Introduction
Rho family GTPases, a part of the Ras superfamily
which are involved in regulating multiple aspects of cell functions
such as cell proliferation, survival, adhesion and migration, are
intimately relevant in tumor initiation and tumor progression
(1–5). Most members of the Rho family can
cycle between an active, GTP-bound conformation and an inactive,
GDP-bound conformation, and this GTP-binding and GTP-hydrolysis
process is tightly regulated by various classes of regulators
including the guanine nucleotide exchange factors and
GTPase-activating proteins. The best characterized members of the
Rho family include Rho (A, B and C), Rac (1, 2 and 3) and Cdc42.
RhoE is a unique member of the Rnd subfamily, a distinct branch of
Rho family proteins that bind, but do not hydrolyze GTP, thus
remains constitutively active upon expression without the
regulation by guanine nucleotide exchange factors and
GTPase-activating proteins (6–9). In
addition, the carboxy-terminal sequence of RhoE is farnesylated
while most Rho family proteins are modified by geranylation. These
observations suggest that the function and regulation of RhoE may
be different from other Rho family members (6).
There are some studies on the role of RhoE in human
malignancies. Several reports have examined RhoE expression
patterns in tumor tissues, but the results were contradictory
(7,10–12).
RhoE expression level does not change in response to serum
stimulation, however, it can be enhanced by DNA-damaging agents
such as cisplatin or UBV (9).
Tumor suppressor p53 can also induce cell cycle arrest at G1 and
allow cells to repair DNA damage induced by ionizing radiation, UV
light or chemotherapeutic drugs before proceeding to DNA
replication (13–15). Ongusaha et al have shown
that RhoE is a transcriptional target of p53 and can inhibit
ROCK1-mediated apoptosis in response to genotoxic stress (16).
In the present study we systematically investigated
the expression levels of RhoE in human gastric, colorectal, lung
and breast cancer tissues by using tissue microarrays and explored
the effect of RhoE expression on the growth and invasion phenotypes
of the cancer cell lines. We further attribute the relatively
reduced RhoE expression to the reduction of p53 transcription
activity in the cancer cells and present evidence that RhoE
expression could be affected by p53 mutation. These findings may
partly explain the mechenism of RhoE downregulation in some
cancers.
Materials and methods
Cell culture
Human embryonic kidney cells 293, gastric cancer
cell lines MKN45 and AGS, colorectal cancer cell lines SW480 and
LOVO, liver cancer cell lines HepG2 and SMMC-7721, lung cancer cell
lines A549 and SPCA1, and the prostate carcinoma cell line PC3,
were preserved at our institute. All cell lines were maintained in
RPMI-1640 medium (Invitrogen Corporation, Gaithersburg, MD, USA)
supplemented with 10% fetal bovine serum (FBS) (Gibco, Carlsbad,
CA, USA), 100 U/ml penicillin G sodium, and 100 μg/ml
streptomycin sulfate (Sigma, St Louis, MO, USA). Cells were grown
at 37°C in a humidified atmosphere containing 5%
CO2.
Plasmids and transfection
Expression vectors pCDNA3.0-RhoE, PEGFP,
MIEG3-wt-p53, p21-promoter-luc and PRL-TK were kindly provided by
Professor Yi Zheng (Cincinnati Children’s Hospital, Cincinnati, OH,
USA). MIEG3-wt-p53 and PEGFP were digested with BamH1 and EcoR1 in
order to construct the expression vector PEGFP-wt-p53. PEGFP-mt-p53
plasmids that mutated at p53 hot points (175, 248, 273, 282,
175/248, 175/273, 175/282, 248/273, 248/282, 273/282) were
constructed on the basis of PEGFP-wt-p53.
PGL3-RhoE-promoter-3000-luc was stored in our laboratory. Cells
were transfected by Lipofectamine 2000 (Invitrogen), and stable
transfection cell lines were selected with G418 (Gibco).
Immunohistochemistry
Tissue microarrays were obtained from Cybrdi Co.
(Xi’an, China), containing 74 cases of gastric carcinoma, 32 cases
of colorectal carcinoma, 62 cases of lung carcinoma and 34 cases of
breast carcinoma. All were matched with the adjacent cancerous
tissues and their clinicopathological grades were evaluated using
WHO standards. The tissue microarrays were evaluated by three
pathologists who did not know the information of the slides. IHC
studies were performed using a standard avidin-biotin complex
immunoperoxidase method. Briefly, tissue sections were
deparaffinized in xylene and rehydrated with a series of decreasing
alcohol concentrations and the endogenous peroxidase activity was
inhibited with a 3% solution of H2O2 for 10
min. For antigen retrieval, tissue slides were treated in citrate
buffer (pH 6.0) with microwave for 10 min. The detection was then
performed following the instruction of mouse
streptavidin-peroxidase immunostaining kit (SP-9000 Histostain-Plus
Kit; Zhong Shan Goldbridge Biotechnology Co., Ltd, Beijing, China)
containing normal goat serum, biotinylated secondary antibody and
streptavidinperoxidase complex. The slides were independently
incubated with anti-RhoE monoclonal antibody (05–723, clone 4,
Upstate Biotechnology, Lake Placid, NY, USA) which were diluted
1:50 in PBS overnight at 4°C. Pre-immune serum, instead of a
primary antibody, was used as a negative control. Then the slides
were stained with DAB (Pierce Biotechnology, Rockford, IL, USA),
dehydrated, cleared and mounted with neutral balsam.
The ratio of positive cells per specimen was
evaluated quantitatively and scored: 0 for staining ≤1%, 1 for
staining of 2 to 25%, 2 for staining of 26 to 50%, 3 for staining
of 51 to 75%, and 4 for staining >75% of the cells examined.
Staining intensity was graded as follows: 0, no signal; 1, weak; 2,
moderate; and 3, strong staining. Both scoring systems were
utilized to evaluate the level of protein expression. A total score
of 0 to 12 was finally calculated and graded as negative (−, 0–1);
weak (+, 2–4); moderate (++, 5–8); and strong (+++, 9–12). This
scoring system has been described previously.
Monolayer growth rate and cell cycle
assay
Cells (500–1,000) were plated in 96-wells cell
culture plate. Cell counting kit-8 (Dojindo, Kumamoto, Japan) was
used to determine the monolayer culture growth rate. Cultures were
assayed at 0, 1, 2, 3, 4, 5 or 6 days. Absorbance values were
determined on the microplate reader (Bio-Rad Laboratories,
Hercules, CA, USA) at 490 and 630 nm. Cell cycle profiles were
measured by flow cytometry using propidium iodide (PI).
Transfectants which were stable transfected with RhoE and pcDNA3.0,
respectively, were starved for 24 h in 0.5% serum-containing medium
and then were stimulated with 10% FBS before fixing with 70%
ethanol at 4°C overnight, washed with PBS, and stained with PI (50
μg/ml) in PBS supplemented with RNase (10 mg/ml) for 30 min.
Flow cytometry was performed on a FACScan (Becton-Dickinson,
Heidelberg, Germany) equipped with cellquest software, and cellular
DNA content was determined for 1×104 cells.
