Introduction
Lung cancer is the leading cause of cancer-related
death world-wide. More than 80% of lung cancers are non-small cell
lung carcinoma (NSCLC) with lung adenocarcinoma being the most
common subtype. Over half of patients with NSCLC have advanced or
metastatic disease at the time of diagnosis. Systemic chemotherapy
is the standard treatment for such patients with advanced NSCLC.
Despite recent improvements in diagnosis and first-line treatment,
the prognosis remains poor, with an overall 5-year survival
probability of only about 15% (1).
Over the last decade, the molecular heterogeneity of
NSCLC has become better understood, and it is now clear that some
tumors are characterized by ‘driver’ oncogene mutations. The most
prevalent mutated oncogenes identified in lung adenocarcinoma are
epidermal growth factor receptor (EGFR), v-Ki-ras2 Kirsten
rat sarcoma viral oncogene homolog (K-ras), and anaplastic
lymphoma kinase (ALK, where translocation and not mutation
is present) (2,3). Several randomized trials demonstrated
that EGFR tyrosine kinase inhibitors (EGFR-TKIs), erlotinib and
gefitinib, are more effective for patients harboring activating
EGFR mutations than standard platinum-based chemotherapy, at
least in terms of response rate, progression-free survival,
toxicity profile and quality of life (4–6). A
multi-targeted tyrosine kinase inhibitor, crizotinib, has also been
reported to exhibit clinical activity against
ALK-translocated NSCLC (7).
Although K-ras mutation is a major driver mutation, there is
no effective treatment that targets the active form of the K-ras
protein.
Several alterations in intracellular signaling are
involved in the development of cancer and tumor progression
(8). The phosphatidylinositol
3-kinase (PI3K)/Akt (also known as protein kinase B) pathway is
believed to be a potential target cancer therapy (9). As a biological function, the
proliferative and anti-apoptotic effects of Akt-mediated signaling
have been established through extensive studies (10,11).
Phosphorylated Akt was detected in 70% of the tumor specimens from
NSCLC patients (12), suggesting a
high incidence of PI3K/Akt pathway activation in NSCLC cells.
Activated Akt is also proposed to contribute to increased
resistance to chemotherapy in NSCLC (13). Accordingly, if Akt also is
activated in the K-ras mutation-harboring cancer,
suppression of Akt may be a strategy for sensitizing cancer cells
against chemotherapeutic agents and to improve treatment
outcomes.
Amrubicin (AMR) is a totally synthetic anthracycline
anticancer drug based on doxorubicin, whose hydroxyl group at
position 9 is replaced by an amino group in AMR to enhance efficacy
(14). In recent years, AMR
monotherapy and combination therapy have been actively studied and
shown promise for the treatment of small-cell lung cancer (SCLC)
(15). In addition, clinical
activity of AMR has been proposed for the treatment of NSCLC
(16). We previously showed that
Akt-suppressing agents synergistically inhibit cell growth when
combined with AMR or sensitize cancer cells to AMR in SCLC cells
(17). Therefore, it is
hypothesized that the combination of Akt-suppressing agent and AMR
can be an effective treatment strategy for NSCLC.
We report here that the combination of AMR and
Akt-suppressing agents, including EGFR-TKIs, show synergistic cell
growth inhibition and that this synergism by the combination of
EGFR-TKIs and AMR may be involved with the K-ras mutation
itself in A549 lung adenocarcinoma cells that have wild-type
EGFR and mutant K-ras genes.
Materials and methods
Chemicals and reagents
AMR (a gift from Dainippon Sumitomo Pharama, Tokyo,
Japan) and pemetrexed (PEM) (a gift from Eli Lilly, Indianapolis,
IN, USA) were dissolved in distilled water and stored at −20°C. A
stock solution of cisplatin (CDDP) (a gift from Nippon Kayaku,
Tokyo, Japan) was reconstituted with water, diluted in 0.9% sodium
chloride solution, and stored at −20°C. Gefitinib (a gift from
AstraZeneca, Chestitre, UK), erlotinib (a gift from F. Hoffmann-La
Roche, Basel, Switzerland), paclitaxel (PTX) (a gift from
Brisol-Meyers-Squibb, Tokyo, Japan),
2-(4-morpholinyl)-8-phenyl-4H-l-benzopiran-4-one (LY294002) (Wako
Pure Chemical Industries, Osaka, Japan), and
4′,5,7-trihydroxy-isoflavone (genistein) (Wako Pure Chemical
Industries) were dissolved in dimethyl-sulfoxide and stored at
−20°C. 3-(4,5-Dimethyl-thiasol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) (Wako Pure Chemical Industries) was dissolved in
phosphate-buffered saline (PBS) and stored at −20°C.
Cells
The sources of A549, Ma10 and PC9 cells, all of
which were lung adenocarcinoma cell lines, were described
previously (18). A549 and Ma10
cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)
supplemented with 10% fetal bovine serum (FBS) and antibiotics (100
U/ml penicillin and 100 μg/ml streptomycin). PC9 cells were
also maintained in RPMI-1640 medium supplemented with 10% FBS and
antibiotics. These cells were grown in a humidified atmosphere of
5% CO2 - 95% air at 37°C.
MTT assay
MTT assay was performed to evaluate cell
proliferation inhibition. Cells were counted with a
hematocytometer, and 104 cells were incubated in 100
μl of medium containing the indicated drugs for 72 h using
96-well flat-bottom multi-plates (Nalge Nunc, Penfield, NY, USA).
After 72 h, 10 μg of MTT in 10 μl PBS was added to
each well and incubation was continued for an additional 4 h.
