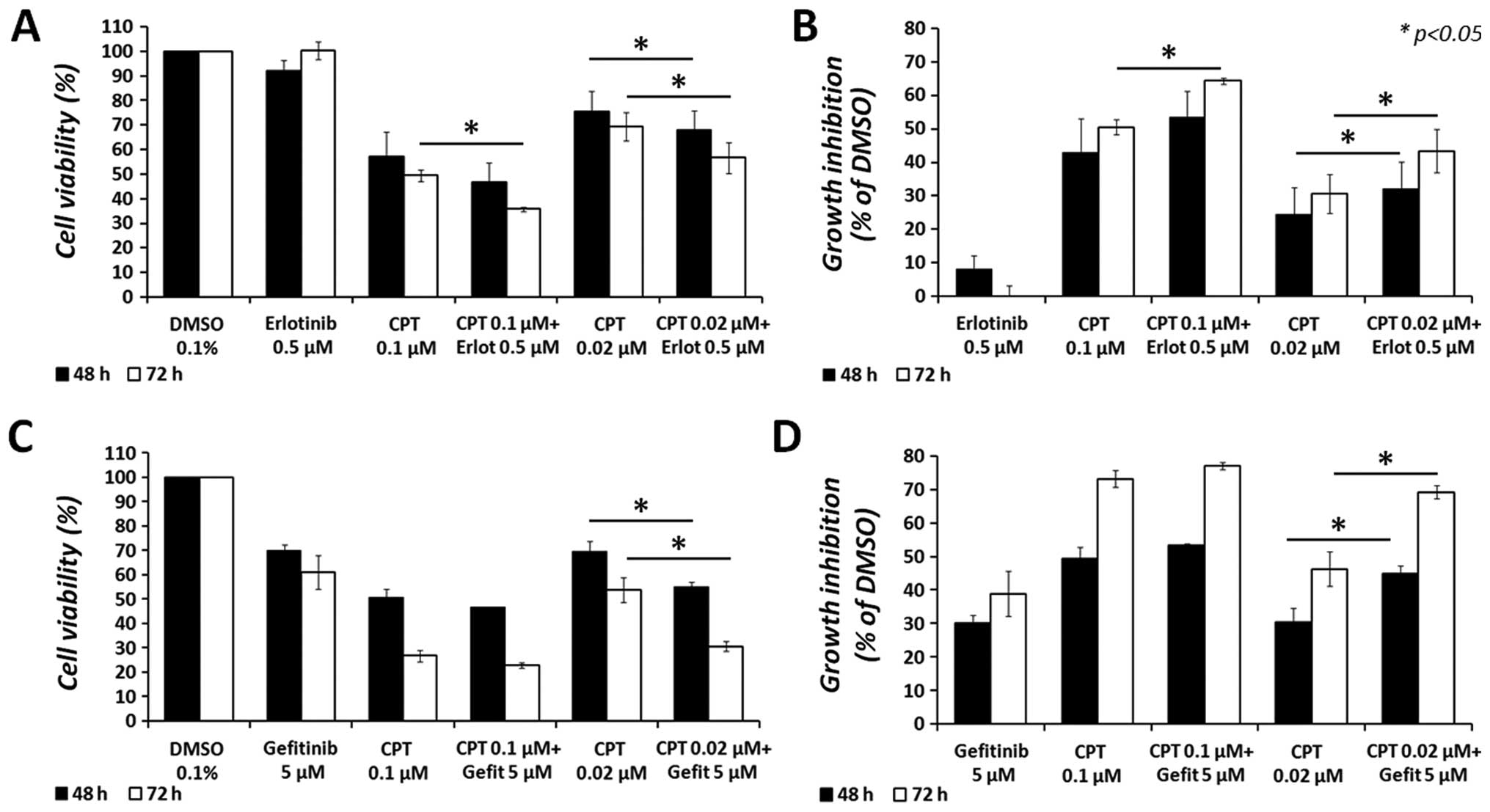
in Fig. 1. After a 48-h treatment,
erlotinib increased the anticancer effect of CPT from 24.3±8.2 to
32.2±7.9% (a 7.9% difference; p<0.05) at the lowest CPT dose
examined (0.02 μM), and from 42.9±10 to 53.3±7.9% (a 10.4%
difference) at 0.1 μM CPT. Moreover, after 72-h treatment,
erlotinib increased the anticancer effect of CPT from 30.7±5.8 to
43.3±6.4% (a 12.7% difference; p<0.05) at 0.02 μM of CPT,
and from 50.4±2.3 to 64.2±1% (a 13.8% difference; p<0.05) at 0.1
μM of CPT. Similarly, the combination of gefitinib with CPT
increased the cytotoxic effect of CPT, at 0.02 μM, from
30.4±4.3 to 45±2.3% (a 14.6% difference; p<0.05) after 48 h, and
from 46.3±5.1 to 69.2±2% (a 22.9% difference; p<0.05) after 72
h. However, no significant difference was observed with gefitinib
at the highest dose of CPT (0.1 μM). It is noteworthy that
all combined treatments significantly (p<0.05) reduced the
cytotoxic effect of erlotinib or gefitinib, as single agents, in
MCF7 cells.
Combined treatment of gefitinib or
erlotinib and CPT shows an increased inhibitory effect on topo I
activity in PC3 and MCF7 cells
Previous data showed that certain tyrosine kinase
antagonists, tyrphostins, can inhibit the activity of cellular topo
I. In order to characterize the mechanism of the combined
treatment, we sought to investigate the possibility that either
erlotinib, or gefitinib, can exert a similar inhibitory effect. As
the combined treatment demonstrated a significant cytotoxic effect
after 48 h, MCF7 and PC3 cells were treated for ≤48 h with either
0.1 or 0.02 μM CPT and 1 μM of erlotinib or
gefitinib.
