Introduction
Prostate cancer (PCa) is one of the most common
neoplasms among aging males. As of 2011, PCa is the second most
frequently diagnosed cancer and the sixth leading cause of cancer
death in male worldwide (1). In
2010 it resulted in 256,000 deaths, up from 156,000 deaths in 1990
(2). PCa is a prevalent malignancy
in American men, with 217,730 estimated new cases that occurred in
2010 and almost 32,050 deaths (American Cancer Society, 2010).
Early stage PCa uniquely relies on androgens for proliferation, and
blockade of the androgen receptor pathway almost invariably induces
tumor regression. However, in later stages, PCa cells become
androgen-independent and no curative therapy exists for this
refractory disease (3,4).
Although improved screening methods allow a
diagnosis of prostate cancer at an early stage, it still remains
one major cause of death in men in industrialized countries. In
particular, no curative treatment is available to date upon
progression to androgen-independent and metastatic disease
(5,6). Although advances in chemotherapy have
improved patient outcome (4,5),
there remains a clear need for effective mechanism-based
therapeutic approaches that can achieve long-term improvements in
patient outcomes (7).
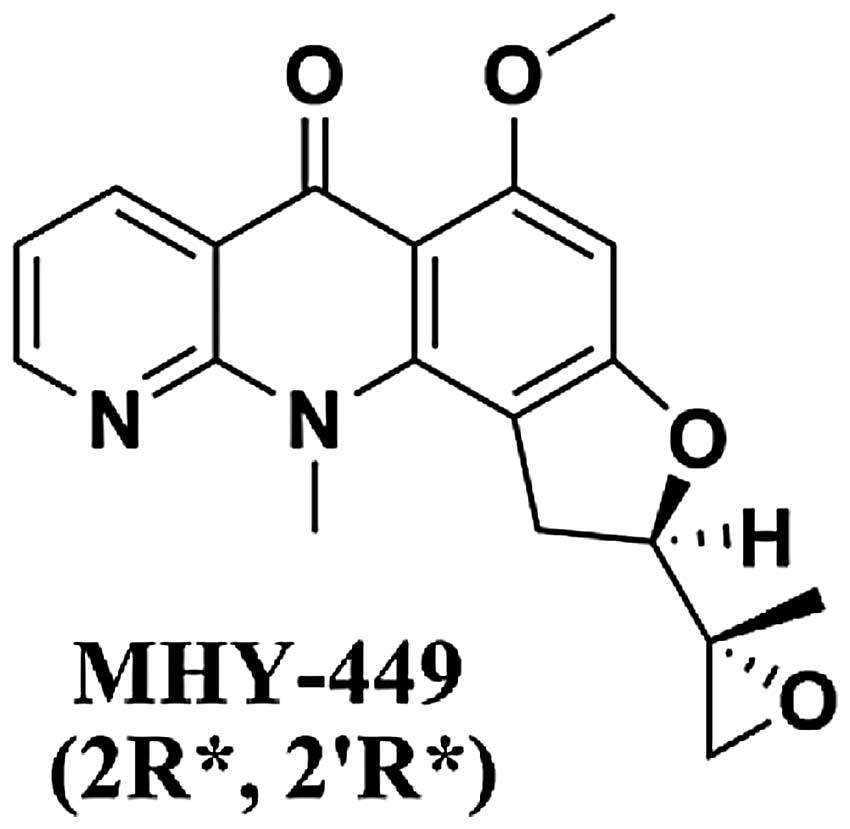
MHY-449,(±)-(R*)-5-methoxy-11-methyl-2-((R*)-2-methyloxiran-2-yl)-1,2-dihydrobenzofuro[4,5-b][1,8]
naphthyridin-6(11H)-one, was designed and synthesized on the basis
of the chemical structure of psorospermin with a xanthone template
and acronycine derivatives with an acri-done template and its
cytotoxicity was evaluated against five human cancer cell lines,
such as prostate cancer cell lines (LNCaP, DU145 and PC3) and
breast cancer cell lines (MCF-7/ADR and MCF-7) (8). MHY-449 has shown cytotoxicity against
human prostate and breast cancer cells and induction of G2/M phase
arrest of the cell cycle in MCF-7/ADR cells (8). Moreover, MHY-449 was shown to inhibit
effectively human colon cancer cell growth by inducing G2/M phase
cell cycle arrest and apoptosis in HCT116 human colon cancer cells
(9). However, the underlying
molecular mechanisms of the cytotoxic effect of MHY-449 in PCa
cells were not fully elucidated. Therefore, the current study was
designed to investigate the cytotoxic effect of MHY-449 on p53
wild-type (p53-wt) LNCaP cells and p53 null type (p53-null) PC3
cells and its molecular mechanisms of action.
Materials and methods
Chemicals
The simplified code name and structure of MHY-449
[(±)-(R*)-5-methoxy-11-methyl-2-((R*)-2-methyloxiran-2-yl)-1,2-dihydrobenzofuro[4,5-b][1,8]naphthyridin-6(11H)-one]
used in this study is shown in Fig.
1. Detailed method for the design and synthesis of this
compound is described elsewhere (9). The compound was dissolved in
dimethylsulfoxide (DMSO) and stored at −20°C before the experiments
and dilutions were made in culture medium. The maximal
concentration of DMSO did not exceed 0.1% (v/v) in the treatment
range, where there was no influence on the cell growth. Antibodies
specific for Bax, Bcl-2, caspase-3, -8, poly (ADP-ribose)
polymerases (PARP), Akt, p-Akt, Forkhead-Box Class O (FoxO) 1, and
p-FoxO1 were obtained from Santa Cruz Biotechnology (Santa Cruz,
CA, USA). Antibodies against for extracellular signal-regulated
kinases (ERK), phospho(p)-ERK, c-Jun N-terminal kinases (JNK),
p-JNK, p38 mitogen-activated protein kinases (MAPK) and p-p38 MAPK
were purchased from Cell Signaling (Beverly, MA, USA). PD98059 was
from Santa Cruz Biotechnology.
Cell culture and cell viability
assay
The human prostate cancer cell lines p53-null PC3
and p53-wt LNCaP cells were cultured in RPMI-1640 (HyClone, Logan,
UT, USA) supplemented with 10% fetal bovine serum (FBS, HyClone), 2
mM glutamine (Sigma-Aldrich Co., St. Louis, MO, USA), 100 U/ml
penicillin (HyClone), and 100 μg/ml streptomycin (HyClone)
at 37°C in a humidified 5% CO2. Cell viability was
determined by MTT assay. For the MTT assay, PC3 and LNCaP cells
were seeded in a 24-well culture plate, cultured for 24 h in the
growth media, and then treated with or without various reagents for
the indicated concentrations. The cells were incubated with 0.5
mg/ml MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide] (Sigma-Aldrich) at 37°C for 2 h. The formazan granules
generated by the live cells were dissolved in DMSO, and the
absorbance at 540 nm was monitored by using a multi-well
reader.
