Introduction
Glioblastoma multiforme (GBM) is one of the most
frequent and devastating primary brain tumors in adults with a high
rate of death and disability (1,2). The
WHO grade III anaplastic astrocytomas and grade IV GBM make up
approximately three-quarters of all gliomas. GBMs make up 17% of
all primary brain tumors in the United States. Despite both the
knowledge of brain tumor biology and current resources for GBM
treatment have greatly developed over the past decade, a large
fraction of brain cancer patients suffer tumor recurrence shortly
after GBM tumor resection. The current 5- and 10-year survival
rates for GBM patients are 4.5 and 2.7%, respectively (3). Therefore, new and more effective
therapies are necessary to improve the prognosis of this
disease.
Brain tumor recurrence due to the resistance to
chemo- and radiotherapy of the cancer cells. Cells that escape
chemo-therapy- and radiotherapy-induced cell death eventually
re-enter the cell cycle and contribute to local tumor recurrence
(4,5). The identification and significance of
cancer stem cell-like glioma cells (CSGCs) reveal the mysterious
mask of drug resistance and tumor recurrence. Most of the CSGCs are
in the resting state, namely the G0 phase of the cell cycle, and
express ATP-binding cassette transporters that detoxify cells by
eliminating exogenous compounds (6–10).
Therefore, understanding the mechanisms associated with ‘escape’
from clinical therapy of tumor stem cells, in conjunction with the
use of new treatments based on the clearance and intervention of
tumor stem cells, are required to advance the treatment of
glioma.
The use of mesenchymal stem cells (MSCs) as
therapeutic vehicles for brain tumors has garnered much attention
over the past decade. This is attributable to the fundamental
ability of MSCs to migrate, or home, to brain tumors irrespective
of the blood-brain barrier and to be manipulated into expressing
various therapeutic molecules, such as interleukins, interferons,
prodrugs, oncolytic viruses, antiangiogenic agents, pro-apoptotic
proteins and growth factor antagonists (11–14).
A number of studies have shown that MSCs express both chemokine
receptors and adhesion molecules enabling their homing function to
target sites in vivo in response to specific chemokine
gradients. Many cytokine/receptor pairs, such as SDF-1/CXCR4,
SCF/c-Kit, HGF/c-Met, VEGF/VEGFR, PDGF/PDGFr, MCP-1/CCR2, and
HMGB1/RAGE (15–18), play roles in inducing stem cells
recruitment to tumors.
Although emerging studies suggest that MSCs may
serve as therapeutic vehicles for the treatment of various tumors,
the tropism character of mesenchymal stem cells for cancer stem
cells has rarely been described. In this study, we tested the
tropism of bone marrow-derived (BM-) and adipose tissue-derived
(AT-) mesenchymal stem cells (MSCs) for cancer stem cell-like
glioma cells (CSGCs). In order to obtain optimal results, we
obtained BM- and AT-MSCs, fibroblast cells, and CSGCs from
homologous tumor-bearing mice. To our surprise, there were almost
no migration of MSCs, including BM- and AT-MSCs, towards CSGCs that
were in the resting state, compared with fibroblast cells. However,
the number of migrated MSCs was noticeably increased with the
proliferation of CSGCs from the resting state into the cell cycle.
In order to further explore the potential of MSCs migration for
CSGCs, we also induced CSGCs differentiation into the terminal
cells. The results displayed that the MSC migration was decreased
significantly when the CSGCs were induced into terminal
differentiation cells. The results were confirmed by further
experiments from analysis of three important cytokine/receptor
pairs, SCF/c-Kit, SDF-1/CXCR4 and VEGF/VEGFR, which play key roles
in mediating the recruitment from MSCs to tumors. Unexpectedly,
fibroblast, as a negative control, also showed a slight curve
change, with the proliferation of CSGCs. This finding seems to be
contradictory with many previous reports, which claimed fibroblasts
have hardly any tropism for tumor cells (19). We speculate that fibroblasts may be
involved in the stem cell migration as a helper cell component.