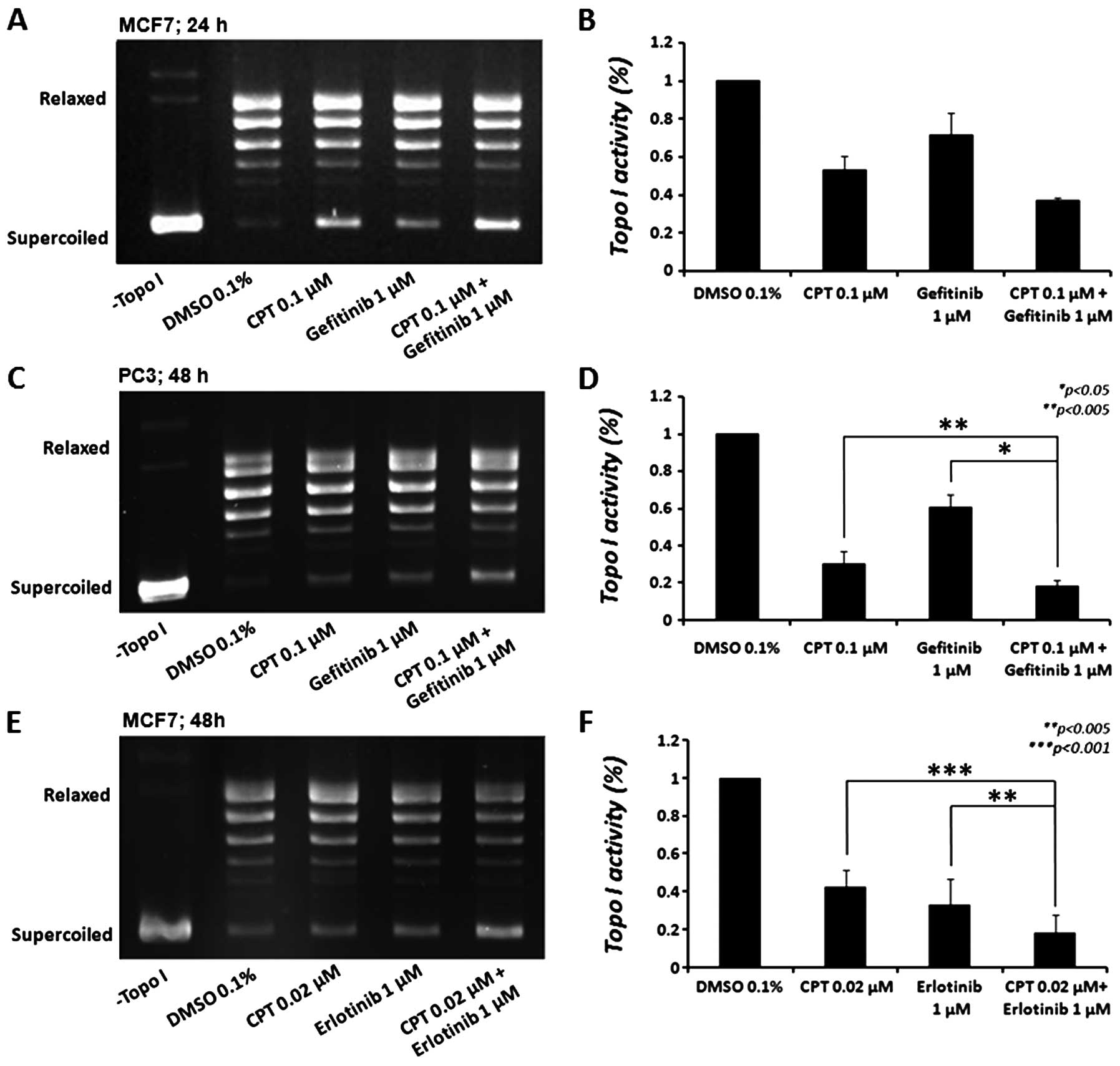
As depicted in Fig.
2, gefitinib reduced topo I activity in MCF7 cells by 28.6±12%
(p<0.05) after 24 h (Fig. 2A and
B); however, this effect was decreased after 48 h (not shown).
CPT (0.1 μM), as expected, reduced topo I activity by
47.1±7.3% (p<0.005). In cells treated with a combination of both
drugs, a 62.8±1.1% decrease in topo I activity was observed, an
additional decrease of 15.8±6.2% in topo I activity, compared to
CPT alone (p<0.06) and a 34.3±10.9% additional decrease,
compared to gefitinib alone (p<0.05), compatible with the
increased cytotoxicity of both drugs (Fig. 1). When we examined the effect of
gefitinib treatment on the PC3 cellular topo I activity (Fig. 2C and D) we observed a
non-significant reduction (∼11%) after 24 h (not shown), which
increased after 48 h to 39.4±6.8% (p<0.005). CPT (0.1 μM)
reduced topo I activity by 69.5±6.4% (p<0.001). In cells treated
with both drugs, 81.7±3.3% decrease in topo I activity was
observed, an additional decrease of 12.2±3.2% in topo I activity,
compared to CPT alone (p<0.005) and 42.3±8.5% additional
decrease, compared to gefitinib alone (p<0.05) was detected.
Erlotinib reduced topo I activity in MCF7 cells by 66.8±7.8%
(p<0.005), 48 h after treatment (Fig. 2E and F); however, it did not reduce
the enzyme activity in PC3 cells. As expected, CPT (0.02 μM)
reduced topo I activity by 57.3±5.3% (p<0.001) in MCF7 cells
after 48 h. Similarly to gefitinib, when we examined the combined
effect of these drugs on cellular topo I activity, an increased
effect was observed (81.6±5.4% reduction). This indicates that the
combined treatment exhibited a 24.2±2.9% additional decrease in
topo I activity compared to CPT alone (p<0.005) and a 14.7±2.6%
additional decrease compared to erlotinib alone (p<0.001). These
effects are compatible with the increased cytotoxicity of both
drugs in MCF7 cells. While the results suggest that erlotinib and
gefitinib, at the indicated doses, significantly reduce topo I
activity, similarly to CPT, the different responses of MCF7 and PC3
cells to these drugs might be attributed to cell-specific
properties.
Erlotinib, but not gefitinib, inhibits
the DNA relaxation activity of purified topo I
As both gefitinib and erlotinib reduced topo I
activity in the cell, we examined the mechanism underlying this
inhibitory effect.