Nuclear staining with Hoechst 33342
Cells were washed with PBS and fixed with 3.7%
paraformaldehyde (Sigma-Aldrich) in PBS for 10 min at room
temperature. Fixed cells were washed with PBS, and stained with 4
μg/ml Hoechst 33342 (Invitrogen, Eugene, OR, USA) for 20 min
at room temperature. The cells were washed two more times with PBS
and analyzed via a fluorescent microscope.
Assessment of DNA fragmentation
Cells were lysed in a buffer, containing 5 mM
Tris-HCl (pH 7.5), 5 mM EDTA, and 0.5% Triton X-100, for 30 min on
ice. Lysates were vortexed and cleared by centrifugation at 27,000
× g for 20 min. Fragmented DNA in the supernatant was treated with
RNase, followed by proteinase K digestion,
phenol/chloroform/isoamyl alcohol mixture (25:24:1, v/v/v)
extraction and isopropanol precipitation. DNA was separated through
a 1.6% agarose gel, and stained with 0.1 μg/ml ethidium
bromide (EtBr, Fluka), and then visualized by UV source.
Flow cytometric analysis
The DNA content was measured following the staining
of the cells with propidium iodide (PI, Sigma-Aldrich). The cells
were treated under the appropriate conditions for 24 h,
subsequently trypsinized, washed once in cold PBS, and then fixed
in 70% ethanol at −20°C overnight. The fixed cells were pelleted
and stained in cold PI solution (50 μg/ml in PBS) at room
temperature for 30 min in the dark. The stained cells were analyzed
by flow cytometry (FC500, Beckman Coulter, Istanbul, Turkey).
Assay of caspase activity
The cells were harvested and washed with cold PBS.
Total cells were lysed with the lysis buffer [40 mM Tris (pH 8.0),
120 mM, NaCl, 0.5% NP-40, 0.1 mM sodium orthovanadate, 2
μg/ml aprotinin, 2 μg/ml leupeptin and 100
μg/ml phenymethylsulfonyl fluoride (PMSF)] at 4°C for 30
min. The lysed cells were centrifuged at 10,000 × g for 2 min, and
100 μg of protein was incubated with 100 μl of
reaction buffer and 10 μl of colorimetric tetrapeptides,
Z-DEVDpNA for caspase-3, Z-IETD-pNA for caspase-8, and Ac-LEHD-pNA
for caspase-9, respectively. The reaction mixture was incubated at
37°C for 30 min and liberated p-nitroaniline (pNA) was measured at
405 nm using a multi-well reader.
Annexin V staining
Annexin V-FITC is used to quantitatively determine
the percentage of cells within a population that are actively
undergoing apoptosis. The cells were treated under the appropriate
conditions for 24 h, subsequently harvested, trypsinized, washed
once in cold PBS, suspended the cells in 1X binding buffer
(Becton-Dickinson, Annexin V-FITC Apoptosis Detection Kit). The
counted cells were stained in PI and Annexin V-FITC solution
(Becton-Dickinson, Annexin V-FITC Apoptosis Detection Kit) at room
temperature for 15 min in the dark. The stained cells were analyzed
by flow cytometry within 1 h.
Gel electrophoresis and western blot
analysis
The cells were treated under the appropriate
conditions, harvested and washed with cold PBS. Total cells were
lysed in lysis buffer [40 mM Tris (pH 8.0), 120 mM NaCl, 0.5%
NP-40, 0.1 mM sodium orthovanadate, 2 μg/ml aprotinin, 2
μg/ml leupeptin and 100 μg/ml phenymethylsulfonyl
fluoride (PMSF)]. Protein extracts were denatured by boiling at
100°C for 5 min in sample buffer (0.5 M Tris-HCl, pH 6.8, 4% SDS,
20% glycerol, 0.1% bromophenol blue, 10% β-mercaptoethanol). Equal
amount of the total proteins were subjected to 6–15% SDS-PAGE and
transferred to PVDF membrane. The membranes were blocked with 5%
non-fat dry milk in Tris-buffered saline with Tween-20 buffer
(TBS-T; 20 mM Tris, 100 mM NaCl, pH 7.5 and 0.1% Tween-20) for 1 h
at room temperature. Then, the membranes were incubated overnight
at 4°C with the primary antibodies. The membranes were washed with
TBS-T buffer and incubated for 1 h with horseradish
peroxidase-conjugated anti-rabbit or anti-mouse immunoglobin (Santa
Cruz Biotechnology). The membranes were washed again with TBS-T
buffer. Antigen-antibody complexes were detected by the enhanced
chemiluminescence (ECL) detection system (Amersham Biosciences Co.,
Little Chalfont, UK).