In the present study, we report for the first time
that BM- and AT-MSCs show little migration towards CSGCs that were
in the resting and differentiated status. Only when induced into
proliferation process, CSGCs secrete chemokines and induce
migration of MSCs. Our findings expose a fatal defect of MSCs-based
tumor therapy, because there always exist a part of cancer stem
cells that are in resting status, and bring new challenge for MSCs
as vehicles of therapeutic agents in the treatment of the CSGCs,
and even other tumor stem cells.
Materials and methods
Animals and the brain tumor model
All animal studies were done in accordance with
institutional guidelines under the approved protocols. For the
intracranial xenografts of glioma, male Sprague-Dawley rats (2-week
old; Animal Center of the Chinese Military Medical Sciences) were
anesthetized with ketamine/xylazine i.p. and stereotactically
inoculated with 4×105 C6 cells (Tianjin Institute of
Neurology) suspension (in 5 μl) into the right frontal lobe
(1.5 mm lateral and 1 mm anterior to bregma, at 2.5 mm depth from
the skull base) using a stereotactic apparatus with a microinfusion
pump. The needle was maintained in place for ≥3 min following
injection and the incision was then sutured. MRI was conducted at
the 7th day after cell transplantation at MRI Testing Center of The
Fifth Central Hospital of Tianjin.
Homologous cell culture
Cancer stem cell-like glioma
cells
Glioblastoma tissue was obtained from the confirmed
tumor-bearing rats. Glioblastoma blocks were rinsed twice with
D-Hanks solution, and then sheared into paste after vessels and
necrotic tissue were eliminated. After it was digested with 0.25%
trypsin (Invitrogen, Carlsbad, CA, USA), and cells were pipetted
into a single cell suspension. Digestion was terminated using 10%
fetal bovine serum (Hangzhou, Sijiqing, China). Tissues were
filtered through a 30-μm mesh and centrifuged at 1,000 rpm
for 5 min. After removal of the supernatant, cells were collected
and counted. Cells (1×106) were suspended with PBE
incubation solution (including 0.5% bovine serum albumin, 0.08%
EDTA in PBS, pH 7.2) to a final concentration of 1×108
cells in 0.5 ml, then incubated with anti-CD133 antibody (antibody
final concentration 20 μg/ml) at 4°C for 30 min and then
with antibody-coated superfine magnetic beads (Miltenyi Biotec) at
10°C for 15 min and suspended with 20 times volume of PBE solution.
The separation column was installed into a magnetic field and
pretreated with 0.5 ml PBE, which was naturally eluted due to
gravity. The incubated cell suspension was added to the separation
column and naturally eluted, then 0.5 ml PBE was added to the
separation column and naturally eluted, the column was rinsed twice
and separated from the magnetic field. The column was then inserted
into a new tube, and 1–2 ml PBE was administered along the needle
core, to remove the CD133 positive cells. The CSGCs spheres were
cultured in Neurobasal medium (Invitrogen) containing 1×B27
(Invitrogen), 2 mM L-glutamine, 30 U/ml penicillin-streptomycin, 1
U/ml heparin (Sigma), 20 ng/ml basic fibroblast growth factor
(bFGF, Miltenyi Biotec) and 20 ng/ml epidermal growth factor (EGF,
Provitro).
BM-MSCs
BM-MSCs were derived from long bone of hind legs in
the same tumor-bearing rats as described previously (20). Briefly, the long bone of hind legs
were dissected and bone marrow plugs were extracted from the bones
by flushing the bone marrow cavity with complete culture medium.
The cells were centrifuged (1,400 rpm, 8 min), resuspended in
complete culture medium, plated (50×106 cells per
75-cm2 culture flask), and incubated at 37°C humidified
atmosphere with 5% CO2. Non-adherent cells were removed
after 24 h and the culture medium was replaced every 3 days.
Adherent cells reached confluence of 90–95% within 10–15 days and
were passaged with 0.25% trypsin (Invitrogen) in the ratio 1:3.
