Introduction
Gastric cancer (GC) remains one of the most
frequently occurring malignancies world-wide. Various surgical and
chemotherapeutic treatments have been developed (1), and surgical operation is highly
effective in early-stage cancers (2). However, GC is still a formidable
disease because many cases of GC are diagnosed at an advanced stage
and recur even after complete resection.
The prevalence of GC is associated with several
factors, including environment, diet, microbial infection and
genetic background (3,4). Intake of fruits, vegetables and high
β-carotene containing foods has been reported to reduce the risk of
GC (5–7), while consumption of salted meats
appears to increase the risk of the disease (8). Despite conflicting results serum
cholesterol level has been suggested as one of the factors
affecting the risk of GC (9,10).
High level of serum cholesterol has been well established as a
major risk factor for coronary heart disease and stroke, and has
been implicated in prostate cancer and breast cancer. In contrast,
several studies of cancer epidemiology have indicated that the
lowest level of total cholesterol may also be dangerous because it
is associated with an increased risk of cancer mortality (11,12).
A number of studies have suggested an apparent inverse association
between serum cholesterol level and incidence and mortality of GC
(13–17). However, cellular responses induced
by environmental cholesterol level have not been studied in
carcinomas. In this study, a cell culture model system was used to
clarify the association of cholesterol level and cell viability in
GC cells.
Materials and methods
Cell culture
SNU601, SNU638 and SNU216 human GC cell lines
obtained from the Korea Cell Line Bank were grown in RPMI-1640
medium (Invitrogen) supplemented with 10% (v/v) fetal bovine serum
and 1% antibiotics at 37°C in 5% CO2.
Hoechst 33342 staining and
monodansylcadaverine (MDC) staining
Treated cells were incubated with 1 μg/ml
Hoechst 33342 at 37°C, 5% CO2 for 15 min in the dark,
and both floating and attached cells were collected by
centrifugation. The pooled cell pellets were washed with ice-cold
phosphate-buffered saline (PBS) and fixed in 3.7% formaldehyde on
ice, then washed again with PBS, re-suspended and a fraction of the
suspension was centrifuged in a cytospinner (Hanil, Korea). The
slides were air dried, mounted in an anti-fade solution, and
examined using a DM5000 fluorescence microscope (Leica, Germany) at
respective excitation/emission wavelengths of 340/425 nm. A total
of 500 cells from randomly chosen fields were counted and the
number of apoptotic cells was expressed as a percentage of the
total number of cells counted. For staining of autophagic vacuoles,
treated cells were incubated with MDC for 30 min and washed with
PBS and observed under a fluorescence microscope at 340/525 nm.
MTT assay
Cells were incubated with MTT solution (0.5 mg/ml)
for 4 h and solubilized using dimethylsulfoxide, and the
solubilized formazan product was quantified using an enzyme-linked
immunosorbant assay (ELISA) plate reader at 595 nm; absorbance of
untreated cells was designated as 100% and cell survival was
expressed as a percentage of this value.
Clonogenic assay
Cells treated with cholesterol for 12 h were
trypsinized, washed and re-plated (2,000 cells/60-mm dish). After
incubation for 14 days in a 37°C/5% CO2 incubator,
colonies were stained using crystal violet and scored (>1
mm).
Immunoblot analysis
Equal amounts of protein were electrophoretically
separated using SDS-PAGE and transferred to a nitrocellulose
membrane using a standard technique. Antibodies were used to probe
for active P-ERK1/2, P-MEK1, MEK1, P-JNK1/2, P-p38, p38, caspase-3
(Cell Signaling Technology), PARP, Fas, caveolin-1, ERK2, JNK1/2,
cytochrome c (Santa Cruz), DR4, DR5 (Pro Sci), ATG5 and
LC3II (MBL International). Anti-α-tubulin (BioGenex) was used as a
loading control. Acquisition of signals was performed using an
image analyzer (Image Station 4000MM, Kodak).
Caspase activity assay
The caspase-8 activity assay was performed using a
FADD-like IL-1β-converting enzyme (FLICE) colorimetric assay kit
(BioVision), according to the manufacturer’s protocol. Briefly, 200
μg protein lysates in a 50-μl volume was mixed with
reaction buffer, mixed with IETD-pNA substrate, incubated for 90
min, and the absorbance was measured at 405 nm. Fold increase in
FLICE activity was determined by comparing the results of treated
samples with the level of the untreated control.
Cell transfection
The constitutively active mutant of MEK1 (CA-MEK1)
was designed by substitution of the regulatory phosphorylation
sites, Ser218 and Ser222, with aspartic acid
(S218D/S222D mutant), as described previously (18), and cloned into the pCMV vector.
Then, cells were transfected transiently for 48 h and the protein
amounts of MEK1 and p-ERKs were confirmed by western blot analysis.
For the RNAi, siRNA were obtained from Bioneer and 5–8 μg/ml
RNA were used for transfection by use of AMAXA nucleofector kit
(Amaxa Biosystems GmbH) according to the manufacturer’s protocol.
Sequences of siRNA are as follows: control sense,
5′-CCUACGCCACCAAUUUCGU(dTdT)-3′; DR5 sense,
5′-CAGACUUGGUGCCCUUUGA(dTdT)-3′.
Statistical analysis
All numerical data are reported as mean ± SE. All
data represent the results of at least three independent
experiments. Groups were compared using Student’s t-test.