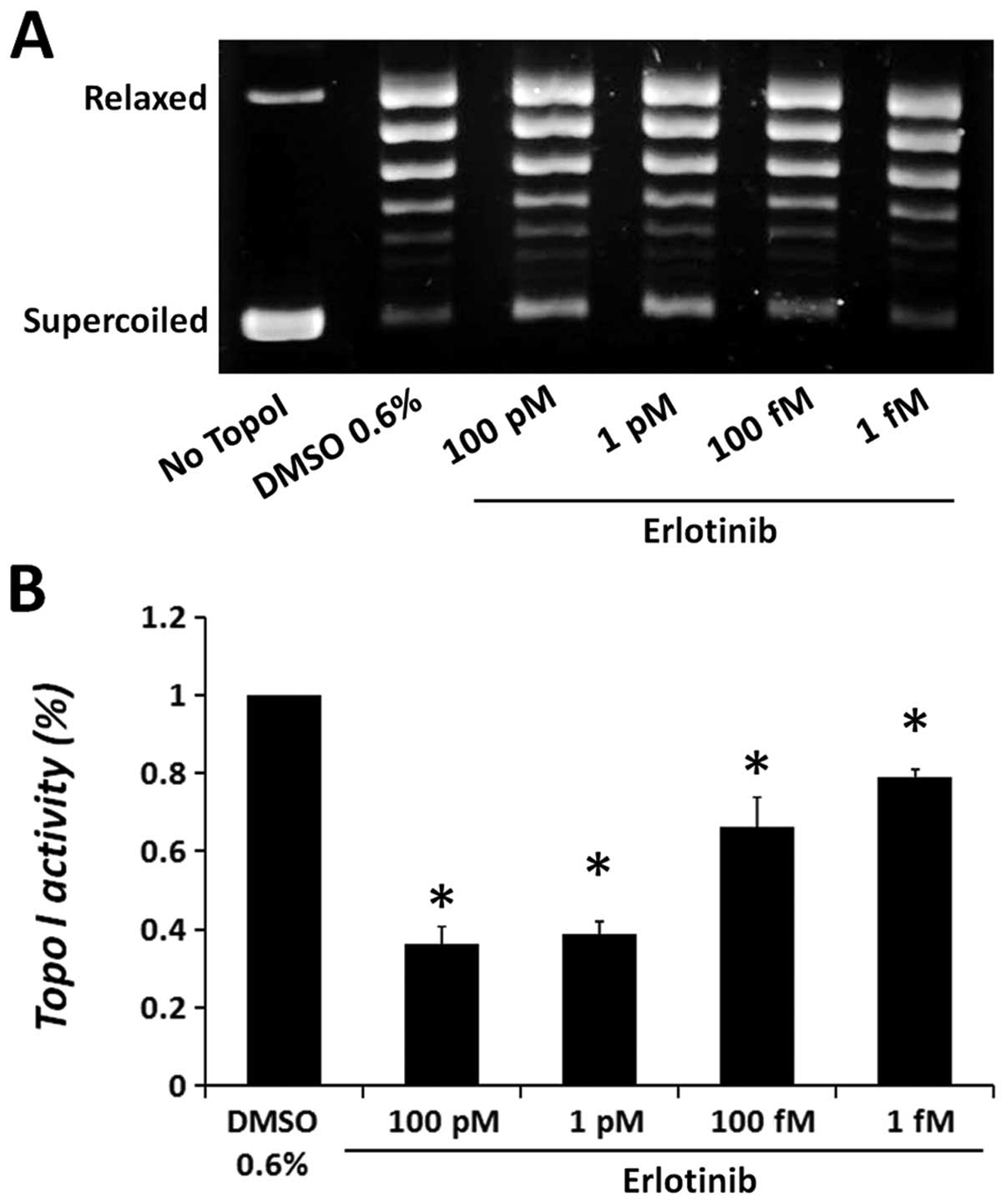
First, we investigated the direct effect of the drug
on purified calf thymus topo I. One unit of the enzyme was added to
a topo I reaction mixture in the presence of various concentrations
of erlotinib or gefitinib and DNA relaxation activity was examined.
As erlotinib exhibited reduced solubility at high concentrations,
under topo I assay conditions, low doses of the drug were examined.
The results depicted in Fig. 3
show that erlotinib significantly reduced the catalytic activity of
purified topo I in a dose-dependent manner, compared to the vehicle
control (DMSO). Erlotinib reduced topo I activity by 63.6±9.4%, at
the highest dose examined, 100 pM, while at the lowest dose of 1
fM, a 20.8±2.2% reduction in the enzyme activity was observed.
The addition of gefitinib to topo I reaction mixture
did not affect the DNA relaxation activity of the enzyme, even at
higher concentrations (data not shown), suggesting its indirect
inhibitory effect on the cellular topo I.