Cells at passages 3 were identified by flow cytometric analysis to
detect the expression of surface antigen CD29, CD31, CD34, CD45,
CD71 and CD105 (Chemicon).
AT-MSCs
AT-MSC were isolated from subcutaneous fat tissue in
the same tumor-bearing mice as described previously (21). Adipose tissue was cut into fine
pieces, and digested with 1 mg/ml collagenase IA (Sigma) for 1 h at
37°C with shaking at a speed of 200 rpm. The released cells were
centrifuged for 15 min at 400 × g to removed them from the tissues.
The cell pellet suspended with PBS was filtered through a
70-μm mesh to remove debris and centrifuged for 5 min at
1,500 rpm each time to wash the cells. The residual cells were
cultured and identified similarly to the BM-MSCs. The CD29, CD34,
CD45, CD71, CD90 and CD105 (Chemicon) were selected for the flow
cytometric analysis.
Fibroblast cells
The tails (4–5 cm) were harvested from the same
tumor-bearing rats and rinsed with PBS three times. The tails were
cut into 1 mm pieces, then were rinsed and cultured in DMEM
supplemented with L-glutamine, 4,500 mg/l D-glucose, 10% FBS, and
1% antibiotic-antimycotic solution. All cells were incubated at
37°C in an incubator in a 5% CO2/95% air atmosphere.
After 5 days, tail pieces were discarded and the fibroblasts were
continued in culture until they reached 90% confluency. Then
8×105 fibroblasts were taken for retroviral
infection.
Preparation and characterization of
the substrate
In order to finite the cells in the resting,
proliferation, and differentiated status, the spheroids of CSGCs
were cultured on the surface of the substrate with different
stiffness, combined with or withdrew bFGF and EGF in medium. The
sheets of polyacrylamide gel were prepared as described by Dembo
and Wang (22). With varied
stiffness, the gel here contained a mixture of acrylamide (10%,
Sigma) and different percents of bis-acrylamide (0.03% for soft and
0.26% for stiff, respectively). The thickness of the polyacrylamide
thin film was 60 μm, and the stiffness was tested as
described by Qin et al (23) and Huang et al (24). For cell culture, the gel was
incubated with type I collagen solution (0.2 mg/ml, c8919,
Sigma-Aldrich) at 4°C overnight sterilized with UV irradiation.
CSGC proliferation and
differentiation
The CSGC spheres with 100–200 cells each sphere were
divided into three groups. The first part of the CSGC spheres were
cultured on surface of the soft gel culture dishes with the
Neurobasal medium containing bFGF and EGF. To limit the CSGCs in
the resting status, the second part of the CSGC spheres were seeded
on surface of the soft gel culture dishes with the same Neurobasal
medium but lacking bFGF and EGF. To induce CSGC differentiation,
the third part of the CSGC spheres were seeded on surface of the
stiff gel culture dishes with the same Neurobasal medium but
lacking bFGF and EGF. The size and morphology of CSGC spheres were
observed. Twenty-four hours after culture, the resulting
conditioned media of the three parts were collected, respectively,
as chemoattractants for the testing of enzyme linked immunosorbent
assay (ELISA) and transwell migration assay. The proliferation
activity of cells was detected by immunocytochemistry for Ki-67,
and the cell differentiation was detected by telomeric repeat
amplification protocol assay and immunocytochemistry for the
expression of CD133, nestin, GFAP, β-tubulin III, and myelin.
Telomeric repeat amplification
protocol assay
Telomeric repeat amplification protocol assay was
carried out using Telomerase PCR kit (Roche) following the
manufacturer’s recommendations and previously described (25).