Results
Cholesterol induces autophagic and
apoptotic death
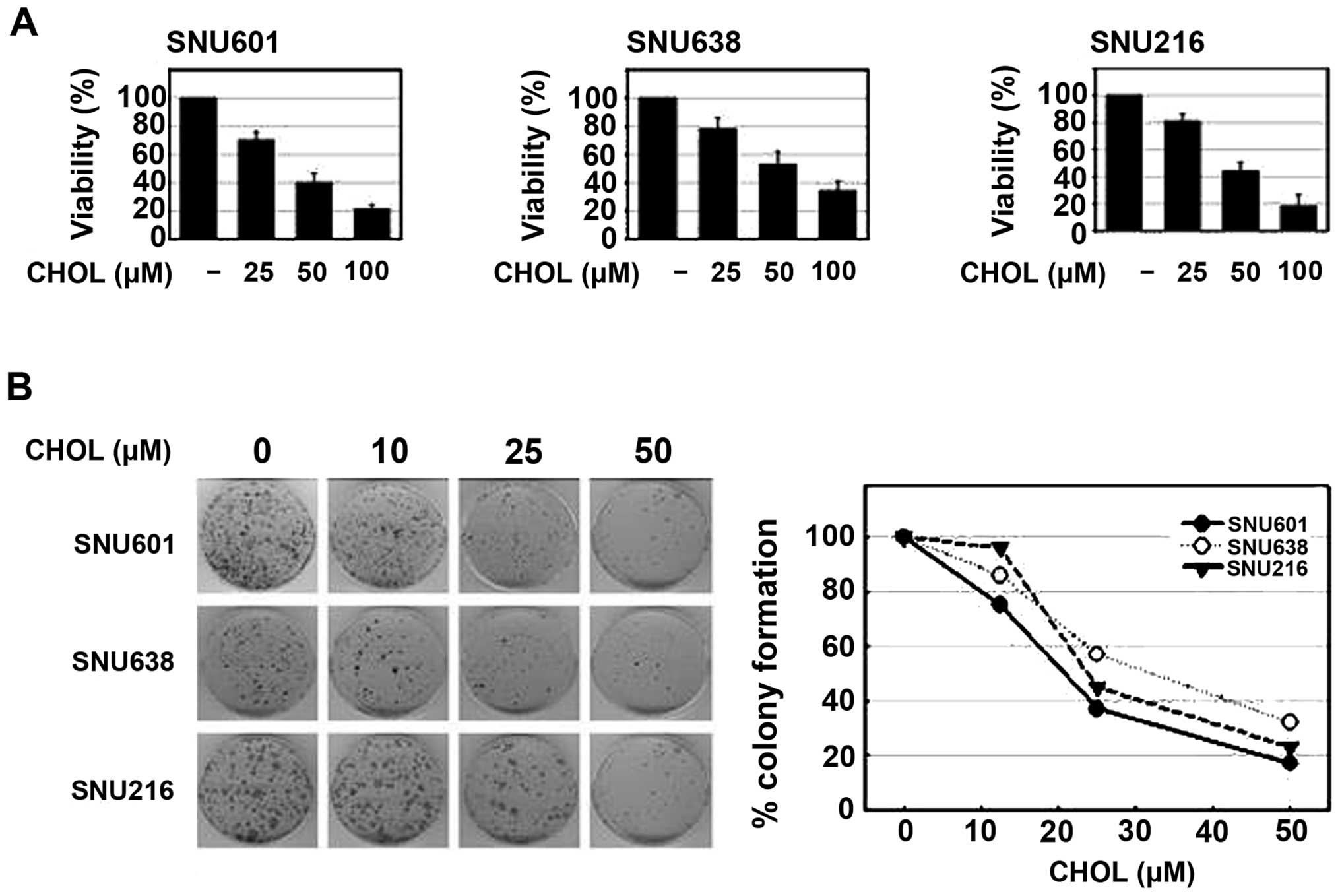
To assess the effect of cholesterol on the viability
of GC cells, a number of different human stomach cancer cell lines,
SNU601, SNU638 and SNU216, were used in the study. Incubation of
these cells in a culture medium with addition of different
concentrations of water-soluble cholesterol resulted in a
dose-dependent decrease in cell viability in all three cell lines,
as determined by the MTT assay. In addition, treatment with
cholesterol also resulted in a marked, dose-dependent decrease in
colony forming ability of these cells (Fig. 1). Thus, cholesterol may exert
antitumor activity in GC cells.