In vitro apoptosis assay
Hoechst/PI staining and TUNEL experiments were
performed to study cell apoptosis. First, cells were trypsinized
and washed with PBS twice. For the Hoechst/PI staining, prepared
cells were fixed in 100 μl PBS containing 0.1 μg
Hoechst 33342 for 10 min at 37°C. After centrifuging, cells were
stained with PI. Next, the apoptotic cells were viewed and counted
by fluorescent microscopy at once. For the TUNEL experiment,
1×105 prepared cells were added into 6-wells cell
culture plates which were coated with thin slides. Cells were
harvested for 48 h and then tested by chromogenic method TUNEL
apoptosis detection kit (Beyotime, Jiangsu, China). Cells were
fixed in 4% paraformaldehyde for 1 h, and then incubated in
Biotin-UTP solution for 1 h at room temperature. After that,
samples were incubated in streptavidin-HRP buffer and colored by
DAB and hematoxylin. TUNEL-positive nuclei stained dark brown and
TUNEL-negative nuclei stained blue. The sections were examined
using light microscopy.
Invasion assay
Millicell hanging cell culture inserts (Millipore,
Billerica, MA, USA) precoated with Matrigel (Becton-Dickinson) were
put in a 24-well plate to perform cell invasion assays. Cells
(1–1.5×104 per well) were suspended in 0.25 ml of
culture medium with 1% FBS and added to the upper chamber. Medium
(1.25 ml) supplemented with 10% FBS was plated in the bottom of the
well. The invasion assay was carried out for 48 h in a humidified
incubator. The cells that traversed the membrane pore and spread to
the lower surface of the filters were stained with eosin for
visualization and then counted in 10 random high power fields
(×200).
Western blot analysis
Cells were lysed in lysis buffer containing 50 mM
Tris-HCl (pH 7.4), 150 mM NaCl, 0.2 g/l sodium azide, 1 g/l SDS, 1%
Triton X-100, together with a protease inhibitor mix (Calbiochem,
La Jolla, CA, USA). Cellular protein was quantified by a BCA
protein assay kit (Pierce). Equal amounts of proteins (30–40
μg) were fractionated by 8–15% sodium dodecyl
sulfate-polyacrylamide gels (SDS-PAGE) and then transferred to PVDF
(Millipore). The sheets were pre-incubated in a mixture of PBS and
5% non-fat milk powder for 2 h at room temperature. For
immunoblotting studies, membranes were incubated overnight at 4°C
with 5% non-fat milk powder containing the primary antibodies as
following: antibodies against RhoE (05–723, clone 4, Upstate
Biotechnology). After a TBS-0.05% Tween-20 wash, the sheets were
incubated with an IRDye 800CW secondary antibody (LI-COR
Biosciences, Lincoln, NE, USA) at room temperature for 1 h in a
cassette. After incubation, the sheets were washed in TBS-0.05%
Tween-20 also in a cassette and then detected by Odyssey infrared
fluorescence scanner (LI-COR).
Dual-luciferase reporter assay
For dual-luciferase reporter assays,
1×105 cells per well in 24-well plates were seeded the
day before transfection. Cells were transfected with 1.0 μg
of p21-promoter-luc together with 0.2 μg PRL-TK or
RhoE-promoter-luc 0.4 μg, wt-p53/mt-p53 1.0 μg
together with 0.2 μg PRL-TK. The cells were harvested for 48
h and then detected by dual-luciferase reporter assay system kit
(Promega, Madison, WI, USA). The luciferase activity was measured
with the Synergy2 instrument (BioTek, Winnoski, VT, USA) equipped
with Gen5 software. The firely luciferase expression was normalized
to Renilla luciferase and reported as relative luciferase
activity. Results are presented as means ± standard deviations (SD)
of data from three dependent experiments.
RNA extraction and p53 mutation
detection
The total RNA of 293, MKN45, AGS, SW480, LOVO,
SPCA1, A549, HepG2 and SMMC-7721 was extracted using TRIzol
(Invitrogen) according to the manufacturer’s protocol. The quantity
and purity of the RNA was determined by electrophoresis and the
ratio of the optical density at 260 nm to that at 280 nm. p53 was
amplified by RT-PCR using the SuperScript™III One-step RT-PCR
system with Platinum® Taq high fidelity kit
(Invitrogen). Primers used for p53 were: forward,
5′-AGTCTAGAGCCACCGTCCA-3′; and reverse, 5′-TCT
GACGCACACCTATTGCAAGC-3′ (17). A
total of 2 μl RNA was brought to a volume of 50 μl
containing 25 μl 2X reaction mix, 1 μl
SuperScript/Platinum Taq high fidelity enzyme mix, 1 μl of
both sense and antisense primer. Amplification was performed as per
the following conditions: one cycle of 55°C for 30 min was
performed to synthesize the cDNA. After denaturation at 94°C for 2
min 40 cycles of PCR were performed to amplify p53 (denaturation at
94°C for 15 sec, annealing at 58°C for 30 sec, extension at 68°C
for 90 sec and a final extension of 68°C for 5 min). The PCR
products were analyzed by agarose gel electrophoresis.
The p53 amplicons were purified and linked with A
tail by DNA A-tailing kit (Takara Bio, Dalian, China), then
subcloned into pMD18-T vector. The DNAs were sequenced by using
vector primers (Invitrogen).
Statistical analysis
SPSS11.0 software package (SPSS, Chicago, IL, USA)
was used to analyze data. Pared-samples t-test was used to compare
the stained value of the immunohistochemistry samples. The
correlation between tumor grades and the stained value and the
positive ratio were tested by χ2 test. The correlation
of molecules was tested by multiple linear regression analysis.
Assays for characterizing phenotype of cells were analyzed by
Student’s t-test. A value of p<0.05 was considered to indicate
statistical significance.