Immunocytochemistry
The proliferation activity of cells in each group
was detected by binding the monoclonal antibody Ki-67 to a nuclear
antigen present in all cells that are in the G1, S, G2, and M phase
of the cell cycle, but not in the G0 phase. The cell
differentiation was detected by immunocytochemistry for markers of
undifferentiated CSGCs (CD133 and nestin), and differentiated CSGCs
(GFAP for astrocyte, β-tubulin III for neuron, and myelin for
oligodendrocyte). Immunocytochemistry was performed as described
previously (26). 96-wells were
fixed with 2% paraformaldehyde for 15 min at room temperature,
treated with 5% normal goat serum (Vector Laboratories), and then
stained with the following antibodies: anti-Ki-67 (mouse monoclonal
IgG1; 1:2,000; Santa Cruz), anti-CD133 (mouse monoclonal IgG1;
1:1,000; Miltenyi Biotec), anti-nestin (mouse monoclonal IgG1;
1:2,000; Chemicon), anti-β-tubulin III (mouse monoclonal IgG1;
1:2,000; Chemicon), anti-GFAP (rabbit polyclonal; 1:1,000;
Chemicon), and anti-myelin (mouse monoclonal; 1:1,000; Chemicon).
The primary antibodies were detected with Tex-Red or
FITC-conjugated donkey anti-mouse IgG and FITC-conjugated donkey
anti-rabbit IgG (1:1,000; Jackson ImmunoResearch). Cells were
counterstained with DAPI (Vector Laboratories). Images from 10
random fields per well were captured. The percentage was calculated
based on the total number of nuclei counted.
ELISA for secreted cytokines by
CSGCs
SCF, SDF-1 and VEGF proteins secreted into the
culture supernatants were analyzed by ELISA accordance with the
instructions. Briefly, 100 μl of each standard and sample
was added into appropriate wells and incubate overnight at 4°C with
gentle shaking. After washing, 100 μl of biotinylated
antibody was added to each well for 1 h at room temperature. Then
100 μl of prepared streptavidin solution was added to each
well, and incubated for 45 min at room temperature. At last, 100
μl of TMB One-Step Substrate Reagent (Item H) was added to
each well for 30 min at room temperature in the dark with gentle
shaking. After stoping the reaction, the result was read at 450 nm
immediately.
Western blotting for expressed
receptors by BM- and AT-MSCs
BM-MSCs or AT-MSCs or fibroblast cells were
cultured, respectively, in the conditioned media of the three
groups containing chemoattractants for 24 h. The expression level
of receptor, c-Kit, CXCR4 and VEGFR, was detected by western
blotting as previously described (27). Proteins were incubated with primary
antibodies against c-Kit (1:2,000, Santa Cruz), CXCR4 (1:1,000,
Santa Cruz), and VEGFR (1:1,000, Zhongshan Bio Corp.) followed by
incubation with an HRP-conjugated secondary antibody (Zymed). The
specific protein was detected using a SuperSignal protein detection
kit (Pierce). The membrane was stripped and re-probed with a
primary antibody against β-actin (1:1,000, Santa Cruz).
In vitro migration studies
The migratory ability of BM- and AT-MSCs was
determined using Transwell plates (Corning Costar) that were 6.5 mm
in diameter with 8 μm pore filters. BM- or AT-MSCs
(1×104) were suspended in serum-free medium containing
0.1% bovine serum albumin (Sigma) and seeded into the upper well,
the fibroblast cells as negative control; 600 μl of
conditioned medium from the above was placed in the lower well of
the Transwell plate, the Neurobasal medium containing or lacking
bFGF and EGF as control. Following incubation for 24 h at 37°C,
cells that had not migrated from the upper side of the filters were
removed with a cotton swab, and filters were stained with the
Three-Step Stain Set (Diff-Quik; Sysmex). The number of cells that
had migrated to the lower side of the filter was counted under a
light microscope with five high-power fields (×400). Experiments
were done in triplicate.
Statistical analysis
All data are expressed as mean ± SE. Statistical
differences between different test conditions were determined by
Student’s t-test. P<0.05 was considered statistically
significant.
Results
Brain tumor model and MRI
The tested rats were listless and had reduced
activity at 7 days following cell transplantation. MRI images
showed intracranial space-occupying lesions (data not shown). The
results from HE staining showed obvious atypical cells in the brain
tissue (data not shown).