To exclude the specific effect of increased water
solubility of cholesterol, we tested the effect of hydrophobic
cholesterol solubilized in acetic acid in these cells;
water-insoluble hydrophobic cholesterol also reduced cell viability
when compared with the control group, the same as water-soluble
cholesterol treatment (data not shown). Then, in order to explore
the cause of cholesterol-induced viability reduction, we attempted
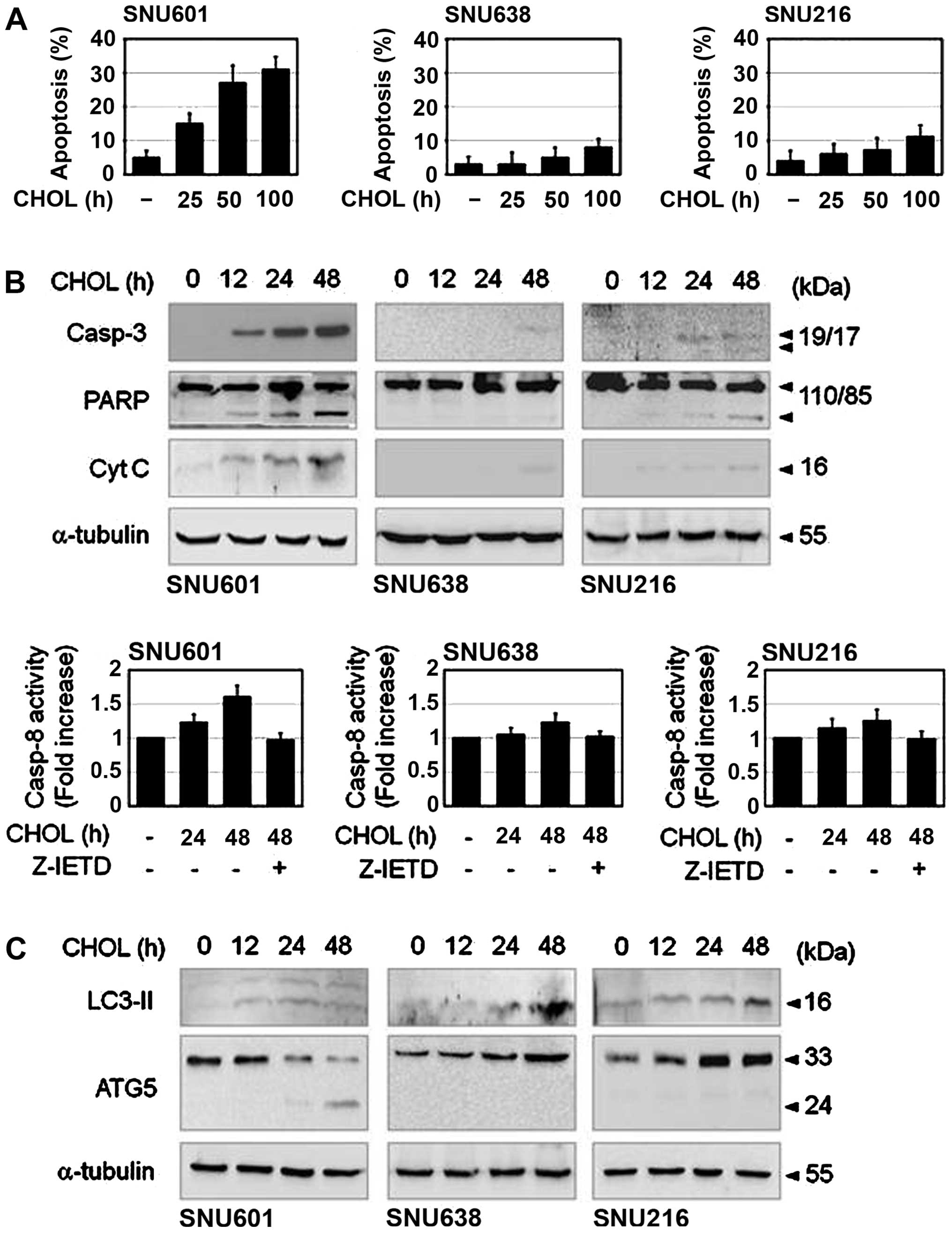
to determine whether cholesterol induces apoptosis in GC cells.
Cells exposed to cholesterol were stained by Hoechst 33342 and
apoptotic nuclei were then counted. Upon exposure to cholesterol,
apoptotic body formation showed a clear increase in SNU601 cells,
however, only a slight increase was observed in SNU638 and SNU216
cells (Fig. 2A). Cholesterol also
strongly induced cleavage of caspase-3, degradation of PARP and
activation of caspase-8 in SNU601 cells, and weakly in SNU638 and
SNU216 cells, as demonstrated by immunoblot analysis and caspase
activity assay (Fig. 2B). Although
viability of all three cell lines was decreased by cholesterol,
active apoptotic induction was observed only in SNU601 cells. Thus,
we also attempted to determine whether other types of cell death
are involved in cholesterol-induced reduction of cell viability.
Because cleavage of LC3I to LC3II is a sign of autophagy, we
examined the question of whether autophagy occurred in this event
by detection of LC3II protein level. Treatment with cholesterol
resulted in a mild increase in LC3II level in SNU601 cells, with a
stronger increase in SNU638 and SNU216 cells (Fig. 2C). Furthermore, essential
autophagic factor, ATG5 was also accumulated in SNU638 and SNU216
cells. However, necrotic features, PI inclusion or LDH release were
not observed in these cell lines by 48 h (laboratory observation).
Therefore, cholesterol seemed to trigger cell killing by apoptosis
and autophagy in GC cells.