Results
Reduced expression of RhoE in human
gastric, colorectal, lung and breast cancer tissues
Our laboratoty, previously, has reported that RhoA
and RhoC expressions are significantly upregulated in gastric
cancer tissues, and RhoE expression is likely regulated by
epigenetic modification. We performed immunohistochemistry staining
of cancer tissue microarrays that contain 74 gastric, 32
colorectal, 62 lung and 34 breast cancer carcinoma samples matched
with adjacent normal tissues, respectively, in an effort to examine
the protein expression of RhoE in human cancer tissues. The
staining examination of the tumor tissues in comparison with the
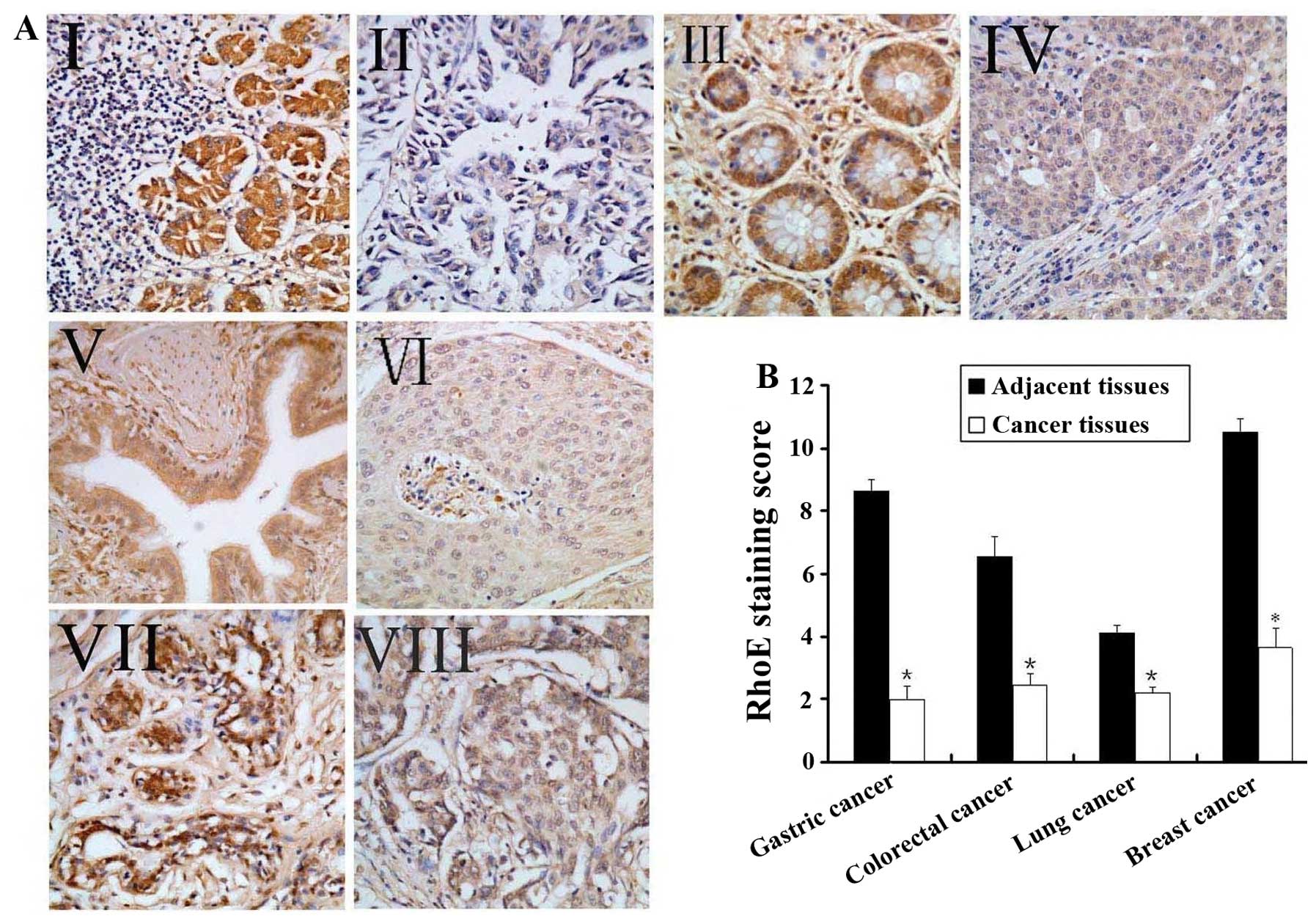
adjacent normal tissues showed that RhoE was abundantly expressed
in the cytoplasm of the normal tissues but is at a low or
non-detectable level in the cancer tissues in general (Fig. 1A). We have attempted to quantify
the relative RhoE expression levels based on parallel
immunohistochemistry staining of the array chips by assigning a
score ranging from ‘−’, ‘+’, ‘++’ to ‘+++’ that correspond to 1–4
in a blank-folded manner. This exercise yielded average staining
scores of 1.99±0.28 (positive ratio was 41.9%), 2.45±0.35 (positive
ratio was 68.7%), 2.19±0.19 (positive ratio was 71.0%) and
3.65±0.62 (positive ratio was 58.8%) for gastric, colorectal, lung
and breast cancers, respectively, while the average staining scores
of the adjacent tissues was 8.64±0.39 (positive ratio was 94.6%),
6.55±0.63 (positive ratio was 90.6%), 4.11±0.24 (positive ratio was
93.5%), 10.53±0.44 (positive ratio was 100.0%). The differences of
the staining scores and positive ratios between the respective
tumor tissues and the adjacent tissues were highly significant
(p<0.005) (Fig. 1B). Therefore,
RhoE expression level appears to be inversely correlated with
tumorigenesis in human gastric, colorectal, lung and breast
cancers.
Since a possible divergence of the relative scores
is related to different cancer progressive grades, we further
examined RhoE expression levels in the tumor tissues based on the
available cancer grades from the patient samples. Among the
gastric, colorectal and lung cancer patient samples with known
cancer grades, RhoE expression correlated well with the cancer
grades (p<0.005). These analyses suggest that RhoE expression is
also associated with cancer progression.
Enhanced RhoE expression inhibits cancer
cell proliferation and invasion
The expression level of RhoE was investigated by
western blot analysis in the ten different cancer cell lines. The
data showed that RhoE protein expression was relatively higher in
the cell lines HEK293, AGS, SMMC7721, LOVO, and A549, as well as
NIH3T3 and COS7, while it was found at a relatively lower level in
MKN45, SW480, HepG2 and SPCA1 cells (data not shown).