Thereafter, 100 μl of 0.04 N HCl in 2-propanol was added and
the multi-plates were incubated overnight to solubilize the MTT
formazan crystal. The absorbance of each well was measured at a
570-nm wavelength (reference 650 nm) using a Sunrise scanning
multi-well spectrometer (TECAN Japan, Kanagawa, Japan). Each
experiment was performed in duplicate or triplicate for each drug
concentration and was independently performed two or three
times.
Flow cytometry with Annexin V
Apoptosis rates were determined by flow cytometry
analysis using an Annexin V-FITC (Beckman Coulter, Fullerton, CA,
USA). A549 cells were plated at a density of 106 cells
per well in 6-well plates and then treated with LY294002 (25
μM) and/or AMR (0.1 μM) for 24 h. Staining was
performed according to the manufacturer’s instructions.
Fluorescence-activated cell sorting (FACS) analysis was performed
immediately after staining using a FACSCalibur flow cytometer
(Becton-Dickinson, Franklin Lakes, NJ, USA).
Western blot analysis
Cells were seeded in 6-well tissue culture plates.
Twenty-four hours after cell seeding, cells were washed with
ice-cold PBS and lysed in lysis buffer [20 mM HEPES, 10 mM EGTA (pH
8), 1% Triton X-100, 40 mM β-glyserophosphate, 2.5 mM
MgCl2, 2 mM Na3VO4] including 1 mM
PMSF, 1 mM DTT, 10 mg/ml leupeptin, 20 μg/ml aprotinin, and
a phosphatase inhibitor cocktail (Nacalai Tesque, Kyoto, Japan).
After 5 min on ice, lysates were centrifuged at 13,000 × g for 10
min at 4°C and the supernatant was collected. Protein was measured
by using the Bio-Rad Protein assay reagent (Bio-Rad Laboratories,
Hercules, CA, USA), and protein lysates containing 20 μg of
total cellular protein were subjected to discontinuous
SDS-polyacrylamide gel electrophoresis. Proteins were
electrotransferred to a polyvinylidene fluoride (PVDF) membrane (GE
Healthcare Japan, Tokyo, Japan) for 40 min at 4°C at 200 V.
Non-specific binding was blocked by incubation with 5% non-fat milk
in tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 h at
room temperature. The following primary antibodies were probed
(1:100 unless otherwise indicated): anti-c-K-ras clone Ab-1
(Calbiochem, San Diego, CA, USA), anti-Akt, anti-phospho-Akt
(Ser473), anti-EGFR, anti-phospho-EGFR (Y1068) (Cell Signaling
Technology, Beverly, MA, USA), and anti-β-actin antibody
(Sigma-Aldrich Japan, Tokyo, Japan), overnight and washed twice
with TBST. After washing, proteins were detected by incubation with
horseradish peroxidase-labeled secondary antibodies (GE Healthcare
Japan). Finally, each protein was detected using an enhanced
chemiluminescence detection system (ECL prime) (GE Healthcare
Japan) and captured with an ImageQuant LAS400 (GE Healthcare
Japan).
Transfection with siRNA
Either 5×104 cells or 5×103
cells were seeded in 6- or 96-well tissue culture plates.
Twenty-four hours after cell seeding, transient small interfering
RNA (siRNA) directed against specific K-ras (ON-TARGETplus siRNA
SMART pool; Thermo Fischer Scientific, Rockford, IL, USA) or
control siRNA-A (sc-37007; Santa Cruz Biotechnology, Santa Cruz,
CA, USA) was transfected to A549 cells according to the
manufacturer’s instructions. Briefly, A549 cells were treated with
the indicated concentration of siRNA using 5 μl
Lipofectamine 2000 transfection reagent in Opti-MEM I reduced serum
medium (both from Invitrogen, Carlsbad, CA, USA) for 6 h. The
medium was removed and replaced with fresh DMEM supplemented with
10% fetal calf serum and antibiotics. Cells were used 24 h after
transfection for western blot analysis or MTT assay.
Assessment of combination effect
To assess the combination effect of the indicated
agents qualitatively, isobologram analysis was utilized as
described previously (19). The
percentage of cell proliferation was calculated as: [(mean
absorbance of drug-treated wells − mean absorbance of cell-free
wells)/(mean absorbance of vehicle cells − mean absorbance of
cell-free wells)] × 100. We used the concentration producing 50%
inhibition of cell growth (IC50) to evaluate
dose-response interactions.
A combination index (CI) was used to compare the
combination effect of the two drugs quantitatively between control
and treated cells. The CI quantitatively depicts synergism (CI
<1), addictive effect (CI = 1), and antagonism (CI >1). The
CI for each fraction-affected value representing the percentage of
proliferation inhibited by a drug was calculated using the Chou and
Talalay method (20). The
fraction-affected value (Fa)/CI plots were constructed in Excel
2007.
Results
Effects of LY294002 on Akt activity and
interactions with chemotherapeutic agents in A549 cells
We tested the interaction between an Akt inhibitor,
LY294002 and representative chemotherapeutic agents including AMR
in A549 cells. A549 cells were treated with the indicated
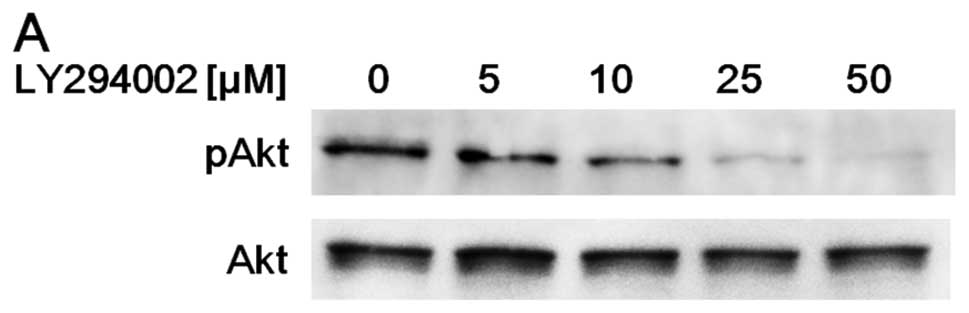
concentration of LY294002 for 1 h. LY294002 at 25 μM
effectively suppressed Akt phosphorylation (Fig. 1A).