Statistical analysis
Results were expressed as the mean ± SD of three
separate experiments and analyzed by Student’s t-test. Means were
considered significantly different at *p<0.05 or
**p<0.01.
Results
MHY-449 has stronger anti-proliferation
effect on PC3 cells than LNCaP cells
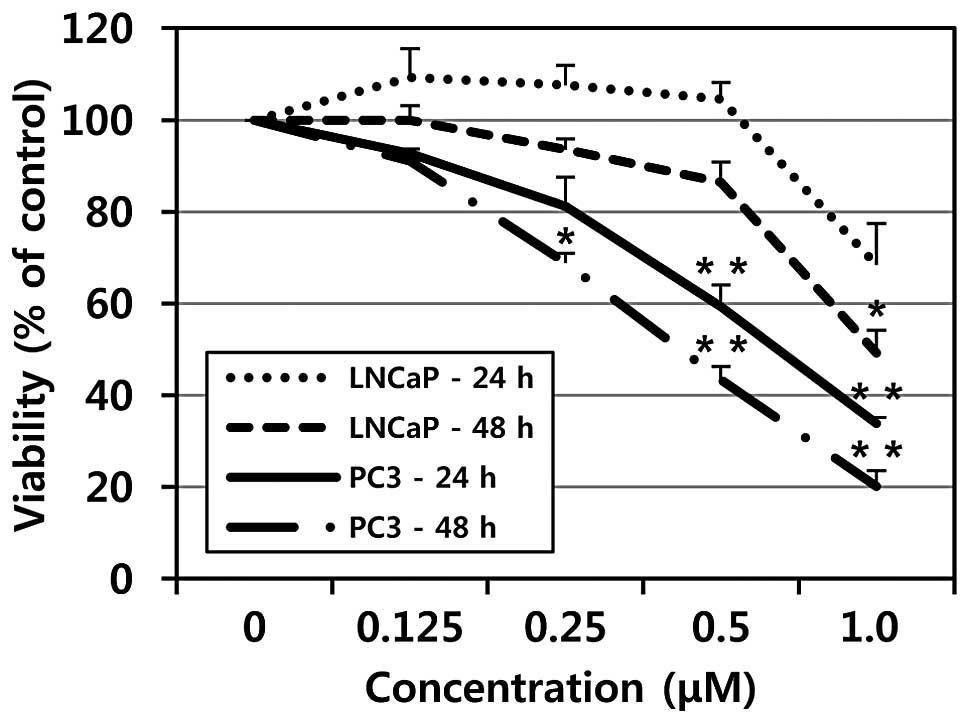
To evaluate the effects of MHY-449 on cell growth of
a human prostate cancer cell line, the MTT assay was performed. As
shown in Fig. 2, treatment with
MHY-449 showed concentration- and time-dependent cytotoxicity on
p53-null PC3 and p53-wt LNCaP cells. However, p53-null PC3 cells
were more sensitive to MHY-449 than p53-wt LNCaP cells. The
IC50 value of MHY-449 was approximately 0.75 and 0.5
μM at 24 and 48 h in PC3 cells, respectively, whereas the
IC50 value of MHY-449 was approximately 1 μM at
48 h in p53-wt LNCaP cells.
MHY-449 induces apoptosis in PC3
cells
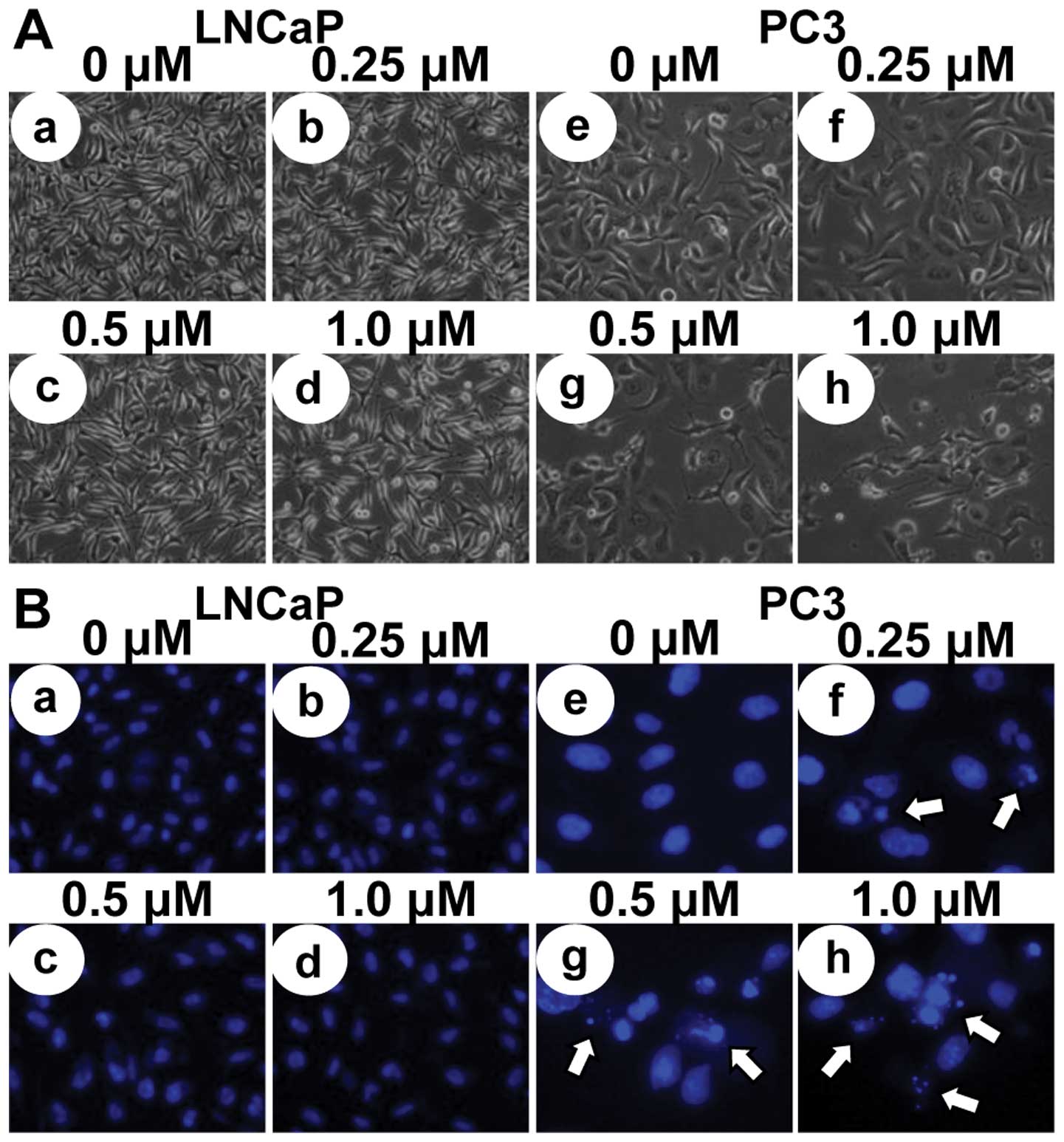
To investigate whether the growth inhibitory effects
of MHY-449 were due to the induction of apoptosis in LNCaP and PC3
cells, micro-photographs were observed. LNCaP and PC3 cells treated
with MHY-449 showed apparently distinct morphological changes
compared with control (Fig. 3A).
Although p53-wt LNCaP cells did not show significant decrease of
cell numbers and morphological changes with MHY-449 treatment
(Fig. 3Aa-d), p53-null PC3 cells
showed prominent decrease of cell numbers and morphological changes
concentration-dependently (Fig.