To explore the effects of RhoE on proliferation and
invasion activities of cancer cells, the four RhoE low-expressed
cell lines, MKN45, SW480, HepG2 and SPCA1, were transfected with
pCDNA3.0-RhoE or pCDNA3.0, and the expression level of RhoE was
detected by western blot analysis (Fig. 2B). We found that the RhoE
expressing cells displayed significantly reduced capability to
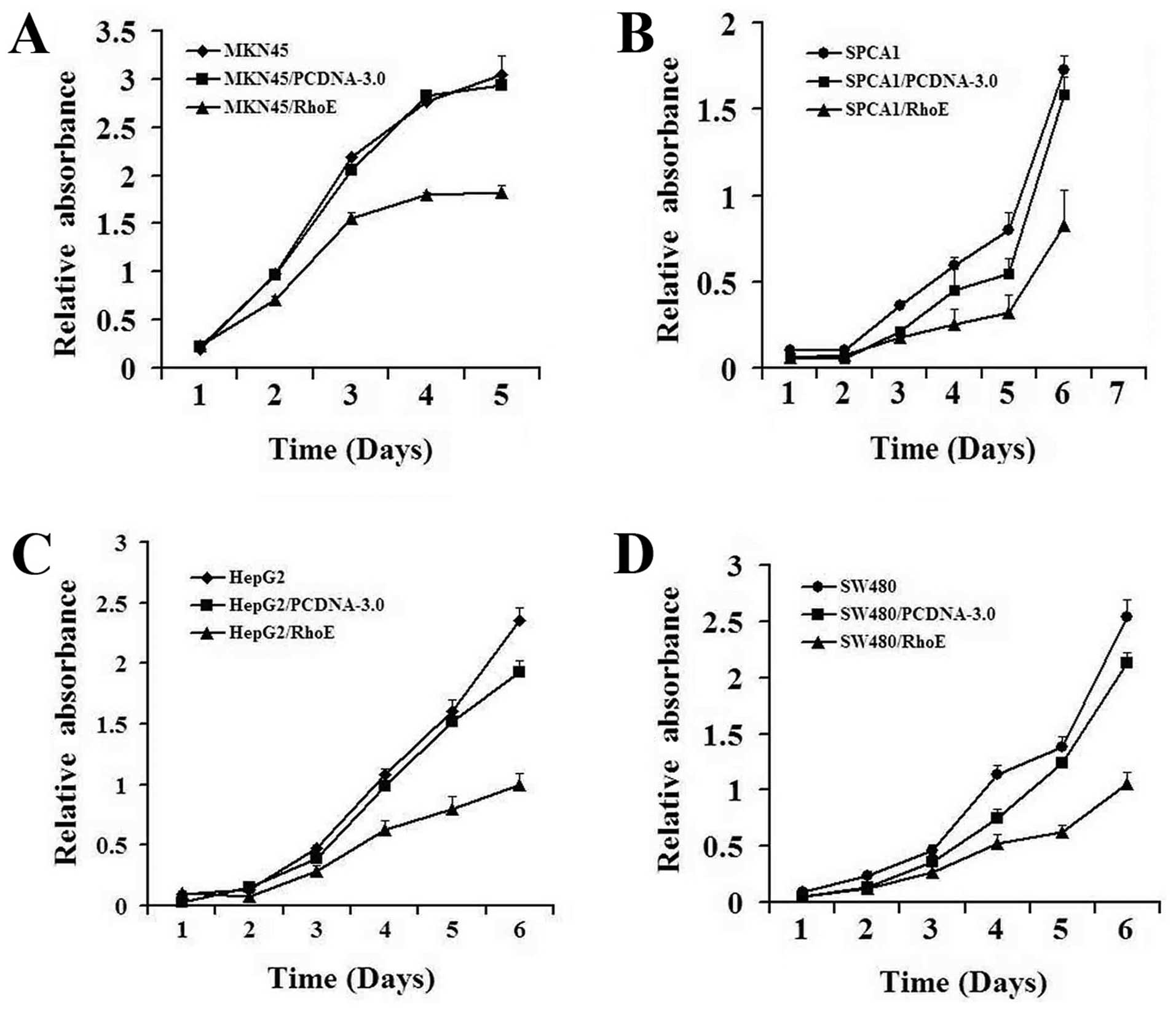
invade through Matrigel-coated Transwells (Fig. 2A). Further, the growth rate of RhoE
transfected cancer cell lines, in each case, was significantly
slower than that of the control cells and the parental cells
(Fig. 3). These data suggest that
RhoE expression can inhibit cell proliferation and suppresses
invasion of the cancer cells.
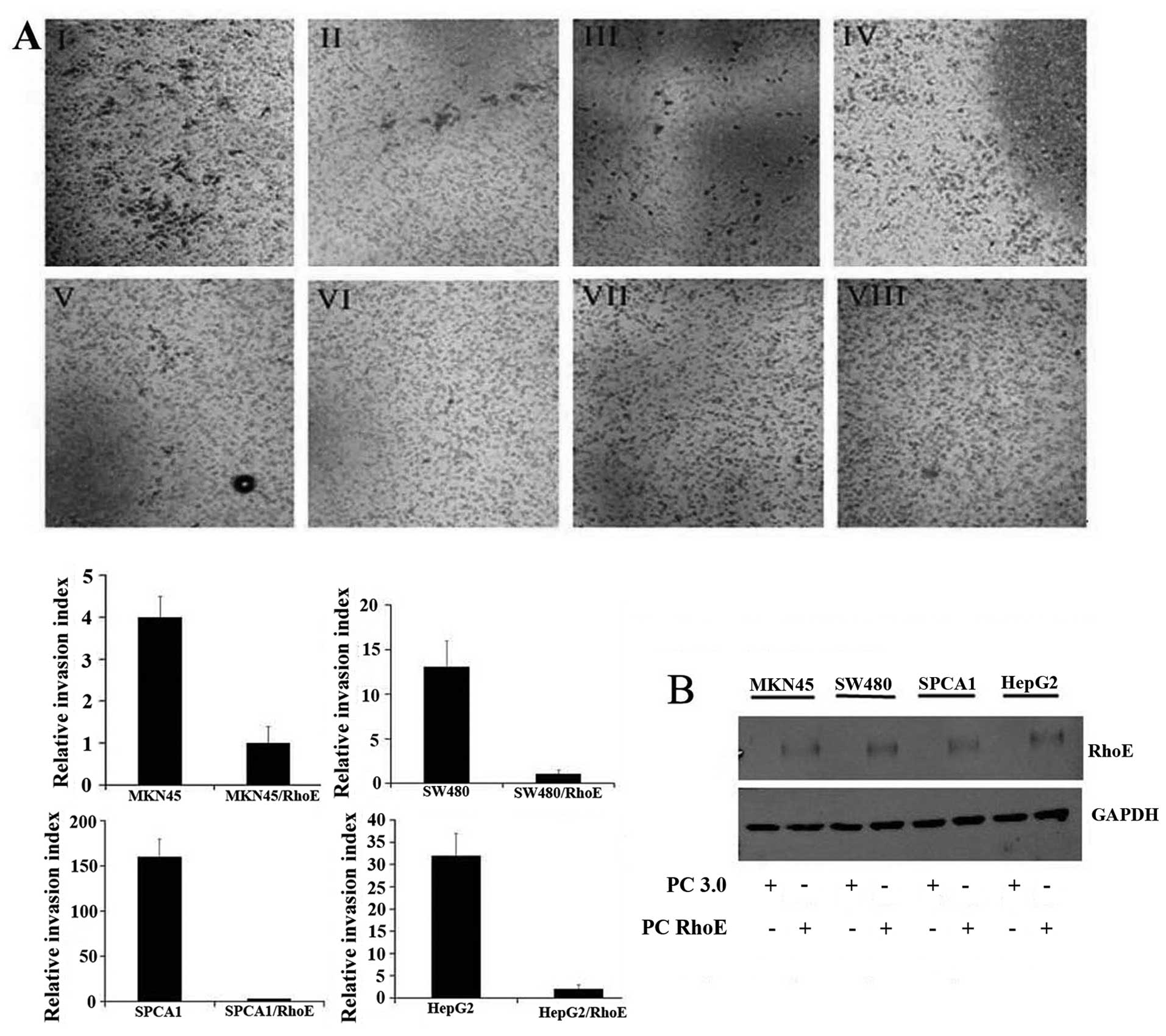 | Figure 2.Enhanced RhoE expression inhibits
cancer cells invasion. (A) The invasion activity of cells was
assayed in Matrigel-coated transwells and cells that succeeded in
invading the Matrigel were quantified 48 h after plating, and then
images were taken. AI-IV were MKN45, SW480, SPCA1 and HepG2,
respectively, transfectants with pcDNA3.0. AV-VIII were the
corresponding pcDNA3.0-RhoE transfectants. Results are presented as
mean ± SEM (n=3, *p<0.05). Magnification, ×200. (B)
Immunoblot analysis of RhoE expression in four RhoE low-expressed
cell lines (SPCA1, SW480, HepG2 and MKN45) that were, respectively,
transfected with pcDNA3.0 and pcDNA3.0-RhoE. GAPDH was used as a
loading control. |
Forced RhoE expression in cancer cells
induces cell cycle arrest and apoptosis
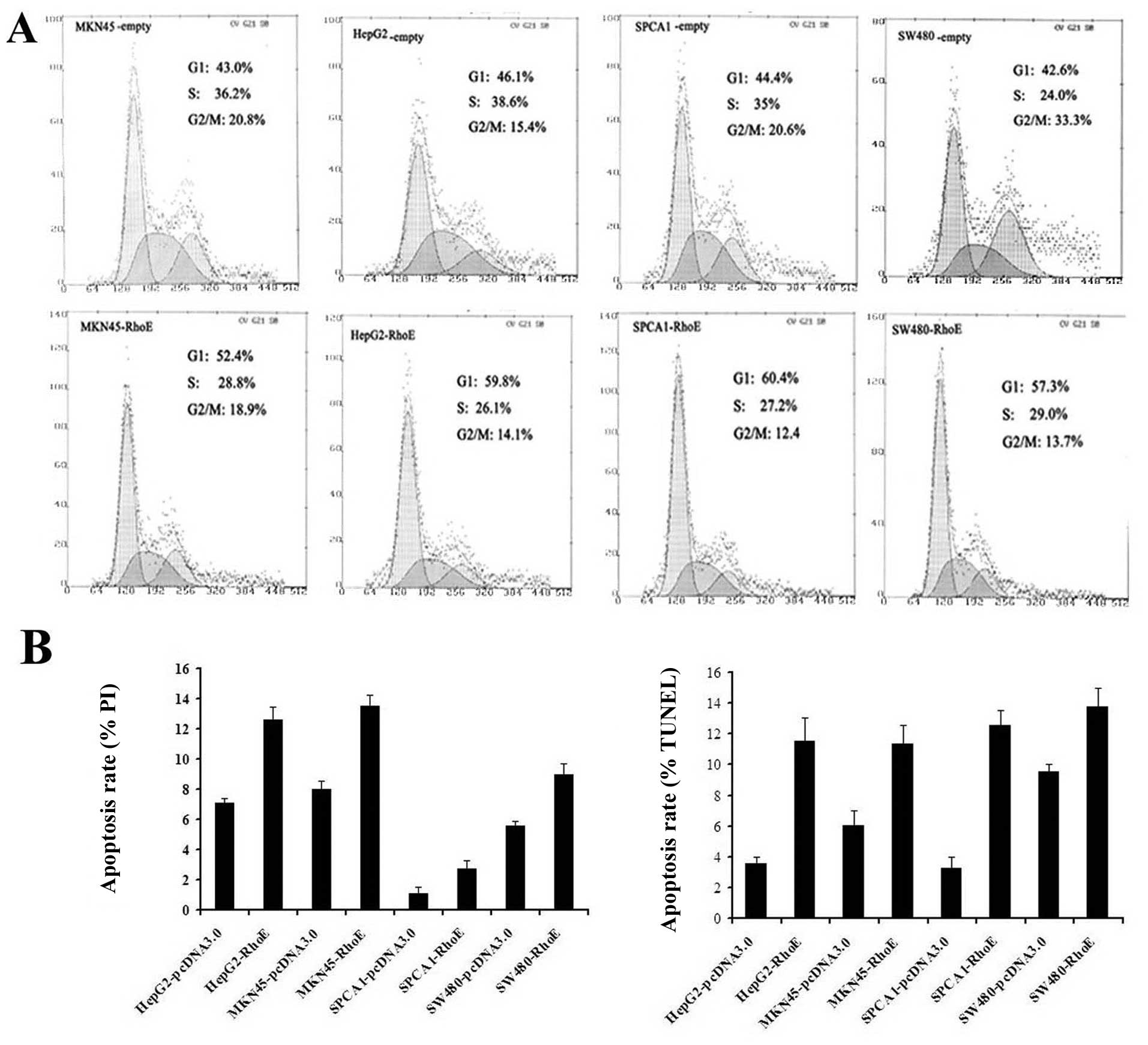
To assess if RhoE expression affects cancer cell