Characterization and identification of
homologous cell lines
Following seeding in mesenchymal culture medium,
typical fibroblastoid colonies could be detected after a few days
rapidly forming a monolayer of adherent cells from both BM- and
AT-MSCs. MSCs cultures contained a homogeneous population of
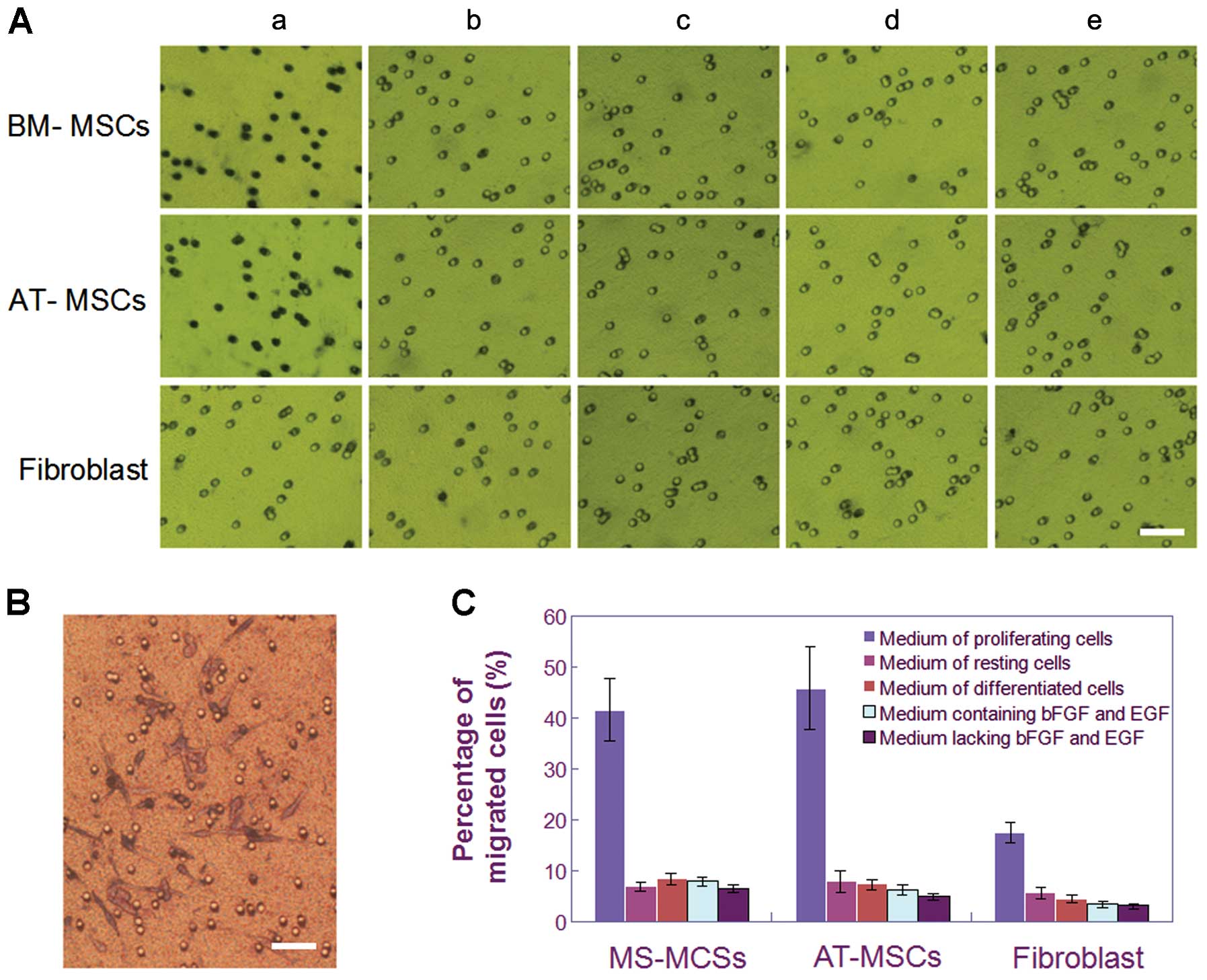
spindle-shaped cells (Fig. 1C and
D). Flow cytometric analysis revealed that BM-MSCs were
positive for the typical mesenchymal markers CD29, CD71 and CD105
but negative for the expression of CD31, CD34 and CD45 (Fig. 1F). While AT-MSCs were positive for
the mesenchymal markers CD29, CD71, and CD105, but negative for the
expression of CD34, CD45, and CD90 (Fig. 1G). Compared with BM- and AT-MSCs,
the fibroblast cells displayed a relatively slow growth rate and a
flat cell shape (Fig. 1E).