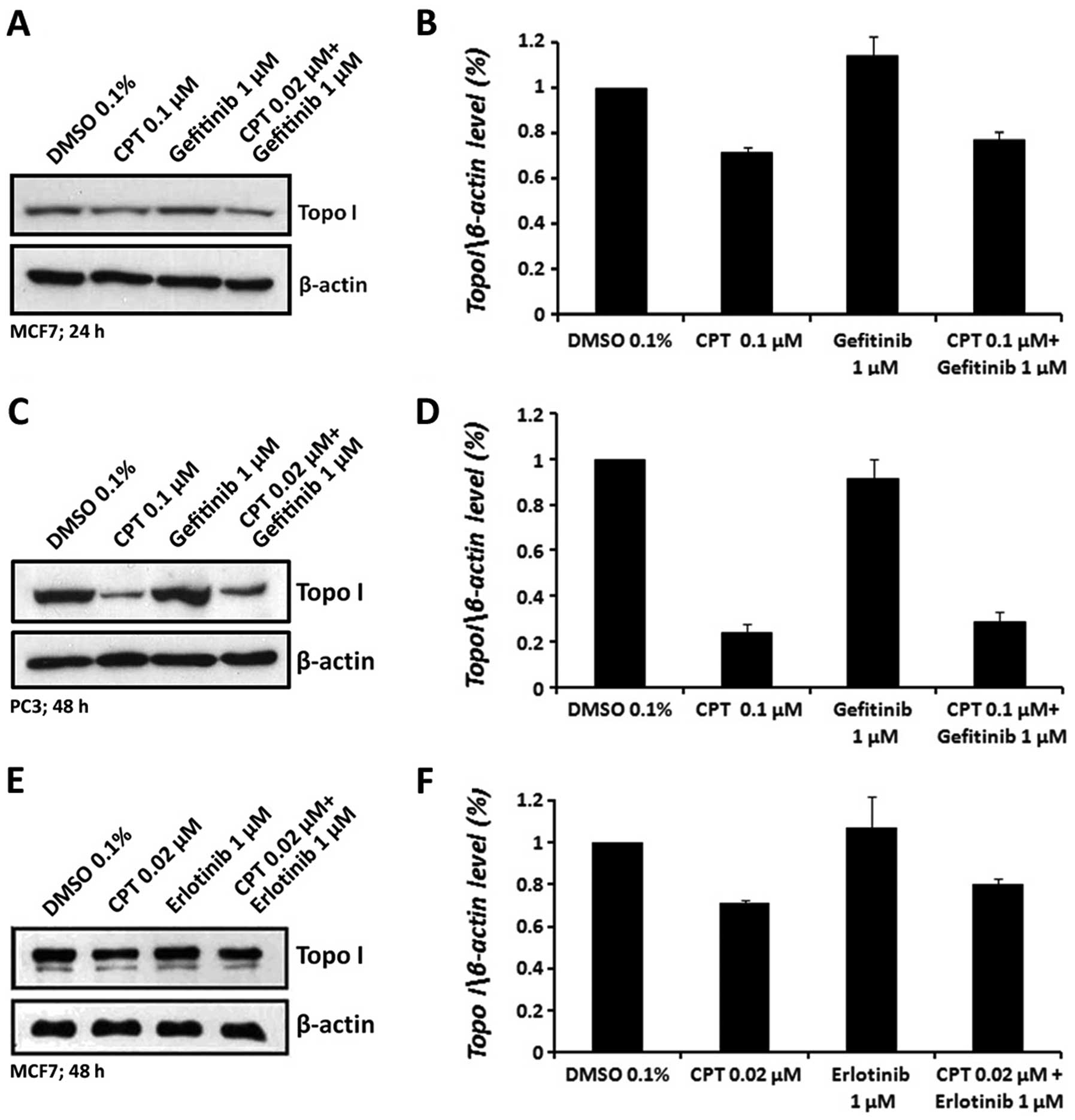
Neither erlotinib nor gefitinib reduces
the level of topo I protein
The reduction in topo I activity in erlotinib or
gefitinib treated cells might be due to: i) reduction in the level
of topo I protein, or ii) conformational changes,
post-translational modifications or otherwise, in topo I protein
that affect its activity. To examine the first possibility, cells
were treated with erlotinib or gefitinib and CPT, as single agents
or in combination, as indicated above. Nuclear extract proteins
were analyzed by western blot analysis using specific anti-topo I
antibody. The results depicted in Fig.
4 show that treatment of MCF7 or PC3 cells with either
erlotinib or gefitinib alone did not alter the level of topo I
protein. CPT treatment, as expected (27–29),
reduced the level of free topo I protein. The combined treatments
of CPT with erlotinib or gefitinib reduced topo I levels to a
similar extent as CPT alone, suggesting that neither erlotinib nor
gefitinib affect the level of topo I protein.
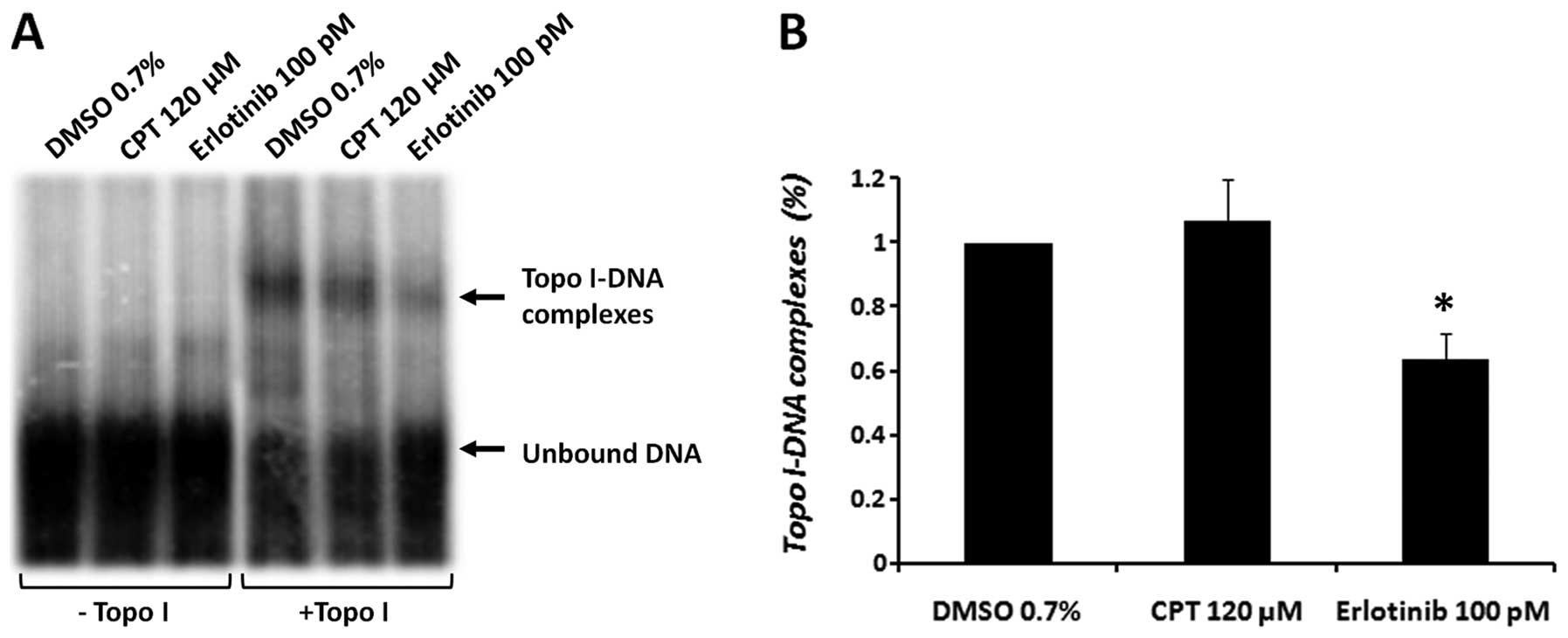
Erlotinib reduces the DNA-binding ability
of topo I
The results obtained thus far suggest that treatment
of cells with erlotinib might cause a transient inhibition of topo
I activity, which alters the inhibitory effect of CPT. Our previous
results demonstrated that other TKIs (certain tyrphostin
derivatives) inhibited topo I activity by decreasing the ability of
this enzyme to bind DNA in vitro (17). Therefore, to determine the effect
of erlotinib on the DNA-binding ability of topo I, an EMSA assay
was performed. A consensus 31-bp topo I DNA-binding sequence was
used. Purified topo I was added to an enzyme-specific reaction
mixture, which contained CPT (120 μM) or erlotinib (100 pM).