Inhibition of autophagy increases
apoptosis
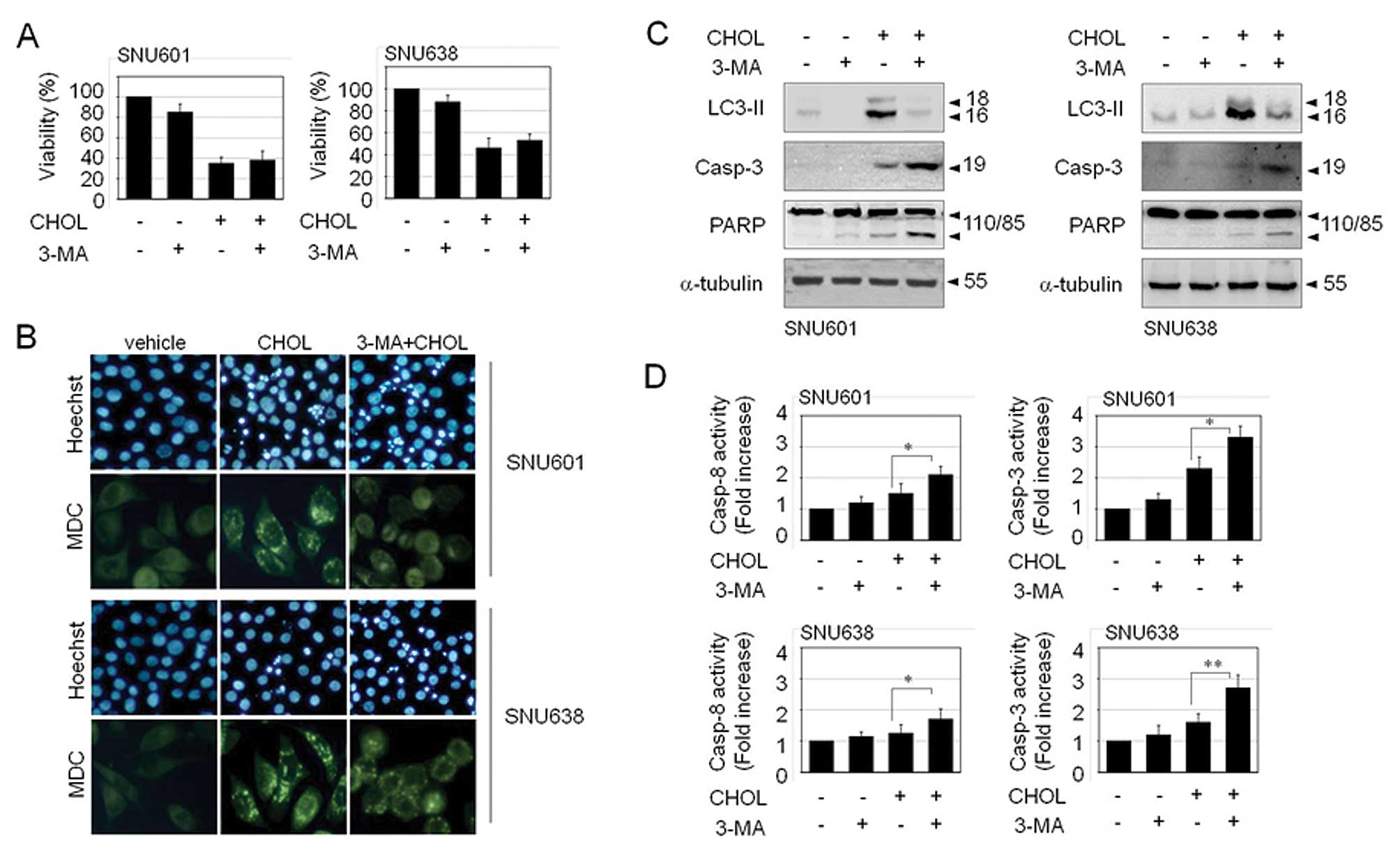
Autophagy may function as a protective mechanism and
as a cell death mechanism. If this cholesterol-induced autophagy
exerts a protective role, prevention of autophagy will further
enhance cell death upon exposure to cholesterol. Nevertheless,
inhibition of autophagy by 3-methyladenine (3-MA) did not aggravate
cholesterol-mediated cell toxicity (Fig. 3A). This implies that
cholesterol-induced autophagy is not a protective autophagy, but a
process of a cell death. However, inhibition of autophagic death
did not restore viability of cholesterol-treated cells either.
Autophagy inhibitor, 3-MA resulted in a decrease in the number of
autophagic vacuoles, as detected by MDC staining. However, instead
of a decrease of autophagic death, 3-MA led to an increase in
apoptosis, as detected by elevated apoptotic body formation,
cleavage of procaspase-3 and PARP, and activation of caspase-3 and
caspase-8 (Fig. 3B–D). Therefore,
it appears that the fate of the cell has already been determined as
death before decision of cell death mode between autophagic death
and apoptosis; thus, blockade of the autophagic pathway may take a
bypass to the apoptotic pathway instead of cell survival.