We previously reported that the combination of
LY294002 and AMR synergistically inhibited the growth of N417
cells, derived from SCLC. In A549 cells, the combination of
LY294002 and AMR also synergistically inhibited cell growth,
whereas only additive interactions were observed in the combination
of LY294002 with CDDP and PEM. In the combination of LY294002 and
PTX, only antagonistic effects were observed, as judged by
isobologram analysis (Fig.
1B).
To evaluate whether the synergism observed in the
combination of AMR and LY294002 is attributable to an enhancement
of apoptotic cell death, the binding of Annexin V to cells was
measured by flow cytometry after treatment with either AMR (0.1
μM), LY294002 (25 μM) or the combination. Although
Annexin V binding did not differ remarkably after treatment with
the single agent compared to untreated cells, a clear increase in
Annexin V binding was observed after the simultaneous combination
of LY294002 and AMR (Fig. 1C).
Effects of genistein on the activity of
Akt and synergistic cell growth inhibition by the combination of
AMR
Because Akt works downstream of tyrosine kinases
(21), we tested whether
genistein, a non-specific tyrosine kinase inhibitor, suppresses Akt
activity and synergistically inhibits cell growth in combination
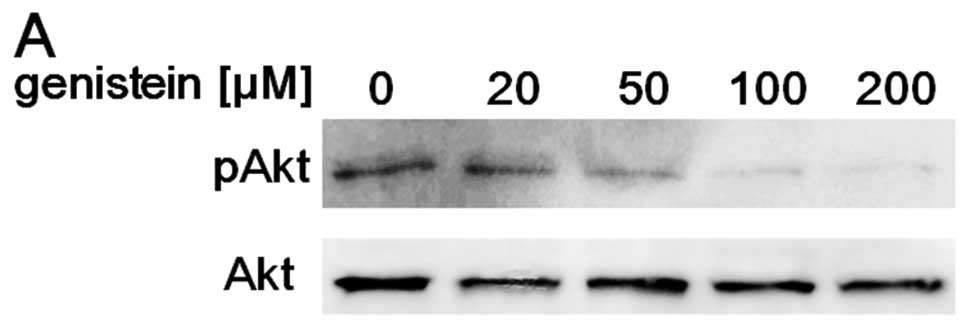
with AMR. A549 cells were treated with the indicated concentration
of genistein for 6 h. As expected, genistein
concentration-dependently suppressed Akt activity in the
concentration range <200 μM (Fig. 2A). Moreover, genistein at these
concentrations showed additive to synergistic growth inhibition in
combination with AMR (Fig.
2B).
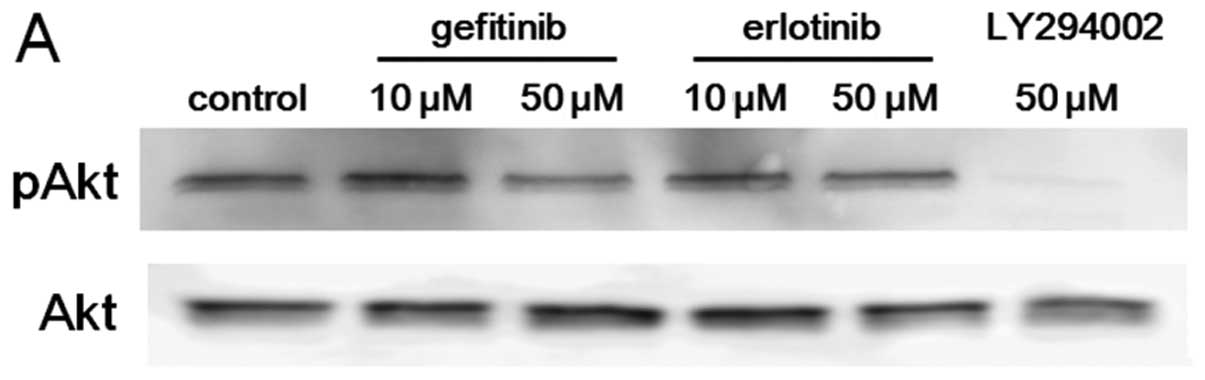
Effects of gefitinib and erlotinib on
cell growth and Akt activity in A549 and PC9 cells
Previously, we reported that A549 and PC9 cells
harbor wild-type and activating mutant (del E746-A750) EGFR