3Ae-h), and they were rounded and more dispersed with
aggregation. Further morphological changes of cellular structures
were assessed with Hoechst 33342 staining. As shown in Fig. 3Be-h, nuclei with chromatin
condensation and formation of apoptotic bodies, which are
characteristics of apoptosis, were seen in p53-null PC3 cells
cultured with MHY-449 concentration-dependently, whereas the
control cells maintained the nuclear structure intact. However,
p53-wt LNCaP cells did not show any morphological changes of the
nuclei after MHY-449 treatment. We also analyzed whether DNA
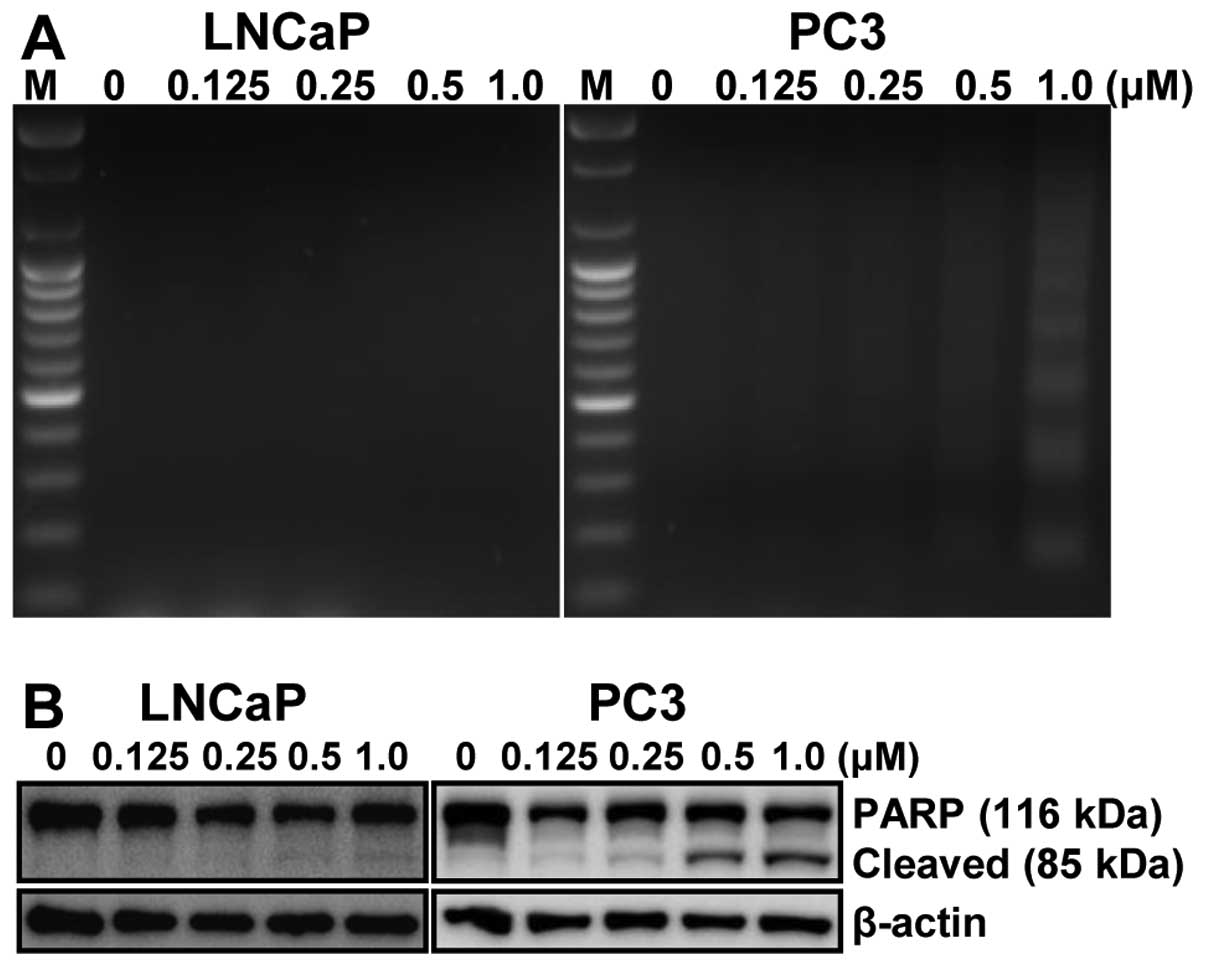
fragmentation, another hallmark of apoptosis, was induced by
MHY-449 treatment on LNCaP and PC3 cells. Following agarose gel
electrophoresis of LNCaP and PC3 cells treated with MHY-449 for 24
h, a typical ladder pattern of internucleosomal fragmentation was
observed in a concentration-dependent manner in p53-null PC3 cells,
but not in p53-wt LNCaP cells (Fig.
4A). Polypeptide degradation, including poly(ADP-ribose)
polymerase (PARP), was examined to see the possible involvement of
apoptosis-associated protease during the growth inhibition of PCa
cells. PARP cleavage was evident by the appearance of the p85 PARP
cleavage fragment and clearly observed in the 0.25, 0.5 and 1.0
μM of MHY-449 treatment in p53-null PC3 cells (Fig. 4B, right panel). However, p53-wt
LNCaP cells did not show any PARP cleavage after MHY-449 treatment
(Fig. 4B, left panel). These
results suggest that MHY-449 induced apoptotic cell death in
p53-null PC3, not in p53-wt LNCaP cells. Therefore, p53-null PC3
cells were used for further studies.