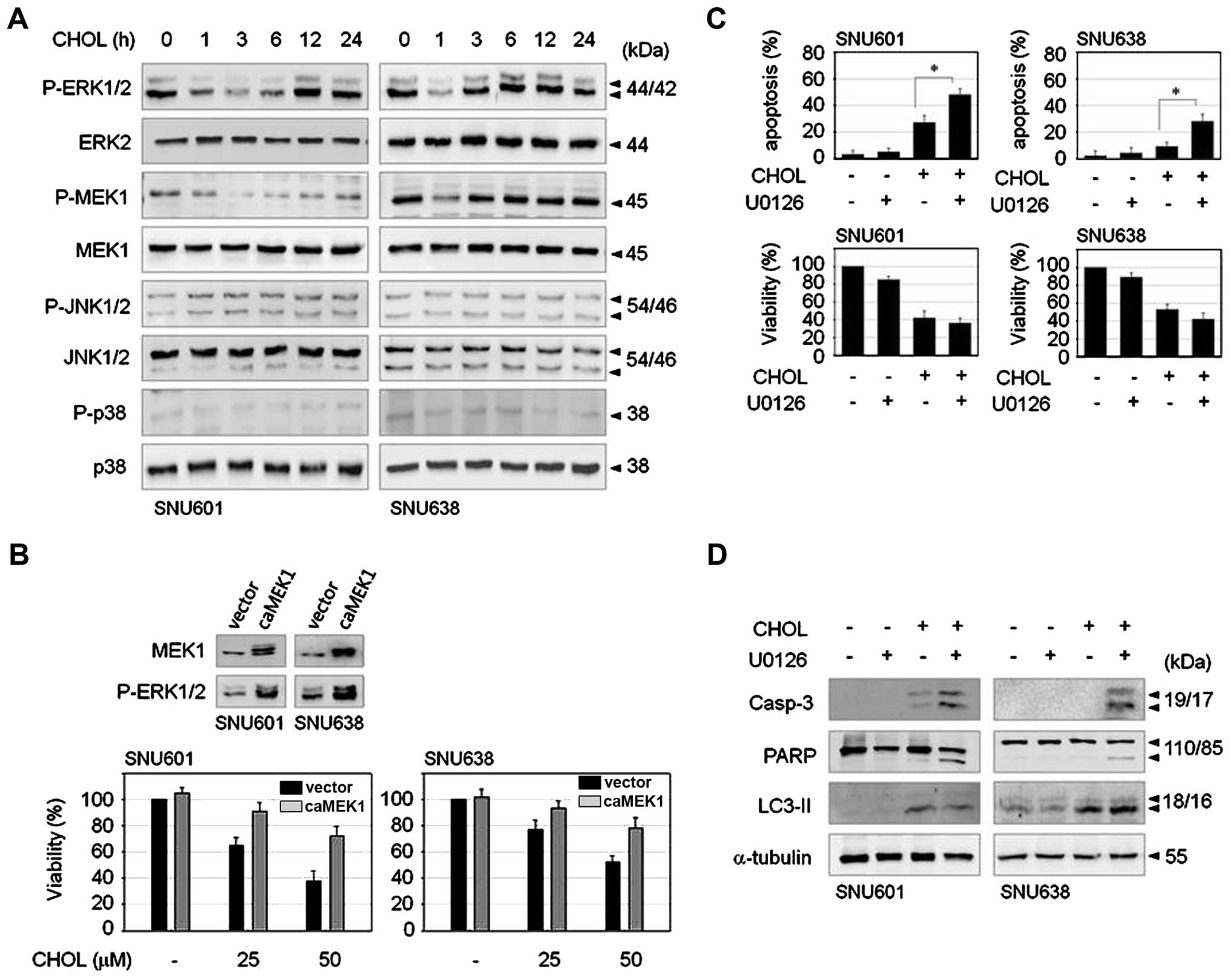
Transient inactivation of the ERK1/2
pathway is triggered by cholesterol
Mitogen-activated protein kinase (MAPK) family
proteins play critical roles in regulation of cell survival and
death; therefore, in order to determine the signaling mechanism
involved in cholesterol-induced GC cell death, we first examined
the effect of cholesterol on activities of MAPK family members.
Exposure to cholesterol triggered an abrupt and transient decrease
of ERK1/2 phosphorylation not affecting JNK or p38MAPK in both
SNU601 and SNU638 cell lines. MEK1, upstream kinase of ERK1/2, was
also transiently dephosphorylated by cholesterol (Fig. 4A). To examine the role of the
ERK1/2 pathway in cholesterol-treated cells, we observed the effect
of cholesterol in CA-MEK1 overexpressed cells. In comparison with
control cells, CA-MEK1 expressed cells demonstrated significant
rescue of cell viability (Fig.
4B). Thus, reduction of ERK activity appears to be responsible
for cholesterol-mediated cell death. Nevertheless, because
phospho-status of ERK was recovered soon after dephosphorylation
upon treatment with cholesterol, we investigated the role of
restored ERK activity. MEK inhibitor, U0126, was added to medium 3
h after treatment with cholesterol in order to inhibit reactivation
of ERK1/2. Combination of U0126 resulted in strongly increased
apoptosis compared to cholesterol treatment alone, as demonstrated
by enhancement of apoptotic body formation and cleavage of
procaspase-3 and PARP (Fig. 4C and
D). However, the level of autophagic marker, LC3II, was not
altered by U0126 in either cell type (Fig. 4D), indicating that inhibition of
the late ERK pathway resulted in activation of strong apoptotic
signaling. These results suggest that cholesterol-induced early
downregulation of ERK signaling evokes loss of cell viability, and
subsequent restoration of ERK activity may be responsible for
inhibition of apoptotic pathway.
Membrane death receptor TRAIL-R2/DR5 is
involved in cholesterol-mediated gastric cancer cell death
Cholesterol is an essential component of the cell
membrane and cholesterol-enriched membrane lipid rafts can affect
membrane receptor-linked signal regulation. Furthermore, in this
study, treatment with cholesterol resulted in activation of
caspase-8, a representative death receptor-mediated apoptotic
enzyme. Thus, we attempted to determine whether the death receptor
pathway is involved in this cholesterol-mediated cell death event.