gene, respectively (18). Using
these cell lines, we evaluated the relationship between
growth-inhibitory activity and Akt suppression by gefitinib or
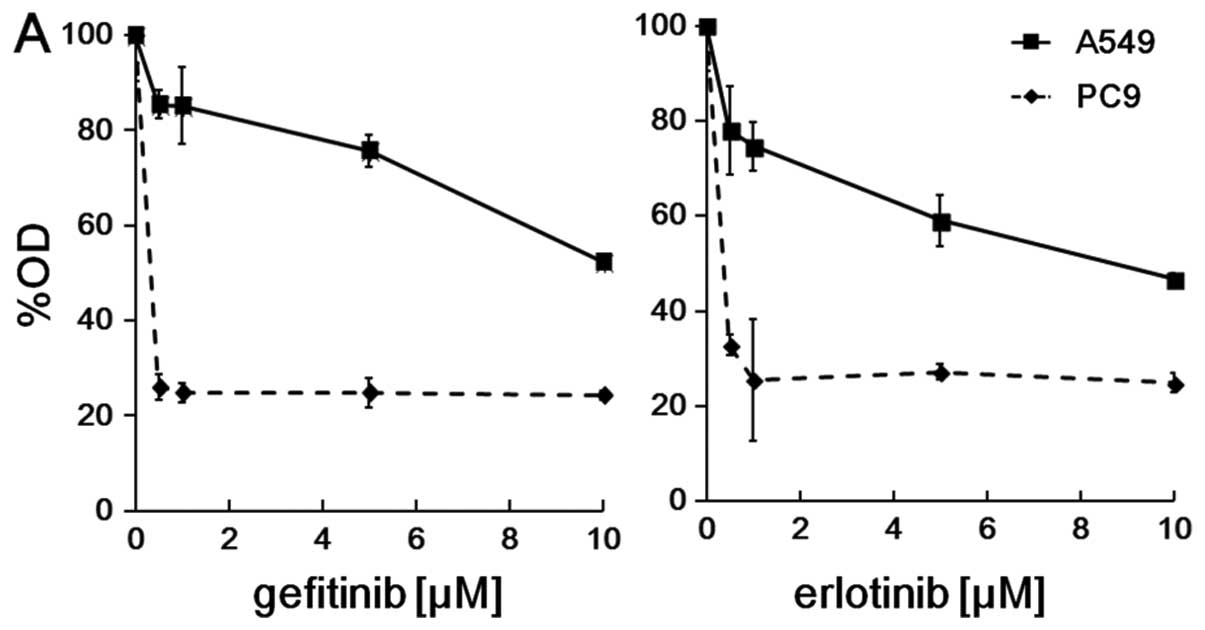
erlotinib. Both gefitinib and erlotinib demonstrated growth
inhibition in both cell lines, but sensitivity to these drugs
varied markedly between them. In PC9 cells with a mutated
EGFR gene, the IC50s for gefitinib and erlotinib
were 10 to 100 nM. On the other hand, compared with PC9 cells, A549
cells were highly resistant in terms of growth inhibition by
EGFR-TKIs with an IC50 of approximately 10 μM
(Fig. 3A).
Akt activity in A549 and PC9 cells was evaluated
after 2 h treatment with gefitinib or erlotinib. In PC9 cells, both
gefitinib and erlotinib suppressed Akt activity at a concentration
of 10 nM or more, indicating that the IC50 and the
Akt-suppressing concentration of EGFR-TKIs are at similar levels.
In A549 cells, on the other hand, although the IC50s for
gefitinib or erlotinib were approximately 10 μM, 100 nM to 1
μM of gefitinib or erlotinib suppressed Akt activity
(Fig. 3B). These observations
suggested that there is a discrepancy between cell growth
inhibition and Akt-suppressing activity by EGFR-TKIs in A549
cells.
Synergistic cell growth inhibition by the
combination of AMR and EGFR-TKIs in A549 cells
Akt-suppressing concentrations of EGFR-TKIs were
apparently lower than the IC50 in A549 cells. It was
postulated that these agents function as Akt inhibitors and
demonstrated AMR-sensitizing activity in A549 cells with wild-type
EGFR. We evaluated the combination effects of EGFR-TKIs and
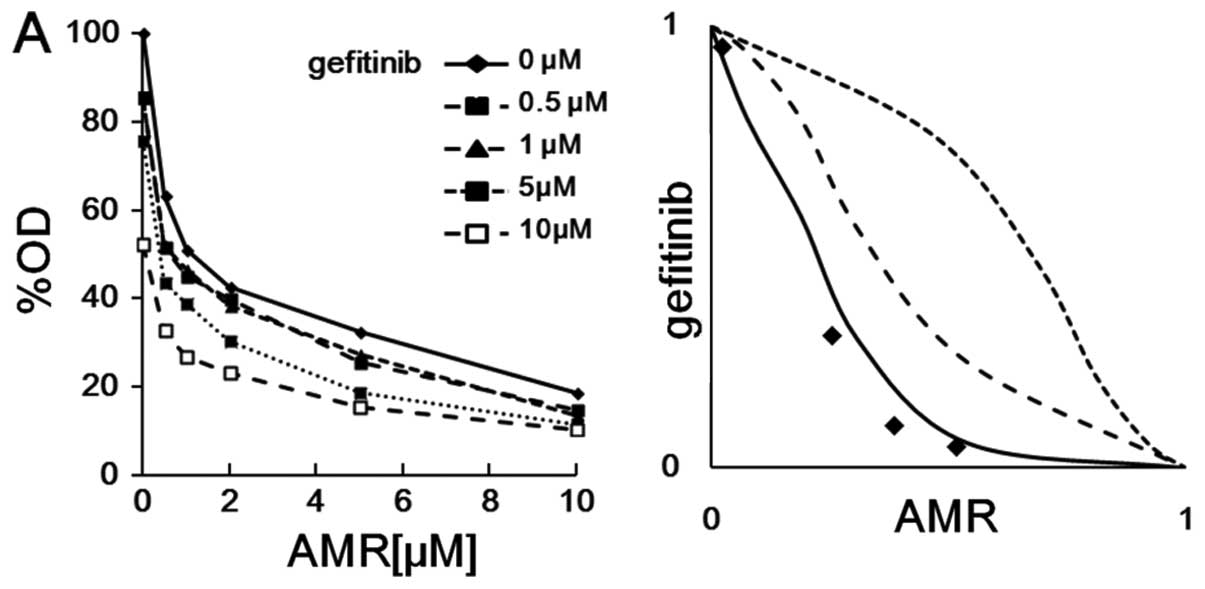
AMR in A549 cells. As shown in Fig.
4A, in the combination of gefitinib and AMR, three of four
experimental points were plotted on the left of the predictor line
of an additive effect when IC50 was taken as the
experimental end-point (indicating a supra-additive effect).