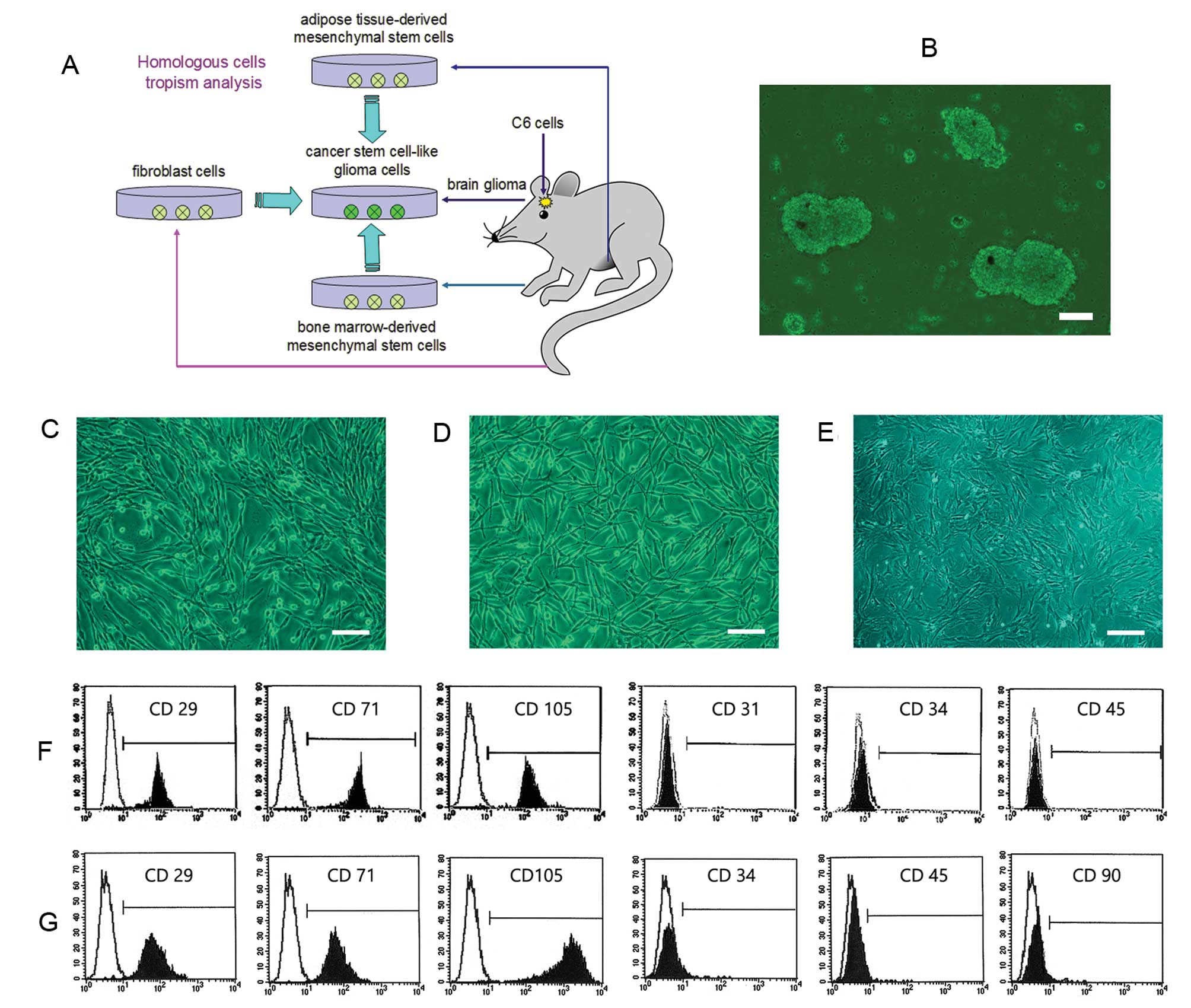 | Figure 1.Characterization of homologous cell
lines from the same rat. (A) A schematic drawing shows that all
four cell lines, the glioma cells, BM-MSCs, AT-MSCs, and
fibroblasts, were obtained from different regions of the same rat.