High concentrations of the drugs are needed to establish a short
time effect in vitro. The results depicted in Fig. 5 reveal a 36.2±7.7% decrease
(p<0.05) in the binding of topo I to the DNA, compared to the
vehicle control (DMSO).
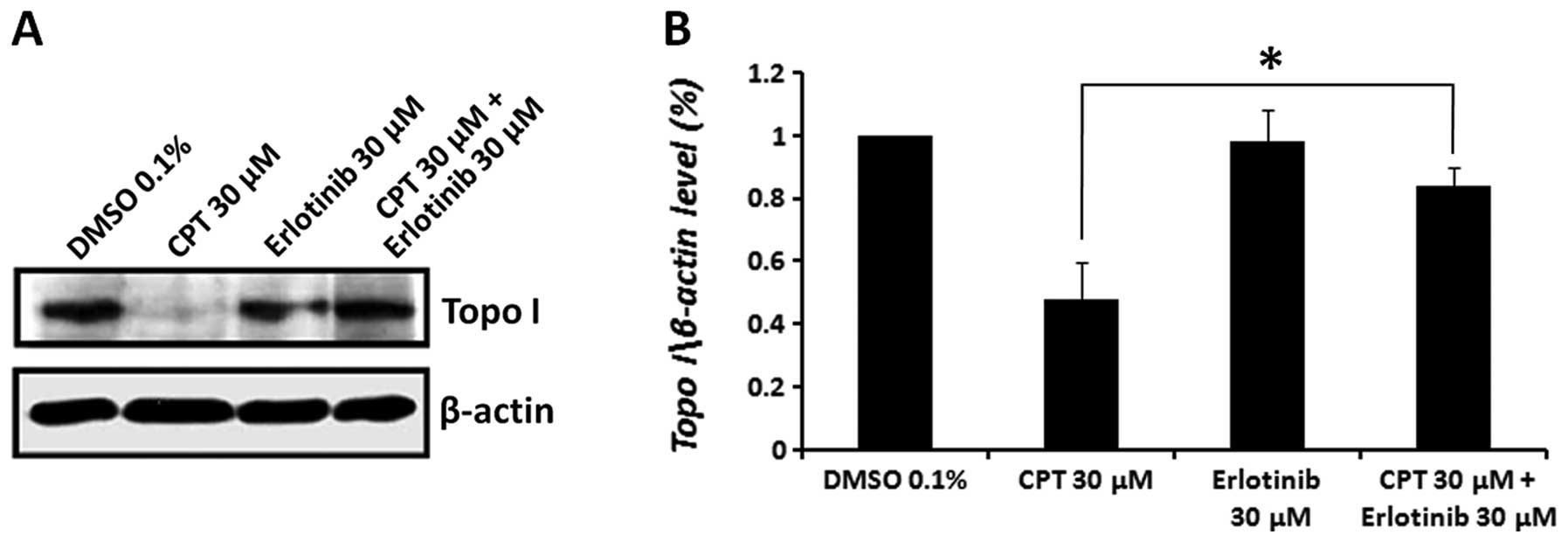
Erlotinib diminishes the CPT induced
cleavable topo I-DNA complex
Since erlotinib reduced the DNA-binding ability of
topo I in vitro, we sought to investigate whether this
inhibitory activity is also exerted within the cell. In a transient
reaction intermediate, termed the cleavable complex, topo I links
covalently to DNA through a tyrosine residue in its active site,
leaving a DNA break with a free 5′-hydroxyl end. Treatment of cells
with CPT results in the stabilization of these cleavable complexes
and the prevention of the DNA-ligation step (30–33).
To examine the effect of erlotinib and the combined treatment with
CPT on the formation of topo I-DNA cleavable complexes, we used the
‘band depletion’ assay (25). This
assay is based on the observation that an increase in the amount of
topo I bound to the DNA, because of CPT treatment, will cause
depletion in the level of free topo I protein. Thus, the prevention
of the enzyme ability to bind DNA will prevent the formation of
topo I-DNA complexes by CPT, which will be manifested in band
depletion reversal and increased level of free topo I. As this
assay is used for short-term treatments of the cells, high drug
doses were employed. MCF7 cells were pre-treated with erlotinib for
3 h, followed by CPT treatment for 1 h, using similar drug
concentrations (30 μM). Cells were immediately lysed with
SDS, and the level of topo I protein was examined by western blot
analysis, using specific anti-topo I antibodies. The results
depicted in Fig. 6 demonstrate a
significant decrease (51.7±12% compared to the vehicle control) in
the level of free topo I in CPT-treated cells, and no effect on the
level of topo I in cells treated with erlotinib alone. Pretreatment
with erlotinib, before administering CPT, abolished the CPT-induced
cleavable complexes and restored the level of topo I enzyme to
84.2±6.1%.
Discussion
Preclinical and clinical studies have demonstrated
the anti-tumor activity of EGFR-TK inhibitors, as single agents or
in combination with other drugs, including topoisomerase
interacting agents (8–11). It has been shown in vitro
and in vivo, that the combined treatment of human colorectal
cancer cells, with anti-EGFR drugs (cetuximab or gefitinib) and
with topoisomerase I inhibitor topotecan (TPT), increased the
antitumor activity of either agent alone (8,9,19).