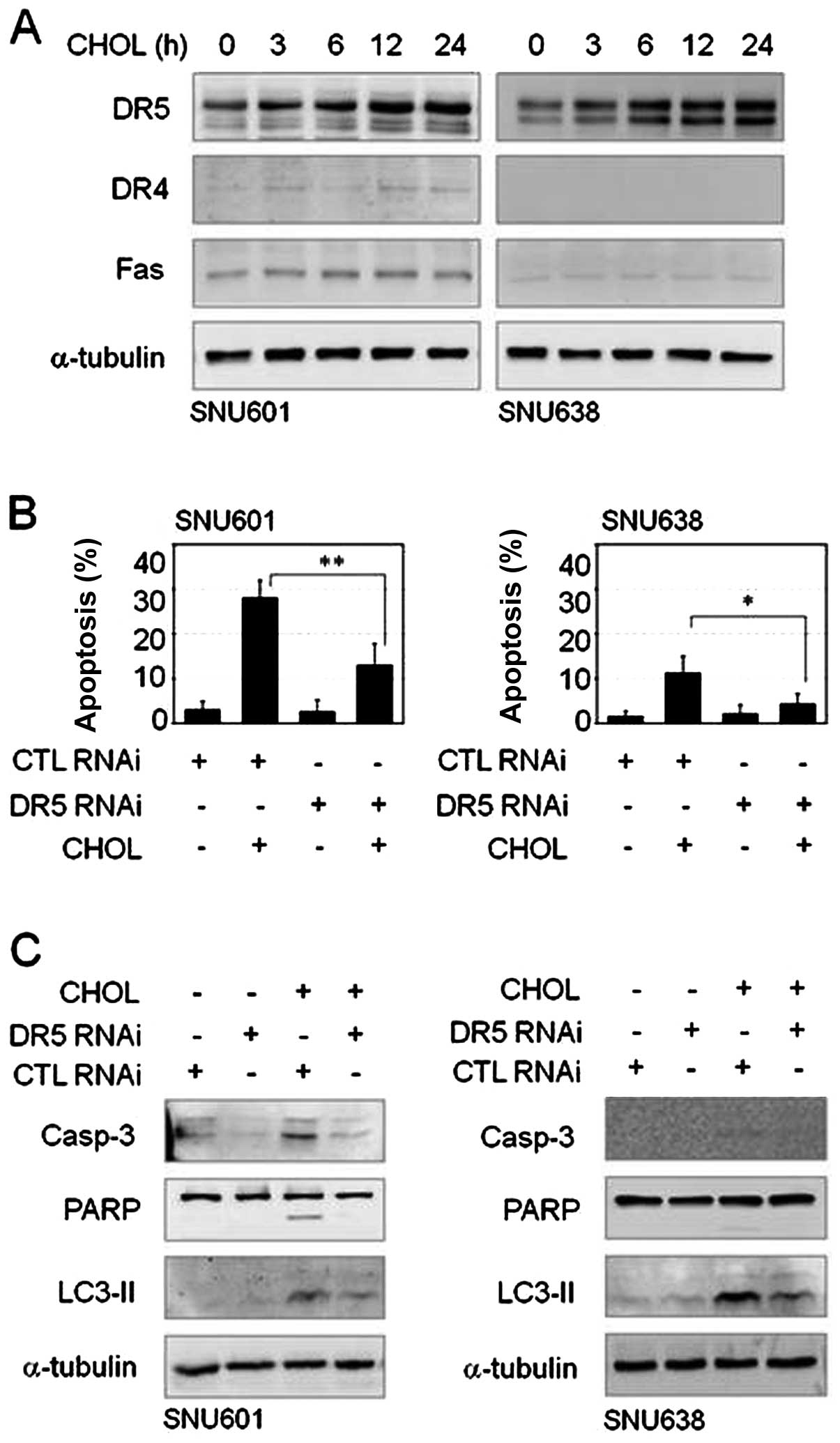
In immunoblot analysis to assess expression of death receptors;
TRAIL-R1/DR4, TRAIL-R2/DR5 and Fas/CD95, increase of TRAIL-R2/DR5
protein was detected after treatment with cholesterol in both cell
lines, however, protein levels of TRAIL-R1/DR4 and Fas/CD95 were
unchanged by cholesterol or were too low to be observed in these
cells (Fig. 5A). In order to
understand the role of TRAIL-R2/DR5 overexpression in
cholesterol-mediated apoptosis, we performed an interference assay
using siRNA of TRAIL-R2/DR5. Compared to scrambled control RNA (CTL
RNAi) transfection, TRAIL-R2/DR5 siRNA transfection resulted in a
decrease in cholesterol-induced apoptosis in both GC cell lines
(Fig. 5B). In addition, knockdown
of TRAIL-R2/DR5 reduced not only cleavage of caspase-3 and PARP but
also elevation of LC3II level (Fig.
5C). Thus, TRAIL-R2/DR5 appeared to be linked to apoptotic and
autophagic death pathways in response to cholesterol.