Similar results were achieved for erlotinib with AMR in A549 cells
(Fig. 4B).
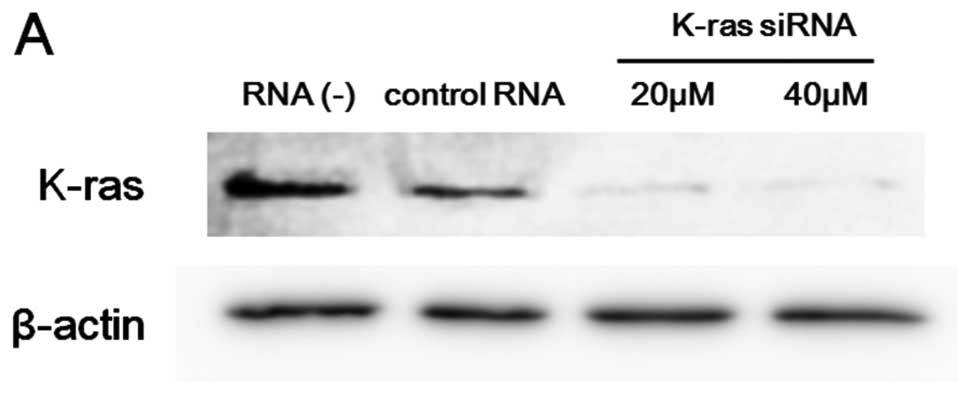
Effects of K-ras knockdown on the
synergism and the activity of EGFR and Akt in A549 cells
In a previous study, we confirmed that A549 cells
harbor oncogenic K-ras mutations (G12S) (18). To investigate whether the active
form of K-ras is responsible for the observed synergism in the
combination of EGFR-TKIs and AMR, we knocked down K-ras by siRNA.
K-ras expression in A549 cells was effectively suppressed by siRNA
as confirmed by immunoblot analysis (Fig. 5A). To compare the combination
effects of EGFR-TKIs and AMR between control and K-ras knockdown
cells, we performed fixed-ratio dilution experiment to calculate CI
(Fig. 5B). As expected, in control
cells the CI-values were constantly less than 1 for every
combination of EGFR-TKIs and AMR. In K-ras knockdown cells, the
curves connecting CI shifted upward. Furthermore, some of the CI
values for the combination of gefitinib and AMR exceeded 1, and
this tendency was remarkable in the combination of erlotinib and
AMR. These results strongly suggested that oncogenic K-ras may be
involved, at least partially, in the synergistic combination effect
of EGFR-TKIs and AMR in A549 cells.
To investigate whether the active form of K-ras
affects the EGFR-mediated signal, we evaluated EGFR and Akt
activity in K-ras knockdown A549 cells. As show in Fig. 5C, both phosphorylated EGFR and Akt
were decreased without a change in the total protein expression
level of these proteins (Fig.
5B).
Combination effects of EGFR-TKIs and AMR
in Ma10 cells with wild-type K-ras
Ma10 cells harbor wild-type EGFR and
K-ras (18). We used these
cells to assess the relationships among Akt activity, EGFR-TKI, and
the synergism in K-ras wild-type cells. In Ma10 cells, both
gefitinib and erlotinib could not suppress Akt activity even at the
high concentration of 50 μM in spite of the clear
suppression by LY294002 (Fig. 6A).
The combination of LY294002 and AMR demonstrated additive to
synergistic cell growth inhibition (Fig. 6B). The combination of EGFR-TKIs
(either gefitinib or erlotinib) and AMR did not exert a synergistic
effect (Fig. 6C), consistent with
the observed synergism by the combination of an Akt-suppressing
agent and AMR.
Discussion
The purpose of the present study was to clarify
whether Akt-suppressing agents have therapeutic potential when
combined with cytotoxic chemotherapeutic agents, including AMR, in
lung adenocarcinoma without potent molecular-targeted therapy. We
found that Akt-suppressing agents including clinically available
EGFR-TKIs synergistically inhibit cell growth in combination with
AMR. This synergism may be attributable, at least in part, to
K-ras mutation in A549 cells.
A549 cells harbor the wild-type EGFR gene
activating K-ras mutation (18). We utilized this lung adenocarcinoma
cell line as a model. In A549 cells, LY294002, an Akt inhibitor,
effectively suppressed Akt activity and synergistically inhibited
cell growth only in the combination with AMR among the
chemotherapeutic agents tested. The increase in Annexin V binding
to cells after simultaneous treatment with LY294002 and AMR
suggests that enhanced apoptotic cell death is a mechanism
underlying this synergism. Although Akt suppression has been
reported to enhance anticancer agent-induced apoptosis (22), only additive effects were observed
in the combination with CDDP or PEM. Even antagonistic effects were
experienced in the combination of LY294002 and PTX in this study.
These observations suggest that Akt inhibition may enhance the
cytotoxicity of chemotherapeutic agents in a drug-specific
manner.
In general, tyrosine kinases are major upstream
regulators of Akt activity, and it is expected that the suppression
of tyrosine kinase activity will lead to Akt inhibition (23). On the other hand, K-ras is also
proposed as a regulator of the PI3K/Akt pathway (24). Thus, we assessed whether a
non-specific tyrosine kinase inhibitor, genistein, would function
as well as LY294002. Similar to LY294002, genistein suppressed Akt
activity and synergistically inhibited cell growth, supporting that
the suppression of certain tyrosine kinases leads to Akt
suppression and enhances AMR cytotoxicity even in
K-ras-mutated A549 cells.
Recent studies have verified that lung
adenocarcinoma with activating EGFR mutations is sensitive
to EGFR-TKIs, and monotherapy with these drugs improved clinical
outcomes (25,26). Conversely, in NSCLC with wild-type
EGFR, the antitumor activity of EGFR-TKIs is limited.