Phase II studies have demonstrated that combined therapy of triple
negative breast cancer (TNBC) with cetuximab (an anti-EGFR
antibody) and cisplatin dramatically increased the patient response
rate, compared to cisplatin treatment alone, and doubled their
progression-free survival duration (20,34).
Indeed, cisplatin has been shown to inhibit the activity and level
of topoisom-erases (35,36). Gefitinib was found to modulate the
ability of SN-38 (the active metabolite of CPT-11) to inhibit topo
I in colorectal tumor cell lines, to accumulate cells in S-phase,
and to induce apoptosis (21).
Erlotinib was found to inhibit tumor growth and metastasis in a
TNBC xenograft model (20) and was
shown to increase the antitumor activity of CPT-11 in colorectal
xenografts, with no increased toxicity (10).
We previously showed that certain tyrosine kinase
antagonists, tyrphostins, inhibit the catalytic activity of the
cellular topoisomerase I (topo I) (17,18).
Since the activity of TKIs is not correlated with EGFR expression
(10,15), and excluding EGFR mutations,
resistance mechanisms have been linked to several cellular
processes, such as activation of alternative tyrosine kinase
receptors (such as IGF-1R) (37),
constitutive ligand-independent activation of ERK or Akt (10,37),
and expression of MDR transporter proteins (38,39),
we examined the mechanism of the combined treatment with
topoisomerases- and EGFR-targeting agents by investigating the
possibility that erlotinib and gefitinib, which are currently used
in chemotherapy treatments (unlike tyrphostins), exert their
anticancer activity by also affecting topoisomerase I.
MCF7 breast cancer and PC3 prostate cancer cell
lines were used to examine the combined effect of topo I and
EGFR-TK inhibitors, and as the source for the cellular topo I.
While the combined treatments with CPT, a known topo I inhibitor,
and neither erlotinib nor gefitinib exhibit any significant
beneficial effects in PC3 cells, an increased cytotoxicity was
observed in MCF7 cells. To investigate the effect of the combined
treatment on topo I, cells were treated with low concentrations of
the indicated drugs to avoid cell toxicity. Interestingly,
gefitinib was shown to reduce the cellular topo I activity in both
MCF7 and PC3 cells, while erlotinib reduced cellular topo I
activity in MCF7 cells only. These differences in cellular response
suggest that both erlotinib and gefitinib exhibit a cell type
dependent inhibition of topo I and the determination of the
combined treatment depends on the cell type and on the assay
conditions. Furthermore, in compatibility with the increased cell
cytotoxicity, the combined treatment increased the inhibitory
effect on topo I activity, compared to that observed by each of the
drugs administered alone.
Examination of the mechanism by which topo I
activity is reduced by either gefitinib or erlotinib, was performed
using various parameters. The reduction of topo I activity in
treated cells was not due to a reduction in the enzyme protein
level, suggesting possible modifications of the enzyme protein that
cause this inhibitory effect. First we examined a possible direct
interaction between the drugs and a purified topo I enzyme and
found that only erlotinib reduced the DNA relaxation activity of
the topo I, in a dose-dependent manner, while gefitinib did not
affect the enzyme activity. The ability of erlotinib to directly
inhibit topo I at very low doses, points to its potential as a
potent topo I inhibitor. It is not yet clear why higher doses of
this drug exerted less inhibitory effect in vitro, however,
we found that erlotinib, at higher concentration, forms
micro-aggregates under our assay conditions, which might interfere
with its binding to the target and may diminish its inhibitory
ability (data not shown). The reduction of topo I activity by
erlotinib, in vitro, is due to modifications of the DNA
binding ability of the enzyme, as observed by the EMSA experiment.
Since no effect on the mobility of the DNA was observed, it is
probable that erlotinib, by itself, did not bind to the DNA.
Therefore, it is possible that erlotinib affects the conformation
of topo I protein by a direct interaction with the enzyme, in a way
that changes its DNA binding activity. Since we previously showed
that tyrphostins, which are tyrosine kinase inhibitors, act also as
topo I antagonists by altering the conformation of topo I and
inhibiting its DNA binding ability (17,18),
our present data strengthen the notion that certain TKIs exert
their inhibitory activity by interacting with topo I protein.
As the in vitro data suggest that erlotinib
interferes with the DNA binding ability of topo I enzyme, it is
possible that this is also its mode of action in the cell. To
examine this notion, we utilized the mode of action of CPT, which
stabilized the DNA-enzyme cleavable complexes, and showed that
pretreatment of the cells with erlotinib, prior to CPT treatment,
diminished the formation of CPT induced DNA-enzyme cleavable
complexes. This result suggests that pretreatment of the cells with
erlotinib modified the ability of topo I to bind to the DNA and
therefore less DNA-enzyme cleavable complexes were stabilized in
the presence of CPT. These data are compatible with the in
vitro results and with our previous finding with other tyrosine
kinase inhibitors (17,18).
Alternatively, it is known that the activity of topo
I in the cell is regulated by phosphorylation of the
serine/threonine or tyrosine residues of the enzyme protein
(1,31,32).
EGFR signal transduction pathway involves activation of
serine/threonine kinases such as PKC (40). PKC and casein kinase II were shown
to phosphorylate topo I and thus regulate its activity (31). Therefore, it is possible to suggest
that the inhibition of EGFR signal transduction pathway by
erlotinib will also affect PKC activity and reduce the
phosphorylation level of topo I, thereby reducing its activity, as
previously suggested (31). This
is supported by the inhibition of cellular topo I in
gefitinb-treated cells, which exhibits a different topo
I-inhibitory mechanism than that of erlotinib. Gefitinib was not
found to directly interact with topo I enzyme in vitro,
however, it was found to modulate EGFR signaling pathways (data not
shown). As topo I protein level is not reduced after gefitinib
treatment, the reduction in the enzyme activity can be a cause of a
pos-translational modification, such as hypo-phosphorylation, that
might be mediated by the blocking of EGFR signaling. Indeed, it has
been suggested that gefitinib can induce inhibition of PKC pathway
(41).
Topo I inhibition could also be dependent on the
inhibition of other cellular proteins. It has been shown that
erlotinib treatment suppresses the activity of certain
cyclin-dependent kinases (CDKs) in breast cancer (42), inhibits the activity of a mutant
JAK2, that is found in polycythemia vera (PV) and other
proliferative disorders (43).