Discussion
Cholesterol plays an important role in the human
body as a precursor of critical biochemical molecules, including
steroid hormones, vitamin D and bile acids. In addition,
cholesterol is an essential component enriched in biological
membranes and participates in control of cellular membrane fluidity
and lipid rafts. Lipid rafts serve as signaling platforms at the
cell membrane and modification of major lipid raft components, such
as cholesterol, sphingolipid and ceramide, play a role in
regulation of cell viability under cytotoxic stimuli (19). Mediation of akt-regulated cell
survival by cholesterol-rich lipid rafts and increased apoptosis by
depletion of cholesterol in prostate cancer cells have been
reported (20,21). In addition, intake of high
cholesterol has been reported to show correlation with an increased
risk of occurrence of certain cancers, including breast, prostate,
and colon cancer. However, low level of cholesterol may also be
harmful in certain cases. Cholesterol depletion prohibits the
effect of chemotherapeutics by inhibition of membrane raft
formation, because certain types of chemotherapeutic drugs require
lipid raft-dependent death receptor activation for induction of
cancer cell death (22,23). In addition, several epidemiological
studies have reported an association of serum cholesterol levels at
the lower end of the distribution with risk of cancer mortality
(10,24–27).
In this study, increased level of cholesterol led to markedly
reduced viability and clonogenicity of stomach cancer cells.
Increased level of cholesterol was previously shown to induce
apoptosis in macrophages through various mechanisms involving the
Fas pathway, the mitochondrial apoptotic pathway and the
endoplasmic reticulum response (28–30).
In measurement of morphological and biochemical features induced by
cholesterol, both apoptotic and autophagic deaths were observed in
GC cells, although dominant type of death was cell line specific.
Cholesterol stimulated accumulation of ATG5 and cleavage of LC3
with a very low level of apoptotic body formation in SNU638 and
SNU216 cells, indicating that autophagic death occurred mainly in
these cells. However, in SNU601 cells, ATG5 was degraded and LC3
cleavage was mild, instead, strong apoptotic features, including
nuclear fragmentation, caspase-3 cleavage and PARP degradation were
detected.
Of particular interest, crosstalk appeared to occur
between the apoptotic pathway and autophagic pathway. In
combination of cholesterol with autophagy inhibitor 3-MA,
autophagic death was reduced and resulted in switching the death
mode to apoptosis without significant effect on cell viability.
Early dephosphorylation of ERK appeared to be responsible for the
cholesterol-mediated cell death since overexpression of CA-MEK1
resulted in significantly restored cell viability. A few hours
after dephosphorylation of ERK, subsequent rephosphorylation was
observed and suppression of ERK reactivation by post-treatment with
MEK inhibitor U0126 resulted in elevation of apoptosis and decrease
of cell viability, indicating that this restoration of ERK activity
is mainly involved in the anti-apoptotic pathway. We also
demonstrated that exposure of GC cells to cholesterol increased
expression of TRAIL-R2/DR5 protein and this was involved in
cholesterol-induced GC cell death. Knockdown of TRAIL-R2/DR5
resulted in reduced activation of caspase-3, cleavage of PARP and
LC3II level in response to cholesterol, thus, TRAIL-R2/DR5 appeared
to be linked to both apoptotic and autophagic death signaling. To
understand the relationship between ERK pathway and TRAIL-R2/DR5
induction, we measured cholesterol-mediated ERK phosphorylation in
a condition of TRAIL-R2/DR5 interference, and knockdown of
TRAIL-R2/DR5 had no effect on reduction of ERK phosphorylation, and
inhibition of ERK reactivation by U0126 did not enhance
TRAIL-R2/DR5 induction (data not shown). Thus, these two events
seem to be independently controlled by cholesterol in GC cells.
Based on these results, our findings demonstrated
that exposure of GC cells to cholesterol resulted in stimulation of
apoptotic and autophagic death through inactivation of ERK and
induction of TRAIL-R2/DR5, and this may be one of the explanations
that serum cholesterol levels at the lower end of the distribution
are related to higher risk of stomach cancer mortality.
Acknowledgements
This research was supported by the
Basic Science Research Program through the National Research
Foundation of Korea (NRF) funded by the Ministry of Education,
Science and Technology (NRF-2011-0014540).
References
|
1.
|
Greenlee RT, Hill-Harmon MB, Murray T and
Thun M: Cancer statistics, 2001. CA Cancer J Clin. 51:15–36. 2001.
View Article : Google Scholar
|
|
2.
|
Roukos DH: Current status and future
perspectives in gastric cancer management. Cancer Treat Rev.
26:243–255. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Fiedorek SC, Malaty HM, Evans DL, Pumphrey
CL, Casteel HB, Evans DJ Jr and Graham DY: Factors influencing the
epidemiology of Helicobacter pylori infection in children.
Pediatrics. 88:578–582. 1991.PubMed/NCBI
|
|
4.
|
Plummer M, Franceschi S and Munoz N:
Epidemiology of gastric cancer. IARC Sci Publ. 2004:311–326.
2004.
|
|
5.
|
Lunet N, Valbuena C, Vieira AL, Lopes C,
Lopes C, David L, Carneiro F and Barros H: Fruit and vegetable
consumption and gastric cancer by location and histological type:
case-control and meta-analysis. Eur J Cancer Prev. 16:312–327.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Kaaks R, Tuyns AJ, Haelterman M and Riboli
E: Nutrient intake patterns and gastric cancer risk: a case-control
study in Belgium. Int J Cancer. 78:415–420. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Palli D, Russo A and Decarli A: Dietary
patterns, nutrient intake and gastric cancer in a high-risk area of
Italy. Cancer Causes Control. 12:163–172. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
De Stefani E, Correa P, Boffetta P,
Deneo-Pellegrini H, Ronco AL and Mendilaharsu M: Dietary patterns
and risk of gastric cancer: a case-control study in Uruguay.