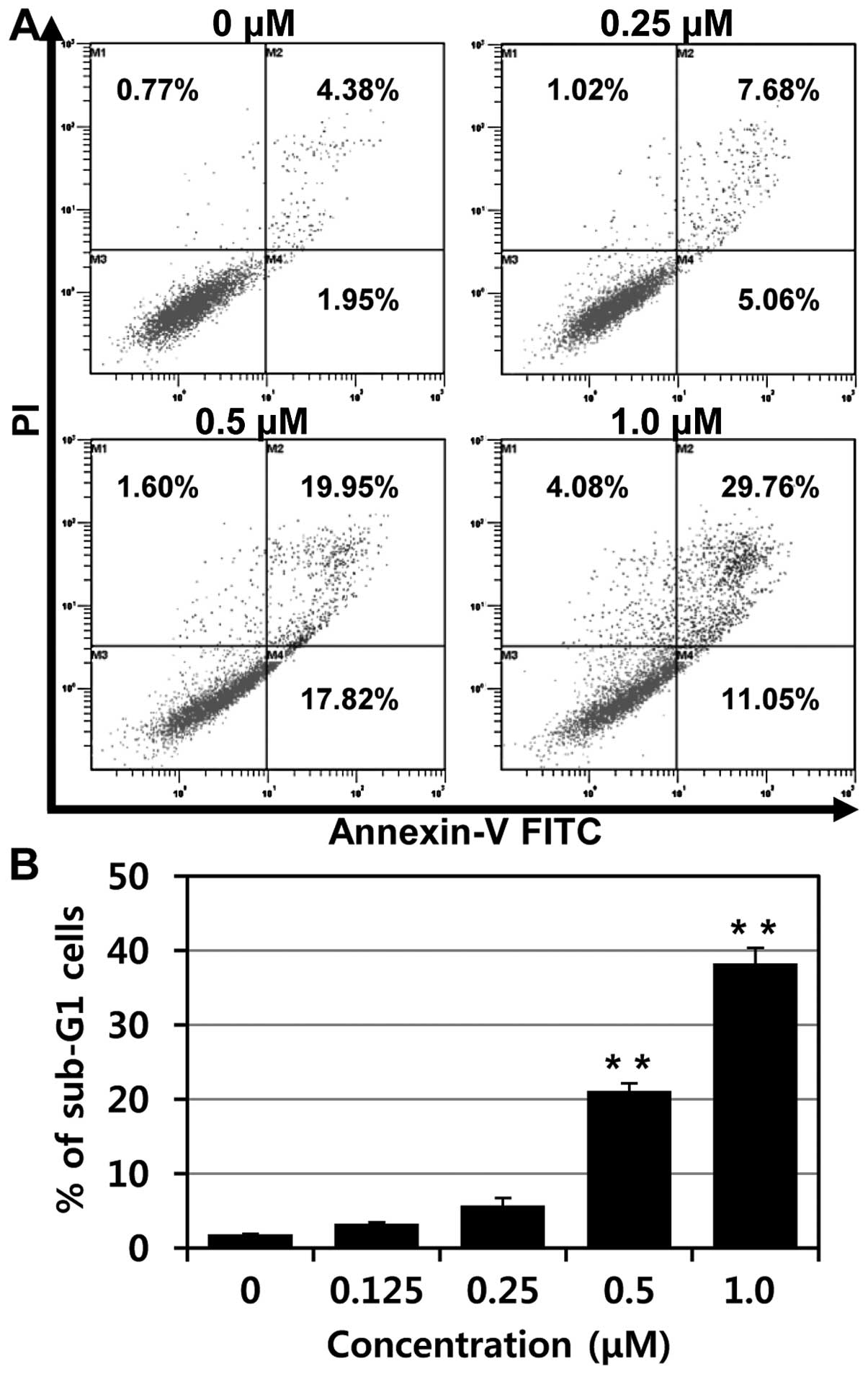
To confirm apoptosis of MHY-449 in p53-null PC3
cells, flow cytometry analysis was performed. As shown in Fig. 5A, increases of early apoptosis
(lower right quadrant) and late apoptosis/death (upper right
quadrant) were clearly observed concentration-dependently.
Moreover, the percentages of apoptotic cells, sub-G1 fraction, were
significantly increased in p53-null PC3 cells treated with MHY-449
in a concentration-dependent manner (Fig. 5B).
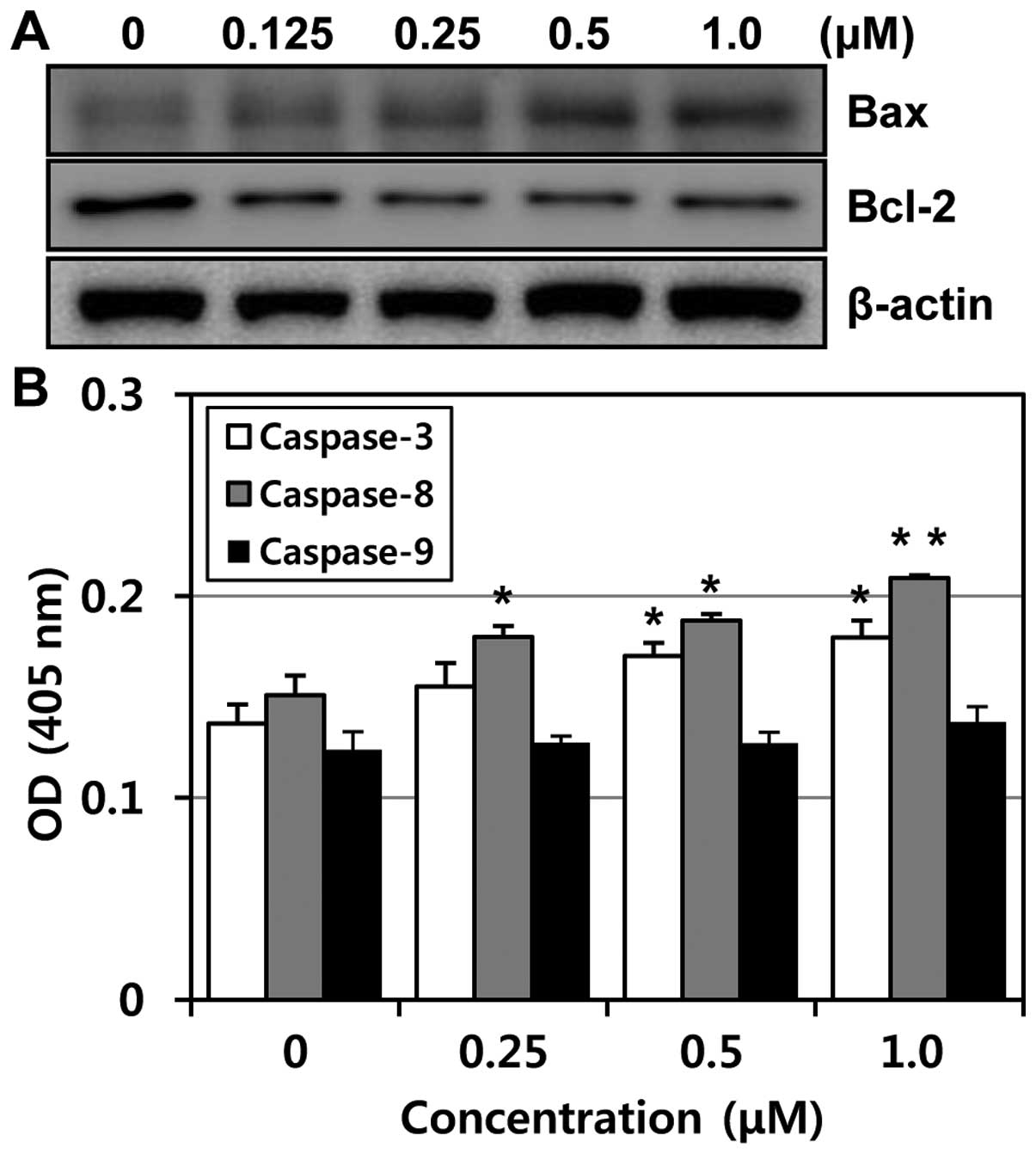
MHY-449 upregulates the expression levels
of apoptosis-related proteins and increases caspase activities in
PC3 cells
To determine whether the expression levels of
apoptosis-related proteins were modulated by MHY-449 in PC3 cells,
western blot analysis was performed. The expression level of Bcl-2
protein was markedly downregulated, while Bax was upregulated in a
concentration-dependent manner (Fig.