Indeed, judging from the IC50 and compared with
EGFR-mutated PC9 cells, A549 cells with wild-type
EGFR were 100-fold resistant to EGFR-TKIs with
IC50s of around 10 μM. Nevertheless, Akt activity
was suppressed at concentrations ranging from 100 nM to 1 μM
of EGFR-TKIs, in contrast to PC9 cells, in which the
Akt-suppressing concentrations of EGFR-TKIs and the
IC50s were at similar levels. The discrepancy in A549
cells with respect to the Akt-suppressing concentrations and
IC50s may be attributable to the K-ras mutation,
which is assumed to function as a driver mutation.
The maximum plasma concentration (Cmax)
of 225 mg/day gefitinib (250 mg/day is administered in clinical
practice) is about 0.7 μM (27). Cmax of 150 mg/day
erlotinib is approximately 4 μM (28). Therefore, the Akt-suppressing
concentration of EGFR-TKIs observed in A549 cells is clinically
achievable.
This finding raises the possibility that Akt
activity can be suppressed even though sufficient antitumor
activity by EGFR-TKIs is absent in lung cancer with wild-type
EGFR. In A549 cells, since both gefitinib and erlotinib
suppressed Akt activity at clinically relevant concentrations, we
evaluated the combination effect of these agents with AMR and
observed synergistic cell growth inhibition. These observations
support the clinical usefulness of the combination therapy by these
drugs.
Since K-ras and EGFR mutations are
mutually exclusive as driver mutations (2), the incidence of K-ras mutation
should be elevated among the subgroup of lung adenocarcinoma
without EGFR mutation. To clarify the role of K-ras
mutation in the observed synergism, the expression of K-ras protein
was suppressed by siRNA, and the combination effects of EGFR-TKIs
and AMR were evaluated. Judging from the CI, the degree of
synergism was decreased, and even antagonism was observed with the
combination of EGFR-TKIs and AMR in K-ras knockdown A549 cells.
Furthermore, in Ma10 cells, in which both the EGFR and
K-ras genes are wild-type (18), only additive to antagonistic
effects, and not synergistic effects, were observed in the
combination EGFR-TKIs and AMR. These findings suggest that
K-ras mutation contributes at least partially to synergistic
cell growth inhibition by the combination treatment of EGFR-TKIs
and AMR.
Akt-suppressing agents consistently demonstrated
synergistic effects in combination with AMR both in A549 cells in
this report and in several SCLC in our previous studies (17). Furthermore, the suppression of Akt
is reported to enhance the cytotoxicity of another anthracycline,
doxorubicin, in other systems (29). These observations support that
anthracyclines, including AMR are suitable cytotoxic drugs for
combination with an Akt-suppressing agent. Actually, the
combination of LY294002 and AMR exerted an additive to synergistic
inhibition also in Ma10 cells.
However, neither gefitinib nor erlotinib suppressed
Akt activity, and the combination of these drugs with AMR was not
synergistic in Ma10 cells, although the expression level of EGFR is
similar to that of A549 cells (18). Recent studies support the linkage
between K-ras mutation and EGFR-mediated signals. Pancreatic
ductal adenocarcinomas driven by K-ras oncogenes are
dependent on EGFR signaling (30).
The transfection of mutated K-ras to head-and-neck cancer
cells induces autocrine production of EGFR ligands such as
amphiregulin and transforming growth factor α, and activates the
PI3K/Akt pathway (31). Activated,
but not wild-type ras, facilitates nucleolin interaction with EGFR
and stabilizes EGFR proteins levels, leading to synergistic
anchorage-independent cell growth in vitro and tumor growth
in vivo (32). In addition,
the expression of constitutively active ras induces ErbB4
phosphorylation (33), and EGFR
inhibitors can prevent the phosphorylation of ErbB4 (34). In the present study, we observed
that the suppression of K-ras protein expression led to the
inhibition of both EGFR and Akt activity in K-ras-mutated
A549 cells, and neither gefitinib nor erlotinib suppressed Akt
activity in K-ras wild-type Ma10 cells. Therefore, we
concluded that oncogenic K-ras induces Akt activation, which can be
suppressed by EGFR-TKIs in A549 cells. In addition, we propose that
the synergistic effect by the combination of EGFR-TKIs and AMR may
be specific in K-ras-mutated lung adenocarcinomas among
those with wild-type EGFR. Further investigation is needed
to clarify the precise mechanism by which active K-ras activates
EGFR-mediated signaling.
The present results may be useful for considering
treatments for NSCLC harboring the K-ras mutation. At
present, molecular-targeted therapy for K-ras is not clinically
available. In addition, it is reported that lung cancer with
K-ras mutation has a poor prognosis (35). Therefore, a novel effective therapy
is strongly desired for the treatment of NSCLC with K-ras
mutation. The results of this study suggest that EGFR-TKI may
function as an Akt inhibitor and enhance the cytotoxicity of AMR,
at least partially, in a K-ras mutation-dependent manner.
AMR has promising antitumor activity not only against SCLC
(36), but also NSCLC (16). Therefore, we propose that the
combination therapy of EGFR-TKI and AMR can be a promising
therapeutic strategy for lung cancer harboring wild-type
EGFR and activating K-ras mutation. Further study,
including a clinical trial, is necessary to establish this
combination therapy as an option for such lung cancer.
In conclusion, the combination of AMR and
Akt-suppressing agents, including EGFR-TKIs, synergistically
inhibits the growth of A549 cells. We propose that the combination
treatment with EGFR-TKI and AMR is promising in NSCLC with
wild-type EGFR and mutated K-ras genes.
References
|
1.