Gefitinib was shown to inhibit and influence the production of
certain growth factors and cytokines, such as VEGF, bFGF and TGF-α
(15). Several studies have shown
that both gefitinib and erlotinib can modulate the activity of
ABC-transporters (44–47) and inhibit the ErbB-2 receptor
(15,48), thus one may assume that other
proteins might be affected by these drugs as well, including
topoisomerases.
The results of this study suggest that topo I is a
novel target of erlotinib, in addition to its known activity as PTK
inhibitor. Therefore, simultaneous inhibition of essential cellular
enzymes, such as topo I and protein tyrosine kinases, may serve as
potent anticancer strategy. Our results point also to the
possibility that a combination of erlotinib and gefitinib with topo
I inhibitors may demonstrate an effective anti-breast cancer
treatment.
Acknowledgements
This study was supported in part by
the Ben-Gurion University Seed Research fund.
References
|
1.
|
Cretaio E, Pattarello L, Fontebasso Y,
Benedetti P and Losasso C: Human DNA topoisomerase IB: structure
and functions. Ital J Biochem. 56:91–102. 2007.PubMed/NCBI
|
|
2.
|
Forterre P, Gribaldo S, Gadelle D and
Serre M: Origin and evolution of DNA topoisomerases. Biochimie.
89:427–446. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Wang JC: DNA topoisomerases. Annu Rev
Biochem. 65:635–692. 1996. View Article : Google Scholar
|
|
4.
|
Corbett KD and Berger JM: Structure,
molecular mechanisms, and evolutionary relationships in DNA
topoisomerases. Annu Rev Biophys Biomol Struct. 33:95–118. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Tomicic MT and Kaina B: Topoisomerase
degradation, DSB repair, p53 and IAPs in cancer cell resistance to
camptothecin-like topoisomerase I inhibitors. Biochim Biophysica
Acta. 1835:11–27. 2013.PubMed/NCBI
|
|
6.
|
Haglof KJ, Popa E and Hochster HS: Recent
developments in the clinical activity of topoisomerase-1
inhibitors. Cancer Ther. 1:117–145. 2006.
|
|
7.
|
Pommier Y: Topoisomerase I inhibitors:
camptothecins and beyond. Nat Rev Cancer. 6:789–802. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Ciardiello F, Caputo R, Bianco R, et al:
Antitumor effect and potentiation of cytotoxic drugs activity in
human cancer cells by ZD-1839 (Iressa), an epidermal growth factor
receptor-selective tyrosine kinase inhibitor. Clin Cancer Res.
6:2053–2063. 2000.
|
|
9.
|
Koizumi F, Kanzawa F, Ueda Y, et al:
Synergistic interaction between the EGFR tyrosine kinase inhibitor
gefitinib (‘Iressa’) and the DNA topoisomerase I inhibitor CPT-11
(irinotecan) in human colorectal cancer cells. Int J Cancer.
108:464–472. 2004.
|
|
10.
|
Chen J, Smith M, Kolinsky K, et al:
Antitumor activity of HER1/EGFR tyrosine kinase inhibitor
erlotinib, alone and in combination with CPT-11 (irinotecan) in
human colorectal cancer xenograft models. Cancer Chemother
Pharmacol. 59:651–659. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Friedmann B, Caplin M, Hartley JA and
Hochhauser D: Modulation of DNA repair in vitro after treatment
with chemo-therapeutic agents by the epidermal growth factor
receptor inhibitor gefitinib (ZD1839). Clin Cancer Res.
10:6476–6486. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Normanno N, Bianco C, De Luca A, Maiello
MR and Salomon DS: Target-based agents against ErbB receptors and
their ligands: a novel approach to cancer treatment. Endocr Relat
Cancer. 10:1–21. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Uberall I, Kolár Z, Trojanec R, Berkovcová
J and Hajdúch M: The status and role of ErbB receptors in human
cancer. Exp Mol Pathol. 84:79–89. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Oda K, Matsuoka Y, Funahashi A and Kitano
H: A comprehensive pathway map of epidermal growth factor receptor
signaling. Mol Syst Biol. 1:2005.0010,. 2005.PubMed/NCBI
|
|
15.
|
Harari PM: Epidermal growth factor
receptor inhibition strategies in oncology. Endocr Relat Cancer.
11:689–708. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Smith J: Erlotinib: small-molecule
targeted therapy in the treatment of non-small-cell lung cancer.
Clin Ther. 27:1513–1534. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Aflalo E, Seri I, Segal S, Gazit A and
Priel E: Inhibition of topoisomerase I activity by tyrphostin
derivatives, protein tyrosine kinase blockers: mechanism of action.
Cancer Res. 54:5138–5142. 1994.PubMed/NCBI
|
|
18.
|
Bendetz-Nezer S, Gazit A and Priel E: DNA
topoisomerase I as one of the cellular targets of certain
tyrphostin derivatives. Mol Pharmacol. 66:627–634. 2004.PubMed/NCBI
|
|
19.
|
Ciardiello F, Bianco R, Damiano V, et al:
Antitumor activity of sequential treatment with topotecan and
anti-epidermal growth factor receptor monoclonal antibody C225.
Clin Cancer Res. 5:909–916. 1999.PubMed/NCBI
|
|
20.
|
Ueno NT and Zhang D: Targeting EGFR in
triple negative breast cancer. J Cancer. 2:324–328. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Azzariti A, Xu J, Porcelli L and Paradiso
A: The schedule-dependent enhanced cytotoxic activity of
7-ethyl-10-hydroxy-camptothecin (SN-38) in combination with
Gefitinib (Iressa, ZD1839). Biochem Pharmacol. 68:135–144. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Johnston MD, Finter NB and Young PA: Dye
uptake method for assay of interferon activity. Methods Enzymol.