6A). The ratio between Bcl-2 and Bax has been suggested as a
primary event in determining the susceptibility to apoptosis
(10). These data suggest that
MHY-449 induces apoptosis by the alteration in expression ratio of
Bax/Bcl-2 protein. In an attempt to further characterize the
mechanisms of apoptosis induced by MHY-449, the activities of
caspase -3, -8 and -9 were determined by colorimetric assay. The
activities of caspase-3 and -8 were increased with the treatment of
MHY-449 concentration-dependently and there was little change in
caspase-9 activity (Fig. 6B).
Therefore, these results suggested that MHY-449 induce
caspase-dependent apoptosis in p53-null PC3 cells.
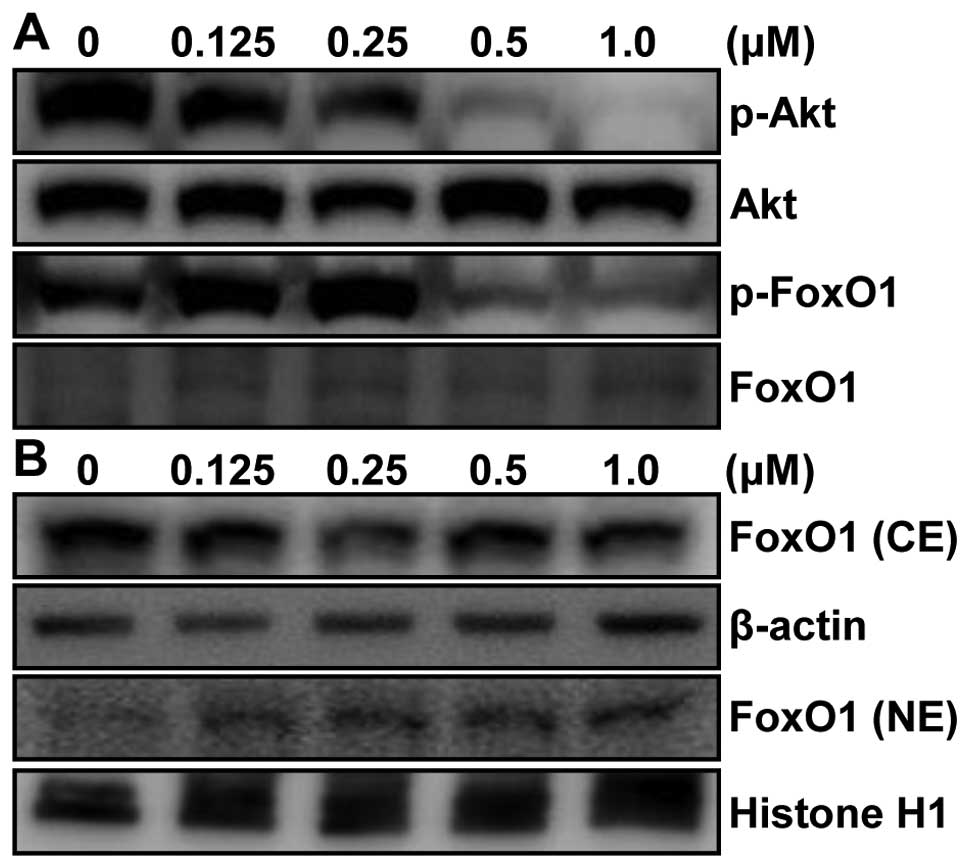
MHY-449-induced apoptosis is associated
with Akt/FoxO pathway
To clarify the molecular mechanism of apoptotic cell
death in p53-null PC3 cells, we hypothesized that MHY-449-induced
apoptosis in p53-null PC3 cells was due to inhibition of Akt/FoxO
pathway. To test our hypothesis, the level of Akt was first
examined in whole lysate by western blot analysis. p53-null PC3
cells are also PTEN-negative via homozygous deletion (11). Therefore, deletion of PTEN in PC3
cells results in increased levels of phosphatidyl-inositol 3,4,5
triphosphate which increases activated Akt (12). As shown in Fig. 7A, phosphorylation level of Akt
(p-Akt) was highly increased in vehicle-treated PC3 cells. However,
p-Akt, not Akt, was drastically suppressed by MHY-449 treatment in
a concentration-dependent manner. Next, we investigated the
downstream molecules of Akt pathway such as FoxO1, which is known
to be critical in carcino-genesis. Our results showed that
phosphorylated level, but not protein level, of FoxO1 was also
decreased in PC3 cells treated with MHY-449. This result clearly
indicates that the reduced phosphorylated level of FoxO1 was not
due to the alteration of protein expression of FoxO1 by MHY-449
treatment (Fig. 7A).
Phosphorylation of FoxO1 by Akt leads to interaction with chaperone
protein, such as 14-3-3, which serve as escort proteins for FoxO1
to move out of the nucleus (13)
leading to transcriptional downregulation of several target
proteins, such as Bim, p21WAF1/CIP1, and p27KIP1 (14). Thus, we assessed whether MHY-449
affects nuclear localization of FoxO1 by immunoblot analysis of
nuclear and cytosolic fractions with FoxO1 antibody. Our results
showed that FoxO1 protein level steadily increased in nuclear
fraction and decreased in cytosolic fraction of MHY-449-treated
p53-null and PTEN-negative PC3 cells (Fig. 7B).