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
2.
|
Ding L, Getz G, Wheeler DA, Mardis ER,
McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan
MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE,
Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis
LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K,
Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR,
Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl
C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby
SD, Sawyer CS, Vickery T, Sander S, Robinson J, Winckler W, Baldwin
J, Chirieac LR, Dutt A, Fennell T, Hanna M, Johnson BE, Onofrio RC,
Thomas RK, Tonon G, Weir BA, Zhao X, Ziaugra L, Zody MC, Giordano
T, Orringer MB, Roth JA, Spitz MR, Wistuba II, Ozenberger B, Good
PJ, Chang AC, Beer DG, Watson MA, Ladanyi M, Broderick S, Yoshizawa
A, Travis WD, Pao W, Province MA, Weinstock GM, Varmus HE, Gabriel
SB, Lander ES, Gibbs RA, Meyerson M and Wilson RK: Somatic
mutations affect key pathways in lung adenocarcinoma. Nature.
455:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Pao W and Girard N: New driver mutations
in non-small-cell lung cancer. Lancet Oncol. 12:175–180. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Mok TS, Wu YL, Thongprasert S, Yang CH,
Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y,
Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL,
Watkins CL, Armour AA and Fukuoka M: Gefitinib or
carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med.
361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Mitsudomi T, Morita S, Yatabe Y, Negoro S,
Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T,
Asami K, Katakami N, Takada M, Yoshioka H, Shibata K, Kudoh S,
Shimizu E, Saito H, Toyooka S, Nakagawa K and Fukuoka M; West Japan
Oncology Group: Gefitinib versus cisplatin plus docetaxel in
patients with non-small-cell lung cancer harbouring mutations of
the epidermal growth factor receptor (WJTOG3405): An open label,
randomised phase 3 trial. Lancet Oncol. 11:121–128. 2010.
View Article : Google Scholar
|
|
6.
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, Lu S, Zhang L, Hu C, Hu C,
Luo Y, Chen L, Ye M, Huang J, Zhi X, Zhang Y, Xiu Q, Ma J, Zhang L
and You C: Erlotinib versus chemotherapy as first-line treatment
for patients with advanced egfr mutation-positive non-small-cell
lung cancer (optimal, CTONG-0802): A multi-centre, open-label,
randomised, phase 3 study. Lancet Oncol. 12:735–742. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Ou SH: Crizotinib: A novel and
first-in-class multitargeted tyrosine kinase inhibitor for the
treatment of anaplastic lymphoma kinase rearranged non-small cell
lung cancer and beyond. Drug Des Devel Ther. 5:471–485.
2011.PubMed/NCBI
|
|
8.
|
Talapatra S and Thompson CB: Growth factor
signaling in cell survival: Implications for cancer treatment. J
Pharmacol Exp Ther. 298:873–878. 2001.PubMed/NCBI
|
|
9.
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase Akt pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Choi Y, Zhang J, Murga C, Yu H, Koller E,
Monia BP, Gutkind JS and Li W: PTEN, but not SHIP and SHIP2,
suppresses the PI3K/Akt pathway and induces growth inhibition and
apoptosis of myeloma cells. Oncogene. 21:5289–5300. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Franke TF, Hornik CP, Segev L, Shostak GA
and Sugimoto C: PI3K/Akt and apoptosis: Size matters. Oncogene.
22:8983–8998. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Lee SH, Kim HS, Park WS, Kim SY, Lee KY,
Kim SH, Lee JY and Yoo NJ: Non-small cell lung cancers frequently
express phosphorylated Akt; an immunohistochemical study. APMIS.
110:587–592. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Brognard J, Clark AS, Ni Y and Dennis PA:
Akt/protein kinase B is constitutively active in non-small cell
lung cancer cells and promotes cellular survival and resistance to
chemotherapy and radiation. Cancer Res. 61:3986–3997.
2001.PubMed/NCBI
|
|
14.
|
Yamaoka T, Hanada M, Ichii S, Morisada S,
Noguchi T and Yanagi Y: Cytotoxicity of amrubicin, a novel
9-aminoanthracycline, and its active metabolite amrubicinol on
human tumor cells. Jpn J Cancer Res. 89:1067–1073. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Ding Q and Zhan J: Amrubicin: Potential in
combination with cisplatin or carboplatin to treat small-cell lung
cancer. Drug Des Devel Ther. 7:681–689. 2013.PubMed/NCBI
|
|
16.
|
Sawa T, Yana T, Takada M, Sugiura T, Kudoh
S, Kamei T, Isobe T, Yamamoto H, Yokota S, Katakami N, Tohda Y,
Kawakami A, Nakanishi Y and Ariyoshi Y: Multicenter phase II study
of amrubicin, 9-amino-anthracycline, in patients with advanced
non-small-cell lung cancer (study 1): West Japan Thoracic Oncology
Group (WJTOG) trial. Invest New Drugs. 24:151–158. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Ueda Y, Igishi T, Hashimoto K, Suyama H,
Araki K, Sumikawa T, Takeda K, Nakazaki H, Matsunami K, Kodani M,
Shigeoka Y, Matsumoto S and Shimizu E: Synergistic cell growth
inhibition by the combination of amrubicin and Akt-suppressing
tyrosine kinase inhibitors in small cell lung cancer cells:
Implication of c-Src and its inhibitor. Int J Oncol. 34:689–696.
2009.PubMed/NCBI
|
|
18.
|
Takata M, Chikumi H, Miyake N, Adachi K,
Kanamori Y, Yamasaki A, Igishi T, Burioka N, Nanba E and Shimizu E:
Lack of Akt activation in lung cancer cells with egfr mutation is a
novel marker of cetuximab sensitivity. Cancer Biol Ther.