(B) CSGC spheres were obtained from a rat confirmed to bear a
tumor. (C) BM-MSCs proliferated rapidly to form a homogeneous
population of spindle-shaped cells. (D) The growth characteristics
of AT-MSCs were similar to those of BM-MSCs. (E) Compared to BM-
and AT-MSCs, fibroblasts exhibited a relatively slow growth rate
and flat cell shape. (F) Flow cytometric analysis revealed that
BM-MSCs were positive for the typical mesenchymal markers CD29,
CD71, and CD105 but negative for CD31, CD34 and CD45. (G) AT-MSCs
were positive for the markers CD29, CD71, and CD105, but negative
for CD34, CD45, and CD90 (bars, 15 μm). |
CSGCs in the proliferation status have
strong tropism on MSCs
Because stiffness of cell adhesion substrates
modulated stem cell proliferation and differentiation, we cultured
the spheroids of CSGCs on the surface of the substrate with
different stiffness, combined or not with bFGF and EGF in medium.
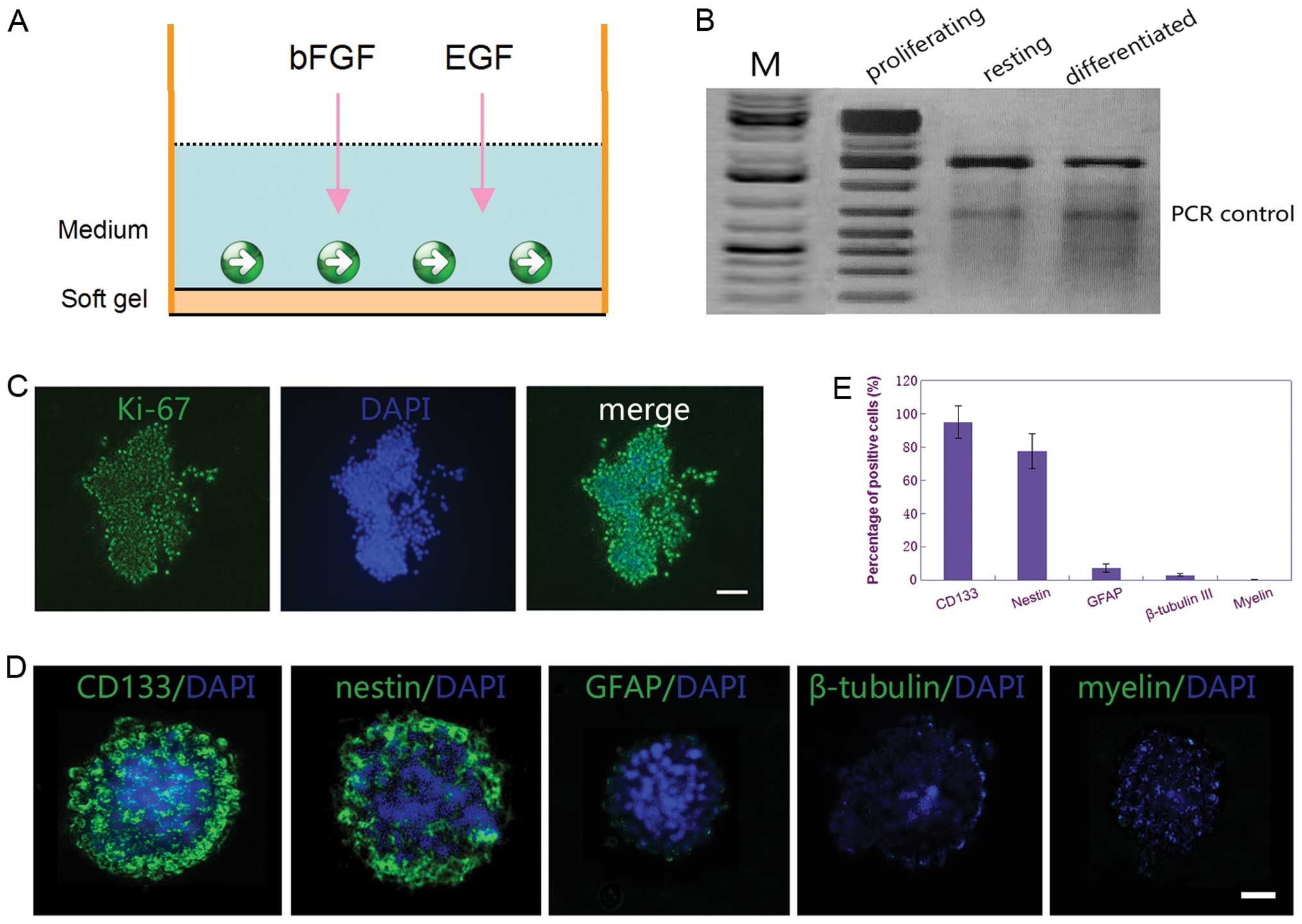
The result of immunocytochemistry for Ki-67 showed that the CSGCs
displayed highly proliferative activity when the CSGCs spheres were
cultured on surface of the soft gel culture dishes with the
Neurobasal medium containing bFGF and EGF (Fig. 2C). Further evidence for the CSGCs
that were in the proliferation status was their expression of
telomerase, as demonstrated in various other stem cell populations
(28) (Fig. 2B). Moreover, immunocytochemical
staining demonstrated that ≥90% of cells were positive for CD133
(marker for glioma stem cells) and ∼70% were positive for nestin
(marker for glioma stem cells or neural precursor cells), whereas