proliferation by modulating cell cycle progression and/or survival,
the RhoE transfectants were analyzed by fluorescence-activated cell
sorting (FACS). The results showed that the percentage of G1 phase
cells increased significantly with RhoE overexpression. Contrary to
the empty plasmid transfected control cells, the RhoE overexpressed
cells failed to transit through the G1/S check point when they were
stimulated by 10% serum (Fig. 4A).
The TUNEL analysis and the Hoechest/PI stained cells also showed
that the apoptosis rate of the RhoE expressing cells was
significantly higher than that of the controls (p<0.01)
(Fig. 4B). Similar observations
were made by visual imaging of caspase-3 positive cells (data not
shown). These results provide evidence that RhoE overexpression not
only induces cell growth arrest through inhibiting the G1/S phase
transition, but also promotes apoptosis in the cancer cells.
RhoE expression is regulated by p53
transcription activity in cancer cell lines
It was reported that RhoE promoter contained several
p53-binding sites and RhoE was one of the p53 transcriptional
target genes (9). At the beginning
of dissecting the mechanism of RhoE regulation in cancer, we
determined a potential correlation of endogenous RhoE expression
with p21-promoter-luc activities driven by cellular p53 in MKN45,
SPCA1, SW480 and HepG2 cancer cell lines which also showed a
relatively lower RhoE expression, and the luciferase/Renilla
(Luc/Ren) ratios were significantly higher in cells with relatively
high RhoE expression (p<0.05) (Fig.
5A). These results suggest that RhoE expression could be
associated with endogenous p53 transcription activity in the
absence of the genotoxic stress.