Gastric Cancer. 7:211–220. 2004.
|
|
9.
|
Asano K, Kubo M, Yonemoto K, Doi Y,
Ninomiya T, Tanizaki Y, Arima H, Shirota T, Matsumoto T, Iida M and
Kiyohara Y: Impact of serum total cholesterol on the incidence of
gastric cancer in a population-based prospective study: the
Hisayama study. Int J Cancer. 122:909–914. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Dessi S, Batetta B, Pulisci D, Spano O,
Anchisi C, Tessitore L, Costelli P, Baccino FM, Aroasio E and Pani
P: Cholesterol content in tumor tissues is inversely associated
with high-density lipoprotein cholesterol in serum in patients with
gastrointestinal cancer. Cancer. 73:253–258. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Jafri H, Alsheikh-Ali AA and Karas RH:
Baseline and on-treatment high-density lipoprotein cholesterol and
the risk of cancer in randomized controlled trials of
lipid-altering therapy. J Am Coll Cardiol. 55:2846–2854
|
|
12.
|
Levy RI: Cholesterol and disease - what
are the facts? JAMA. 248:2888–2890. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Eichholzer M, Stahelin HB, Gutzwiller F,
Ludin E and Bernasconi F: Association of low plasma cholesterol
with mortality for cancer at various sites in men: 17-y follow-up
of the prospective Basel study. Am J Clin Nutr. 71:569–574.
2000.PubMed/NCBI
|
|
14.
|
Kritchevsky SB and Kritchevsky D: Serum
cholesterol and cancer risk: an epidemiologic perspective. Annu Rev
Nutr. 12:391–416. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Schatzkin A, Hoover RN, Taylor PR, Ziegler
RG, Carter CL, Larson DB and Licitra LM: Serum cholesterol and
cancer in the NHANES I epidemiologic follow-up study. National
Health and Nutrition Examination Survey. Lancet. 2:298–301. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Tornberg SA, Holm LE, Carstensen JM and
Eklund GA: Cancer incidence and cancer mortality in relation to
serum cholesterol. J Natl Cancer Inst. 81:1917–1921. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Wannamethee G, Shaper AG, Whincup PH and
Walker M: Low serum total cholesterol concentrations and mortality
in middle aged British men. BMJ. 311:409–413. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Brunet A, Pages G and Pouyssegur J:
Constitutively active mutants of MAP kinase kinase (MEK1) induce
growth factor-relaxation and oncogenicity when expressed in
fibroblasts. Oncogene. 9:3379–3387. 1994.PubMed/NCBI
|
|
19.
|
Edidin M: The state of lipid rafts: from
model membranes to cells. Annu Rev Biophys Biomol Struct.
32:257–283. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Zhuang L, Kim J, Adam RM, Solomon KR and
Freeman MR: Cholesterol targeting alters lipid raft composition and
cell survival in prostate cancer cells and xenografts. J Clin
Invest. 115:959–968. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Zhuang L, Lin J, Lu ML, Solomon KR and
Freeman MR: Cholesterol-rich lipid rafts mediate akt-regulated
survival in prostate cancer cells. Cancer Res. 62:2227–2231.
2002.PubMed/NCBI
|
|
22.
|
Gajate C, Gonzalez-Camacho F and Mollinedo
F: Involvement of raft aggregates enriched in Fas/CD95
death-inducing signaling complex in the antileukemic action of
edelfosine in Jurkat cells. PLoS One. 4:e50442009. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Mollinedo F and Gajate C: Fas/CD95 death
receptor and lipid rafts: new targets for apoptosis-directed cancer
therapy. Drug Resist Updat. 9:51–73. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Iribarren C, Reed DM, Chen R, Yano K and
Dwyer JH: Low serum cholesterol and mortality. Which is the cause
and which is the effect? Circulation. 92:2396–2403. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Kitahara CM, Berrington de Gonzalez A,
Freedman ND, Huxley R, Mok Y, Jee SH and Samet JM: Total
cholesterol and cancer risk in a large prospective study in Korea.
J Clin Oncol. 29:1592–1598
|
|
26.
|
Schuit AJ, Van Dijk CE, Dekker JM,
Schouten EG and Kok FJ: Inverse association between serum total
cholesterol and cancer mortality in Dutch civil servants. Am J
Epidemiol. 137:966–976. 1993.PubMed/NCBI
|
|
27.
|
Tornberg SA, Carstensen JM and Holm LE:
Risk of stomach cancer in association with serum cholesterol and
beta-lipoprotein. Acta Oncol. 27:39–42. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Tabas I: Consequences of cellular
cholesterol accumulation: basic concepts and physiological
implications. J Clin Invest. 110:905–911. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Yao PM and Tabas I: Free cholesterol
loading of macrophages induces apoptosis involving the fas pathway.
J Biol Chem. 275:23807–23813. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Yao PM and Tabas I: Free cholesterol
loading of macrophages is associated with widespread mitochondrial
dysfunction and activation of the mitochondrial apoptosis pathway.
J Biol Chem. 276:42468–42476. 2001. View Article : Google Scholar : PubMed/NCBI
|