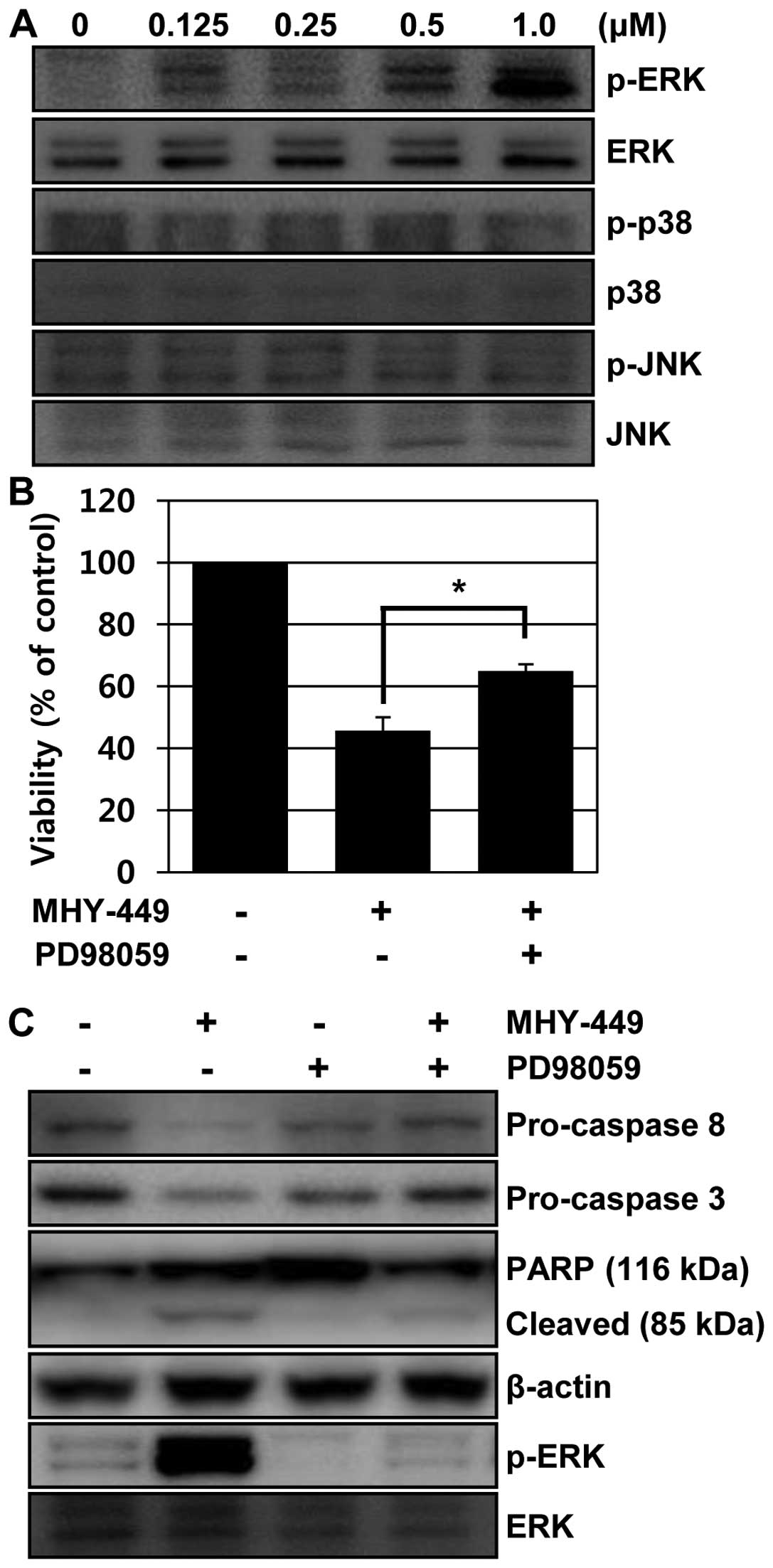
MHY-449 induces ERK phosphorylation
To explore other intracellular signaling pathways
that might be involved in MHY-449-induced apoptosis, we monitored
activation of mitogen-activated protein kinases (MAPK), such as
ERK, p38 MAPK and JNK by western blot analysis with
phospho-specific antibodies. MHY-449 treatment of PC3 cells
resulted in activation of ERK, but not in p38 MAPK and JNK, in a
concentration-dependent manner (Fig.
8A). To determine whether MHY-449-induced ERK activation was
required for cell toxicity, PC3 cells pretreated with PD98059, an
ERK inhibitor, was subsequently exposed to MHY-449 for 24 h, and
then cell death was measured by MTT assay. Pretreatment with the
ERK inhibitor protected against MHY-449-induced cytotoxicity
(Fig. 8B). Similarly, pretreatment
of ERK inhibitor abolished MHY-449-induced downregulation of
pro-caspase-8 and -3 and PARP cleavage (Fig. 8C). These results suggest that cell
death by MHY-449 treatment requires at least activation of the ERK
pathway.
Discussion
The striking new findings reported here show that
two PCa cell lines with different growth dependencies and p53
and/or PTEN status reacted totally differently to the toxic effects
of MHY-449. As compared to androgen-independent, p53-null and
PTEN-negative PC3 cells, androgen-dependent, p53-wt and mutated
PTEN (frame-shift mutation) LNCaP cells (15) were less sensitive to MHY-449 and
did not show any evidence of apoptotic cell death. However, MHY-449
killed PC3 cells primarily by an apoptotic mechanism through
inhibition of Akt/FoxO1 pathway and activation of ERK pathway.
FoxO factors are dysregulated in several tumor types
including breast cancer (16),
prostate cancer (17),
glioblastoma (18),
rhabdomyosarcoma (19) and
leukemia (20). There are four
human FoxO genes, FoxO1, 3, 4 and 6 (21). Phosphorylation of FoxOs by Akt
inhibits transcriptional functions of FoxOs and contributes to cell
survival, growth and proliferation (22,23).
The FoxO signaling is regulated by their interactions with other
intracellular proteins as well as their post-translational
modifications such as phosphorylation (24,25).
FoxOs can induce apoptosis not only by stimulating
expression of death receptor ligands, such as Fas ligand and tumor
necrosis factor-related apoptosis-inducing ligand (TRAIL) but also
by inducing expression of multiple pro-apoptotic members of the
Bcl-2 family of mitochondria-targeting proteins. FoxOs can also
promote cell cycle arrest via upregulation of the cell cycle
inhibitor p27KIP1 which can induce G1 arrest or GADD45 which can
cause G2 arrest (13,26). In PCa, FoxO1 and the androgen
receptor form a complex, which blocks the interaction between FoxO1
and its DNA response element and consequently interfering with its
ability to induce apoptosis and cell cycle arrest of the PCa cells
(27). Overexpression of FoxO1 and
FoxO3 in PCa cells induces apoptosis and upregulation of TRAIL
expression (17). Our results
showed that the protein level of FoxO1 steadily increased in
nuclear fraction and decreased in cytosolic fraction of
MHY-449-treated PC3 cells (Fig.
7B) and consequently induced apoptotic cell death.
The MAPK pathway exists in all eukaryotes, and
controls such fundamental cellular processes as proliferation,
differentiation, survival and apoptosis. Mammalian MAPK can be
divided into three groups based on their structure and function:
ERKs, p38 MAPK and JNKs. Recent research suggests that
phosphorylation of MAPKs plays an important role in the processes
of apoptosis (28). MAPKs are
activated by many stimuli and one of their major functions is to
connect cell surface receptors to transcription factors in the
nucleus, which consequently triggers long-term cellular responses
(29). Depending upon the stimulus
and cell type, ERK signaling pathway can transmit signals, which
result in the prevention or induction of apoptosis or cell cycle
progression (30). MHY-449 was
shown to activate the ERK, but not p38 MAPK and JNK, and the ERK
specific inhibitor PD98059 inhibited MHY-449-induced cell death and
apoptosis. Therefore, it appears that MHY-449-induced apoptosis via
ERK/caspase-3-dependent signaling pathway in PC3 cells.