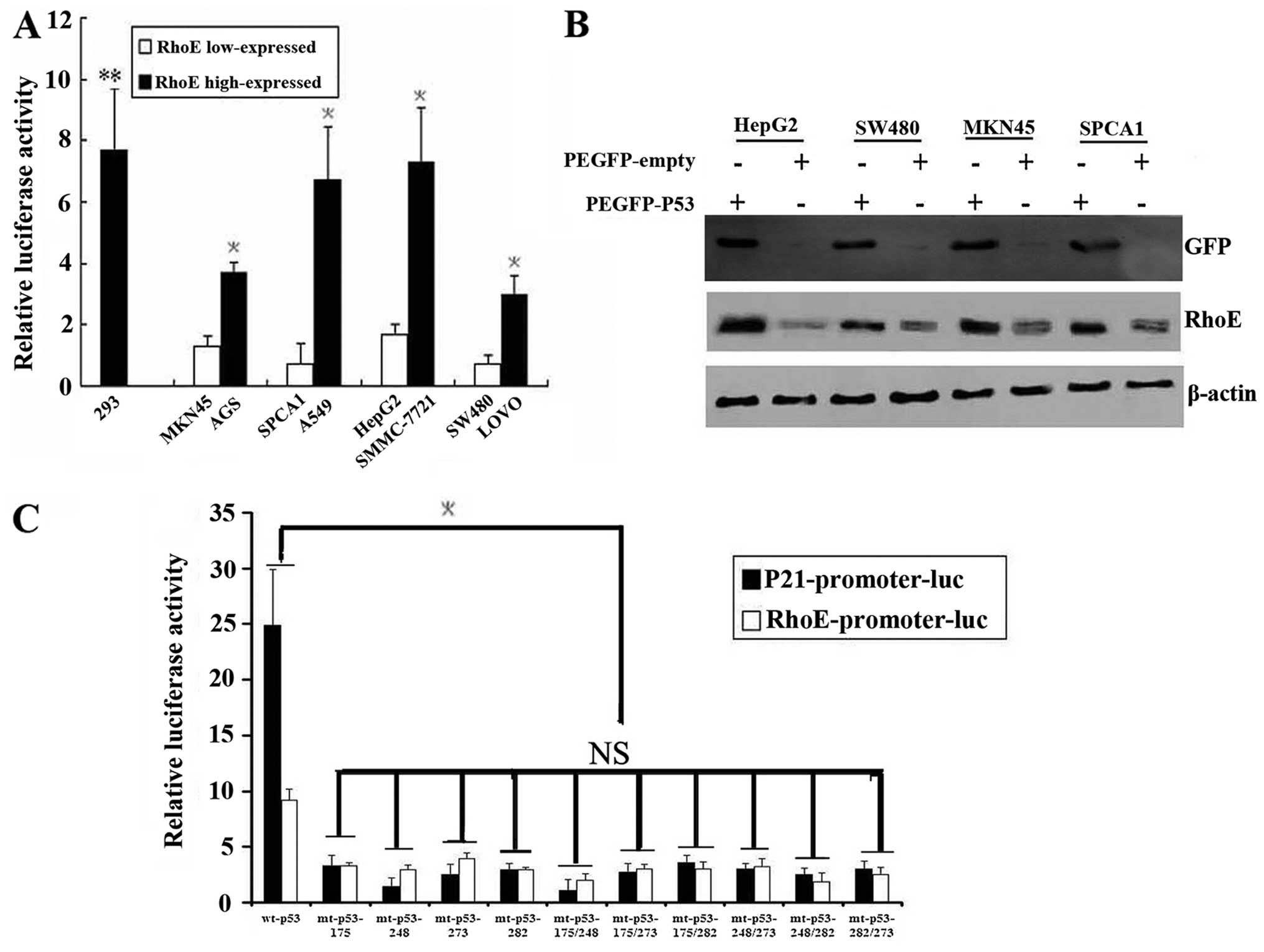 | Figure 5.RhoE transcriptional activity is
increased by wild p53 and decreased by p53 mutation. (A) P53
activity decreased in the RhoE low-expressing cell lines. All of
the four cell lines were co-transfected transiently with the p53
reporter (p21-promoter-luc) and the internal parameter (PRL-TK)
construct. The samples were collected after 48-h transfection.
Error bars represent the standard errors of the means, and the
asterisk (*) indicates statistical difference of pairs
of samples that are significant (p<0.05) while the NS indicates
no statistical difference between normal cell lines and the cancer
cell lines (p>0.05). (B) PEGFP-wt-p53 and the PEGFP-empty were
transiently transfected into the 4 cell lines which RhoE were
low-expressed. Transfection efficiency was determined by counting
the number of GFP-expressing cells per randomly chosen field of 100
cells at 24 h after transfection, and the mean number was
determined as 45%. Cells were collected and lysed after 48 h and
RhoE expression was detected by western blot analysis.
Overexpression of p53 significantly enhances RhoE protein level in
the 4 cell lines. Results are presented as mean ± SEM (n=3,
*p<0.05) (C) P53 mutation decreased the activity of
RhoE promoter. EGFP-mt-p53 constructs which contained p53 hot point
mutations (175, 248, 273, 282, 175/248, 175/273, 175/282, 248/273,
248/282, 273/282) together with RhoE-promoter-3000-luc and PRL-TK
were co-transfected into PC3 cells (p53 null). P21-promoter-luc was
taken as positive control. Luc/Ren ratios were significantly
decreased in the cells which were transfected with EGFP-mt-p53 than
that of EGFP-wt-p53, but there was no difference among the mutants.
Results are presented as mean ± SEM (n=3,
*p<0.05). |
To determine whether forced p53 protein expression
could cause the protein expression of RhoE, plasmids expressing
PEGFP-wt-p53 or PEGFP-empty were transiently transfected into 4
cell lines with relatively low RhoE levels. Transfection efficiency
was determined by counting the number of GFP-expressing cells per
randomly chosen field of 100 cells at 24 h after transfection
(multiple fields), and the mean number was determined as 45%. RhoE
expression level was detected by western blot analysis while the
trans fection efficiency was monitored by GFP expression (Fig. 5B). The results revealed that the
endogenous RhoE expression was increased significantly upon p53
expression in these cells. These results provide evidence that RhoE
might be subject to p53 transcription regulation.
p53 mutation decreases the activity of
RhoE promoter
To determine whether low RhoE expression was the
effect of p53 mutation or deletion, we sequenced the coding regions
of the endogenous p53 gene by RT-PCR in these RhoE low-expressed
cancer cell lines. However, we only found one meaningful mutation
in SW480 cells (codon 273) in which both the activity of p53 and
the expression level of RhoE were lowest among the four cancer cell
lines. These results indicated the possibility that the decreased
p53 activity is related to the detected mutations in the cancer
cells.
To confirm if p53 mutation could affect the RhoE
expression, we generated EGFP-mt-p53 constructs which contained p53
hot point mutations (175, 248, 273, 282, 175/248, 175/273, 175/282,
248/273, 248/282, 273/282). These mutants together with
RhoE-promoter-3000-luc and PRL-TK were co-transfected into PC3
cells (p53 null) and P21-promoter-luc was taken as positive
control. We found that Luc/Ren ratios were significantly decreased
in the cells which were transfected with EGFP-mt-p53 compared with
that of EGFP-wt-p53. However, there was no difference between the
mutants (Fig. 5C). These results
suggest that the p53 gene status is also a key factor affecting
RhoE expression and p53 mutation could partly account for RhoE low
expression.
Discussion
Rho GTPases are important regulators of cell
cytoskeleton organization and gene transcription, and deregulation
of Rho family members has been shown to closely associate with the
malignant phenotypes of cancer (18,19).
While most Rho proteins are geranylgeranylated and are subject to
GTP-hydrolysis regulation, RhoE is farnesylated and does not act as
a classical GTPase switch without a detectable GTPase activity
(6). These biochemical findings
suggest that RhoE may employ a unique regulatory mechanism and
exhibits different function from other Rho family members.