13:369–378. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Steel GG and Peckham MJ: Exploitable
mechanisms in combined radiotherapy-chemotherapy: The concept of
additivity. Int J Radiat Oncol Biol Phys. 5:85–91. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Chou TC and Talalay P: Quantitative
analysis of dose-effect relationships: The combined effects of
multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 22:27–55.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Ono M and Kuwano M: Molecular mechanisms
of epidermal growth factor receptor (EGFR) activation and response
to gefitinib and other EGFR-targeting drugs. Clin Cancer Res.
12:7242–7251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Oh SJ, Erb HH, Hobisch A, Santer FR and
Culig Z: Sorafenib decreases proliferation and induces apoptosis of
prostate cancer cells by inhibition of the androgen receptor and
Akt signaling pathways. Endocr Relat Cancer. 19:305–319. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ma PC, Tretiakova MS, Nallasura V,
Jagadeeswaran R, Husain AN and Salgia R: Downstream signalling and
specific inhibition of c-met/HGF pathway in small cell lung cancer:
Implications for tumour invasion. Br J Cancer. 97:368–377. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Khwaja A, Rodriguez-Viciana P, Wennstrom
S, Warne PH and Downward J: Matrix adhesion and Ras transformation
both activate a phosphoinositide 3-OH kinase and protein kinase
B/Akt cellular survival pathway. EMBO J. 16:2783–2793. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J and
Haber DA: Activating mutations in the epidermal growth factor
receptor underlying responsiveness of non-small-cell lung cancer to
gefitinib. N Engl J Med. 350:2129–2139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Fukuoka M, Yano S, Giaccone G, Tamura T,
Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S,
Rischin D, Eek R, Horai T, Noda K, Takata I, Smit E, Averbuch S,
Macleod A, Feyereislova A, Dong RP and Baselga J:
Multi-institutional randomized phase II trial of gefitinib for
previously treated patients with advanced non-small-cell lung
cancer (The IDEAL 1 Trial) [corrected]. J Clin Oncol. 21:2237–2246.
2003.
|
|
27.
|
Nakagawa K, Tamura T, Negoro S, Kudoh S,
Yamamoto N, Yamamoto N, Takeda K, Swaisland H, Nakatani I, Hirose
M, Dong RP and Fukuoka M: Phase I pharmacokinetic trial of the
selective oral epidermal growth factor receptor tyrosine kinase
inhibitor gefitinib (‘Iressa’, ZD1839) in japanese patients with
solid malignant tumors. Ann Oncol. 14:922–930. 2003.
|
|
28.
|
Hidalgo M, Siu LL, Nemunaitis J, Rizzo J,
Hammond LA, Takimoto C, Eckhardt SG, Tolcher A, Britten CD, Denis
L, Ferrante K, Von Hoff DD, Silberman S and Rowinsky EK: Phase I
and pharmacologic study of OSI-774, an epidermal growth factor
receptor tyrosine kinase inhibitor, in patients with advanced solid
malignancies. J Clin Oncol. 19:3267–3279. 2001.
|
|
29.
|
Fujiwara Y, Kawada K, Takano D, Tanimura
S, Ozaki K and Kohno M: Inhibition of the PI3 kinase/Akt pathway
enhances doxorubicin-induced apoptotic cell death in tumor cells in
a p53-dependent manner. Biochem Biophys Res Commun. 340:560–566.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Navas C, Hernandez-Porras I, Schuhmacher
AJ, Sibilia M, Guerra C and Barbacid M: EGF receptor signaling is
essential for K-ras oncogene-driven pancreatic ductal
adenocarcinoma. Cancer Cell. 22:318–330. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Minjgee M, Toulany M, Kehlbach R, Giehl K
and Rodemann HP: K-RAS(V12) induces autocrine production of EGFR
ligands and mediates radioresistance through EGFR-dependent Akt
signaling and activation of DNA-PKcs. Int J Radiat Oncol Biol Phys.
81:1506–1514. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Farin K, Schokoroy S, Haklai R, Cohen-Or
I, Elad-Sfadia G, Reyes-Reyes ME, Bates PJ, Cox AD, Kloog Y and
Pinkas-Kramarski R: Oncogenic synergism between erbb1, nucleolin,
and mutant ras. Cancer Res. 71:2140–2151. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Tal-Or P, Erlich S, Porat-Shliom N,
Goldshmit Y, Ben-Baruch G, Shaharabani E, Kloog Y and
Pinkas-Kramarski R: Ligand-independent regulation of ErBb4 receptor
phosphorylation by activated ras. J Cell Biochem. 98:1482–1494.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Carrasco-Garcia E, Saceda M, Grasso S,
Rocamora-Reverte L, Conde M, Gomez-Martinez A, Garcia-Morales P,
Ferragut JA and Martinez-Lacaci I: Small tyrosine kinase inhibitors
interrupt EGFR signaling by interacting with erbB3 and erbB4 in
glioblastoma cell lines. Exp Cell Res. 317:1476–1489. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Eberhard DA, Johnson BE, Amler LC, Goddard
AD, Heldens SL, Herbst RS, Ince WL, Janne PA, Januario T, Johnson
DH, Klein P, Miller VA, Ostland MA, Ramies DA, Sebisanovic D,
Stinson JA, Zhang YR, Seshagiri S and Hillan KJ: Mutations in the
epidermal growth factor receptor and in KRAS are predictive and
prognostic indicators in patients with non-small-cell lung cancer
treated with chemotherapy alone and in combination with erlotinib.
J Clin Oncol. 23:5900–5909. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Ohe Y, Negoro S, Matsui K, Nakagawa K,
Sugiura T, Takada Y, Nishiwaki Y, Yokota S, Kawahara M, Saijo N,
Fukuoka M and Ariyoshi Y: Phase I–II study of amrubicin and
cisplatin in previously untreated patients with extensive-stage
small-cell lung cancer. Ann Oncol. 16:430–436. 2005.
|