78:394–399. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Auer B, Vosberg HP, Buhre U, Klocker H,
Hirsch-Kauffmann M and Schweiger M: Intracellular distribution of
DNA topoisomerase I in fibroblasts from patients with Fanconi’s
anaemia. Hum Genet. 61:369–371. 1982.PubMed/NCBI
|
|
24.
|
Sambrook J, Fritsch EF and Maniatis T:
Molecular Cloning: A Laboratory Manual. Cold Spring Harbor
Laboratory Press; Cold Spring Harbor, NY: 1989
|
|
25.
|
Kaufmann SH and Svingen PA: Immunoblot
analysis and band depletion assays. Methods in Molecular Biology,
DNA topoisomerase protocols: DNA Topoisomerase Protocols. Bjornsti
MA and Osheroff N: 94. Humana Press Inc; Totowa, NJ: pp. 253–268.
1999, View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Stevnsner T, Mortensen UH, Westergaard O
and Bonven BJ: Interaction between eukaryotic DNA topoisomerase I
and a specific binding sequence. J Biol Chem. 264:10110–10113.
1989.PubMed/NCBI
|
|
27.
|
Desai SD, Li TK, Rodriguez-Bauman A, Rubin
EH and Liu LF: Ubiquitin/26S proteasome-mediated degradation of
topoisomerase I as a resistance mechanism to camptothecin in tumor
cells. Cancer Res. 61:5926–5932. 2001.PubMed/NCBI
|
|
28.
|
Desai SD, Liu LF, Vazquez-Abad D and
D’Arpa P: Ubiquitin-dependent destruction of topoisomerase I is
stimulated by the antitumor drug camptothecin. J Biol Chem.
272:24159–24164. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Desai SD, Zhang H, Rodriguez-Bauman A, et
al: Transcription-dependent degradation of topoisomerase I-DNA
covalent complexes. Mol Cell Biol. 23:2341–2350. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Wang HK, Morris-Natschke SL and Lee KH:
Recent advances in discovery and development of topoisomerase
inhibitors as antitumor agents. Med Res Rev. 17:367–425. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Pommier Y, Pourquier P, Fan Y and
Strumberg D: Mechanism of action of eukaryotic DNA topoisomerase I
and drugs targeted to the enzyme. Biochim Biophys Acta.
1400:83–105. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Wang JC: Cellular roles of DNA
topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol.
3:430–440. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Wang JC: DNA topoisomerases as targets of
therapeutics: an overview. Adv Pharmacol. 29A:1–19. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Baselga J, Gomez P, Awada A, et al: The
addition of cetuximab to cisplatin increases overall response rate
(ORR) and progression-free survival (PFS) in metastatic
triple-negative breast cancer (TNBC): results of a randomized phase
II study (BALI-1). Ann Oncol. 21(Suppl 8): 27402010.
|
|
35.
|
Wu X, Yalowich JC and Hasinoff BB:
Cisplatin inhibits the catalytic activity of DNA topoisomerase II
by binding to critical protein thiol groups and by binding to DNA.
AACR Meeting: Experimental and Molecular Therapeutics 31:
Topoisomerase, Telomerase, and Nucleosides/Nucleotides. Abst.
3079,. 2004
|
|
36.
|
Aoe K, Kiura K, Ueoka H, et al: Cisplatin
down-regulates topoisomerase I activity in lung cancer cell lines.
Anticancer Res. 24:3893–3897. 2004.PubMed/NCBI
|
|
37.
|
Camp ER, Summy J, Bauer TW, Liu W, Gallick
GE and Ellis LM: Molecular mechanisms of resistance to therapies
targeting the epidermal growth factor receptor. Clin Cancer Res.
11:397–405. 2005.PubMed/NCBI
|
|
38.
|
Chen YJ: Mechanisms underlying resistance
to epidermal growth factor receptor inhibitors in non-small cell
lung cancer. Biol Biomed Rep. 2:141–148. 2012.
|
|
39.
|
Chen YJ, Huang WC, Wei YL, et al: Elevated
BCRP/ABCG2 expression confers acquired resistance to gefitinib in
wild-type EGFR-expressing cells. PLoS One. 6:e214282011. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Jorissen RN, Walker F, Pouliot N, Garrett
TP, Ward CW and Burgess AW: Epidermal growth factor receptor:
mechanisms of activation and signalling. Exp Cell Res. 284:31–53.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Kim H, Kim SH, Kim MJ, et al: EGFR
inhibitors enhanced the susceptibility to NK cell-mediated lysis of
lung cancer cells. J Immunother. 34:372–381. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Ling YH, Li T, Yuan Z, Haigentz M Jr,
Weber TK and Perez-Soler R: Erlotinib, an effective epidermal
growth factor receptor tyrosine kinase inhibitor, induces
p27KIP1 up-regulation and nuclear translocation in
association with cell growth inhibition and G1/S phase arrest in
human non-small-cell lung cancer cell lines. Mol Pharmacol.
72:248–258. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Li Z, Xu M, Xing S, et al: Erlotinib
effectively inhibits JAK2V617F activity and polycythemia
vera cell growth. J Biol Chem. 282:3428–3432. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Shi Z, Peng XX, Kim IW, et al: Erlotinib
(Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B
member 1 and ATP-binding cassette subfamily G member 2-mediated
drug resistance. Cancer Res. 67:11012–11020. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Yang CH, Huang CJ, Yang CS, et al:
Gefitinib reverses chemotherapy resistance in gefitinib-insensitive
multidrug resistant cancer cells expressing ATP-binding cassette
family protein. Cancer Res. 65:6943–6949. 2005. View Article : Google Scholar
|
|
46.
|
Yanase K, Tsukahara S, Asada S, Ishikawa
E, Imai Y and Sugimoto Y: Gefitinib reverses breast cancer
resistance protein-mediated drug resistance. Mol Cancer Ther.
3:1119–1125. 2004.PubMed/NCBI
|
|
47.
|
Marchetti S, de Vries NA, Buckle T, et al:
Effect of the ATP-binding cassette drug transporters ABCB1, ABCG2,
and ABCC2 on erlotinib hydrochloride (Tarceva) disposition in in
vitro and in vivo pharmacokinetic studies employing
Bcrp1−/−/Mdr1a/1b−/− (triple-knockout) and
wild-type mice. Mol Cancer Ther. 7:2280–2287. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Normanno N, Maiello MR and De Luca A:
Epidermal growth factor receptor tyrosine kinase inhibitors
(EGFR-TKIs): simple drugs with a complex mechanism of action? J
Cell Physiol. 194:13–19. 2002. View Article : Google Scholar : PubMed/NCBI
|