RhoA has been implicated in the regulation of cell
morphology, motility, transformation, proliferation and
tumorigenicity (20–23). Previous studies suggest that RhoE
can function as an antagonist of RhoA signaling in two ways: one is
to bind directly to ROCK1 (a RhoA target effector) and inhibit its
activity; the other is to interact with p190B RhoGAP (a RhoA
negative regulator) to reduce cellular level of RhoA-GTP (18,24,25).
Since RhoA is likely to play a positive role in cancer progression,
we propose that RhoE might work in an opposite way. In our previous
study, we found that RhoA was highly expressed in gastric cancer
tissues and RhoA-specific small interfering RNA could partially
reversed the malignant phenotypes of gastric cancer (26). There are several studies on RhoE,
and substantial research shows RhoE is downregulated in most
cancers (7,11,27–29),
and there are also some studies indicated that RhoE is
overexpressed and acts as a metastasis promoter in certain cancers
(10,12). Here, we have evaluated RhoE protein
expression patterns in four types of human cancer that are compared
with the matched, adjacent tissues by immunohistochemistry, and the
results showed that RhoE expression levels decreased significantly
in each cancer, and in some cases RhoE appears to be completely
absent from the tumors. Additionally, we found that RhoE expression
level was inversely associated with the tumor differentiation
grade. The differences seem to be more significant in breast and
gastric cancer tissues, and less in lung cancer tissues.
To explore the effect of enhanced RhoE expression on
the phenotypes of cancer cells, four cancer cell lines (MKN45,
HepG2, SW480 and SPCA1) which show relatively lower RhoE expression
were transfected with pcDNA3.0-RhoE. Overexpression of RhoE not
only significantly inhibited the cancer cell proliferation,
migration and invasion, but also induced apoptosis. These
observations indicate that RhoE is intimately involved in many
aspects of tumor progression and suggest the possibility that RhoE
and its signaling cascade could serve as potential therapeutic
target in anticancer treatment.
Since RhoE has a prominent effect on cancer cell
proliferation, we thought that it might be involved in cell cycle
and/or survival regulation. Our FACS analysis has shown that
enhanced RhoE expression inhibited G1/S cell cycle transition and
promoted apoptosis in the four tested cell lines. Previously,
Villalonga et al (9) and
Poch et al (30) also
reported that RhoE could block G1 phase cell cycle progression in
NIH3T3 cells and human glioblastoma U87 cells, while Bektic et
al (31) found that RhoE could
also induce a G2 block in prostate cancer cells (31,32).
The differences may be due to different cancer types or cell lines
used in the experiments.
Our unpublished data show that RhoE level does not
change in response to serum stimulation. Since the study of
Ongusaha et al indicated that RhoE was a pro-survival target
gene of p53 which inhibits ROCK1-mediated apoptosis in response to
genotoxic stress (16), we
subsequently investigated the p53 activity by dual-luciferase
reporter assay, and found that the transcription activity of p53
was low in the RhoE low-expressed cancer cell lines and high in the
high RhoE expressed cancer cell lines. Furthermore, in each of the
four examined cell lines with relatively low RhoE expressions
(MKN45, HepG2, SW480 and SPCA1), we also found exogenous p53
expression significantly enhanced the RhoE expression. Taken
together, these findings confirm that the expression of RhoE was
regulated by p53.
p53 activity can be regulated by numerous factors in
cells, and loss of function mutations is prominent in the reduction
of its tumor surveillance function. p53 gene mutation was found in
>50% of all types of human cancers and >80% were missense
mutations that lead to the synthesis of a stable full-length
protein (33). At the beginning of
understanding the reason of the significant differences of p53
activities in different cancer cell lines, we have examined p53
mutations in 9 cancer cell lines and found only one meaningful
mutation in SW480, and the activity of p53 and the expression level
of RhoE were both the lowest in this cell line. These results,
coupled with the demonstration that increased functional p53
expression could increase RhoE protein level in cells, suggest that
p53 mutations in cancer cells may be related to the low expression
of RhoE. To demonstrate this hypothesis, 10 kinds of PEGFP-mt-p53
vectors with the hot points of p53 mutation were generated. We
found that Luc/Ren ratios were significantly lower in the cells
which were transfected with EGFP-mt-p53 than that of EGFP-wt-p53,
and there was no difference between the mutants. These results
suggest that RhoE expression could not only be associated with p53
transcription but also be affected by p53 mutation.
Previous cell biology studies have suggested that
RhoE was able to bind to ROCK1 and to inhibit ROCK-induced myosin
phosphatase phosphorylation (25).
ROCK1 is a RhoA downstream target known to mediate actin stress
fiber assembly, whereas RhoE was found to antagonize RhoA to induce
stress fiber disassembly (18,19).
RhoE could also be phosphorylated by ROCK1, leading to an increased
stability and altered cellular localization. Another study has
shown that expression of RhoE leads to decreased cellular RhoA-GTP
by increasing the GAP activity of p190B RhoGAP (18). Here we have shown that RhoE
expression can be regulated by p53 transcription activity. We have
verified that p-MLC level, an indicator of endogenous RhoA-ROCK
signal, decreased in the RhoE-transfectant cancer cells (data not
shown). Therefore, it is possible that RhoE mediate signals from
p53 and serves as an antagonism of RhoA signaling in these cancer
cells through a direct or indirect interaction with p190B RhoGAP or
ROCK1.
In summary, RhoE expression was found significantly
reduced in human gastric, colorectal, lung and breast carcinoma
tissues compared to that of the adjacent normal tissues. Increased
RhoE expression in these cancer cell lines resulted in an
inhibition of proliferation, migration and invasion, and induced
cell apoptosis. RhoE expression could be regulated by p53
transcription and decreased by p53 mutation. Our findings strongly
suggest that RhoE can act as a candidate tumor suppressor and serve
as a marker of cancer classification or progression.
Abbreviations:
|
Rho
|
Ras homology;
|
|
ROCK
|
Rho-associated coiled-coil forming
protein serine-threoine protein kinases;
|
|
SDS
|
sodium dodecyl sulfate;
|
|
PAGE
|
polyacylamide gel electrophoresis cdc
division cycle
|
Acknowledgements
This study was supported by the
National Natural Science Foundation of China (nos. 81071640 and
30672378), the National Basic Research Program of China
(2011CB935800) and the Funds of Health Department of Sichuan
Province (no. 4241218R).
References
|
1.
|
Aznar S, Valeron PF, Rincon SV, Perez LF,
Perona R and Lacal JC: Simultaneous tyrosine and serine
phosphorylation of Stat3 transcription factor is involved in a Rho
GTPase oncogenic transformation. Mol Biol Cell. 12:3282–3294. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Mackay D J and Hall A: Rho GTPases. J Biol
Chem. 273:20685–20688. 1998.PubMed/NCBI
|
|
3.
|
Sahai E and Marshall CJ: Rho-GTPases and
cancer. Nat Rev Cancer. 2:133–142. 2002. View Article : Google Scholar
|
|
4.
|
Van Aelst L and D’Souza-Schorey C: Rho
GTPases and signaling networks. Gene Dev. 11:2295–2322.