In summary, MHY-449 suppressed growth and
proliferation of androgen-independent, p53-null and PTEN-negative
PC3 cells in a concentration-dependent manner by causing induction
of apoptosis. Taken together, these results suggest that the novel
compound MHY-449 might be used to develop adjunct treatments for a
certain type of prostate cancer.
Acknowledgements
This study was supported by the
National Research Foundation of Korea (NRF) grant funded by the
Korea government (MSIP) (no. 2009-0083538). We thank Aging Tissue
Bank for providing research information.
References
|
1.
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2.
|
Lozano R, Naghavi M, Foreman K, et al:
Global and regional mortality from 235 causes of death for 20 age
groups in 1990 and 2010: a systematic analysis for the Global
Burden of Disease Study 2010. Lancet. 380:2095–2128. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Gelmann EP: Molecular biology of the
androgen receptor. J Clin Oncol. 20:3001–3015. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Berry WR: The evolving role of
chemotherapy in androgen-independent (hormone-refractory) prostate
cancer. Urology. 65:2–7. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Pienta KJ and Smith DC: Advances in
prostate cancer chemotherapy: a new era begins. CA Cancer J Clin.
55:300–325. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Petrylak DP: New paradigms for advanced
prostate cancer. Rev Urol. 9(Suppl 2): S3–S12. 2007.
|
|
7.
|
LoPiccolo J, Blumenthal GM, Bernstein WB
and Dennis PA: Targeting the PI3K/Akt/mTOR pathway: effective
combinations and clinical considerations. Drug Resist Updat.
11:32–50. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Kang JA, Yang Z, Lee JY, et al: Design,
synthesis and anticancer activity of novel
dihydrobenzofuro[4,5-b][1,8]naphthyridin-6-one derivatives. Bioorg
Med Chem Lett. 21:5730–5734. 2011.PubMed/NCBI
|
|
9.
|
Hwang HJ, Kang YJ, Hossain MA, et al:
Novel dihydrobenzofuro[4,5-b][1,8]naphthyridin-6-one derivative,
MHY-449, induces apoptosis and cell cycle arrest in HCT116 human
colon cancer cells. Int J Oncol. 41:2057–2064. 2012.
|
|
10.
|
Harris MH and Thompson CB: The role of the
Bcl-2 family in the regulation of outer mitochondrial membrane
permeability. Cell Death Differ. 7:1182–1191. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Vlietstra RJ, van Alewijk DC, Hermans KG,
van Steenbrugge GJ and Trapman J: Frequent inactivation of PTEN in
prostate cancer cell lines and xenografts. Cancer Res.
58:2720–2723. 1998.PubMed/NCBI
|
|
12.
|
McCubrey JA, Steelman LS, Chappell WH, et
al: Roles of the Raf/MEK/ERK pathway in cell growth, malignant
transformation and drug resistance. Biochim Biophys Acta.
1773:1263–1284. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Greer EL and Brunet A: FOXO transcription
factors at the interface between longevity and tumor suppression.
Oncogene. 24:7410–7425. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Essafi A, Fernandez de Mattos S, Hassen
YA, et al: Direct transcriptional regulation of Bim by FoxO3a
mediates STI571-induced apoptosis in Bcr-Abl-expressing cells.
Oncogene. 24:2317–2329. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Li J, Yen C, Liaw D, et al: PTEN, a
putative protein tyrosine phosphatase gene mutated in human brain,
breast, and prostate cancer. Science. 275:1943–1947. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Hu MC, Lee DF, Xia W, et al: IkappaB
kinase promotes tumorigenesis through inhibition of forkhead
FOXO3a. Cell. 117:225–237. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Modur V, Nagarajan R, Evers BM and
Milbrandt J: FOXO proteins regulate tumor necrosis factor-related
apoptosis inducing ligand expression. Implications for PTEN
mutation in prostate cancer. J Biol Chem. 277:47928–47937. 2002.
View Article : Google Scholar
|
|
18.
|
Seoane J, Le HV, Shen L, Anderson SA and
Massague J: Integration of Smad and forkhead pathways in the
control of neuroepithelial and glioblastoma cell proliferation.
Cell. 117:211–223. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Galili N, Davis RJ, Fredericks WJ, et al:
Fusion of a fork head domain gene to PAX3 in the solid tumour
alveolar rhabdomyosarcoma. Nat Genet. 5:230–235. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Parry P, Wei Y and Evans G: Cloning and
characterization of the t(X;11) breakpoint from a leukemic cell
line identify a new member of the forkhead gene family. Genes
Chromosomes Cancer. 11:79–84. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Tuteja G and Kaestner KH: Forkhead
transcription factors II. Cell. 131:1922007. View Article : Google Scholar
|
|
22.
|
Lam EW, Francis RE and Petkovic M: FOXO
transcription factors: key regulators of cell fate. Biochem Soc
Trans. 34:722–726. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Burgering BM and Kops GJ: Cell cycle and
death control: long live Forkheads. Trends Biochem Sci. 27:352–360.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Zhang X, Tang N, Hadden TJ and Rishi AK:
Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta.
1813:1978–1986. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Accili D and Arden KC: FoxOs at the
crossroads of cellular metabolism, differentiation, and
transformation. Cell. 117:421–426. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Datta SR, Dudek H, Tao X, et al: Akt
phosphorylation of BAD couples survival signals to the
cell-intrinsic death machinery. Cell. 91:231–241. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Li P, Lee H, Guo S, Unterman TG, Jenster G
and Bai W: AKT-independent protection of prostate cancer cells from
apoptosis mediated through complex formation between the androgen
receptor and FKHR. Mol Cell Biol. 23:104–118. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Leger DY, Liagre B and Beneytout JL: Role
of MAPKs and NF-kappaB in diosgenin-induced megakaryocytic
differentiation and subsequent apoptosis in HEL cells. Int J Oncol.
28:201–207. 2006.PubMed/NCBI
|
|
29.
|
Bost F, Aouadi M, Caron L and Binetruy B:
The role of MAPKs in adipocyte differentiation and obesity.
Biochimie. 87:51–56. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Chang F, Steelman LS, Lee JT, et al:
Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from
cytokine receptors to transcription factors: potential targeting
for therapeutic intervention. Leukemia. 17:1263–1293. 2003.
View Article : Google Scholar
|