1997.PubMed/NCBI
|
|
5.
|
Pruitt K and Der CJ: Ras and rho
regulation of the cell cycle and oncogenesis. Cancer Lett.
171:1–10. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Foster R, Hu KQ, Lu Y, Nolan KM, Thissen J
and Settleman J: Identification of a novel human rho protein with
unusual properties: GTPase deficiency and in vivo farnesylation.
Mol Cell Biol. 16:2689–2699. 1996.PubMed/NCBI
|
|
7.
|
Nobes CD, Lauritzen I, Mattei MG, Paris S,
Hall A and Chardin P: A new member of the Rho family, Rnd1,
promotes disassembly of actin filament structures and loss of cell
adhesion. J Cell Biol. 141:187–197. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Riento K, Totty N, Villalonga P, Garg R,
Guasch R and Ridley AJ: RhoE function is regulated by Rock
1-mediated phosphorylation. EMBO J. 24:1170–1180. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Villalonga P, Guasch RM, Riento K and
Ridley AJ: RhoE inhibits cell cycle progression and ras-induced
transformation. Mol Cell Biol. 24:7829–7840. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Cuiyan Z, Jie H, Fang Z, Kezhi Z, Junting
W, Susheng S, Xiaoli F, Ning L, Xinhua M and Zhaoli C:
Overexpression of RhoE in non-small cell lung cancer (NSCLC) is
associated with smoking and correlates with dna copy number
changes. Cancer Biol Ther. 6:335–342. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Ma W, Wong CC, Tung EK, Wong CM and Ng IO:
RhoE is frequently down-regulated in hepatocellular carcinoma (HCC)
and suppresses HCC invasion through antagonizing the
Rho/Rho-Kinase/Myosin phosphatase target pathway. Hepatology.
57:152–161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Katiyar P and Aplin AE: FOXD3 regulates
migration properties and Rnd3 expression in melanoma cells. Mol
Cancer Res. 9:545–552. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Lowe SW, Schmitt EM, Smith SW, Osborne BA
and Jacks T: p53 is required for radiation-induced apoptosis in
mouse thymocytes. Nature. 362:847–849. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Lane DP: Cancer. p53, guardian of the
genome. Nature. 358:15–16. 1992. View
Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Clarke AR, Purdie CA, Harrison DJ, Morris
RG, Bird CC, Hooper ML and Wyllie AH: Thymocyte apoptosis induced
by p53-dependent and independent pathways. Nature. 362:849–852.
1993. View
Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Ongusaha PP, Kim HG, Boswell SA, Ridley
AJ, Der CJ, Dotto GP, Kim YB, Aaronson SA and Lee SW: RhoE is a
pro-survival p53 target gene that inhibits ROCK1-mediated apoptosis
in response to genotoxic stress. Curr Biol. 16:2466–2472. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Liu Y and Bodmer WF: Analysis of p53
mutations and their expression in 56 colorectal cancer cell lines.
Proc Natl Acad Sci USA. 103:976–981. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Wennerberg K, Forget MA, Ellerbroek SM,
Arthur WT, Burridge K, Settleman J, Der CJ and Hansen SH: Rnd
proteins function as RhoA antagonists by activating p190 RhoGAP.
Curr Biol. 13:1106–1115. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Etienne-Manneville S and Hall A: Rho
GTPases in cell biology. Nature. 420:629–635. 2002. View Article : Google Scholar
|
|
20.
|
Li X, Liu L, Tupper JC, Bannerman DD, Winn
RK, Sebti SM, Hamilton AD and Harlan JM: Inhibition of protein
geranylgeranylation and RhoA/RhoA kinase pathway induces apoptosis
in human endothelial cells. J Biol Chem. 277:15309–15316. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Ghosh PM, Ghosh-Choudhury N, Moyer ML,
Mott GE, Thomas CA, Foster BA, Greenberg NM and Kreisberg JI: Role
of RhoA activation in the growth and morphology of a murine
prostate tumor cell line. Oncogene. 18:4120–4130. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Kamai T, Kawakami S, Koga F, Arai G,
Takagi K, Arai K, Tsujii T and Yoshida KI: RhoA is associated with
invasion and lymph node metastasis in upper urinary tract cancer.
BJU Int. 91:234–238. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Yoshioka K, Nakamori S and Itoh K:
Overexpression of small GTP-binding protein RhoA promotes invasion
of tumor cells. Cancer Res. 59:2004–2010. 1999.PubMed/NCBI
|
|
24.
|
Guasch RM, Scambler P, Jones GE and Ridley
AJ: RhoE regulates actin cytoskeleton organization and cell
migration. Mol Cell Biol. 18:4761–4771. 1998.PubMed/NCBI
|
|
25.
|
Riento K, Guasch RM, Garg R, Jin B and
Ridley AJ: RhoE binds to ROCK1 and inhibits downstream signaling.
Mol Cell Biol. 23:4219–4229. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Liu N, Bi F, Pan Y, Sun L, Xue Y, Shi Y,
Yao X, Zheng Y and Fan D: Reversal of the malignant phenotype of
gastric cancer cells by inhibition of RhoA expression and activity.
Clin Cancer Res. 10:6239–6247. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Grise F, Sena S and Bidaud-Meynard A:
Rnd3/RhoE is down-regulated in hepatocellular carcinoma and
controls cellular invasion. Hepatology. 55:1766–1775. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Luo H, Dong Z, Zou J, Zeng Q, Wu D and Liu
L: Down-regulation of RhoE is associated with progression and poor
prognosis in hepatocellular carcinoma. J Surg Oncol. 105:699–704.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Zhao H, Yang J, Fan T, Li S and Ren X:
RhoE functions as a tumor suppressor in esophageal squamous cell
carcinoma and modulates the PTEN/PI3K/Akt signaling pathway. Tumour
Biol. 33:1363–1374. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Poch E, Miñambres R, Mocholí E, Ivorra C,
Pérez-Aragó A, Guerri C, Pérez-Roger I and Guasch RM: RhoE
interferes with Rb inactivation and regulates the proliferation and
survival of the U87 human glioblastoma cell line. Exp Cell Res.
313:719–731. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Bektic J, Pfeil K, Berger AP, Ramoner R,
Pelzer A, Schafer G, Kofler K, Bartsch G and Klocker H: Small
G-protein RhoE is underexpressed in prostate cancer and induces
cell cycle arrest and apoptosis. Prostate. 64:332–340. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Trojan L, Schaaf A, Steidler A, Haak M,
Thalmann G, Knoll T, Gretz N, Alken P and Michel MS: Identification
of metastasis-associated genes in prostate cancer by genetic
profiling of human prostate cancer cell lines. Anticancer Res.
25:183–191. 2005.PubMed/NCBI
|
|
33.
|
Soussi T and Béroud C: Assessing TP53
status in human tumours to evaluate clinical outcome. Nat Rev
Cancer. 1:233–239. 2001. View Article : Google Scholar : PubMed/NCBI
|