<12% of cells were positive for GFAP (marker for glial
differentiation), and <5% were positive for β-tubulin III
(marker for neuronal differentiation) and <1% for myelin (marker
for oligodendrocyte differentiation) (Fig. 2D and E). These results displayed
that the CSGCs were in highly proliferative status when cultured on
surface of the soft gel with the Neurobasal medium containing bFGF
and EGF.
After the cells were incubated for 24 h, the
resulting conditioned media were collected and the chemoattractants
were tested by ELISA. The results showed that the proliferating
CSGCs could secreted much higher level of chemotactic factors into
the culture supernatants, as tested SCF, SDF-1 and VEGF, compared
with that in the resting or differentiated status, or the
fibroblast cells (Fig. 3A–C). As
chemoattractants, they could induce expression of respective
receptor, c-Kit, CXCR4 and VEGFR, in both BM- and AT-MSCs (Fig. 3D–I). These findings were consistent
with the results from the in vitro migration assay by
transwell system, that displayed the maximum number of cell
migration of BM- and AT-MSCs towards the supernatants (Fig. 4), compared with the homologous
fibroblast cells. Taking into account the potential impact of bFGF
and EGF, we set up control of the Neurobasal medium containing or
lacking bFGF and EGF in the lower well of the Transwell plate. The
results showed bFGF and EGF could slightly induce MSC migration,
but insufficient to affect the experimental conclusion that the
proliferating CSGCs have stronger tropism on BM- and AT-MSCs than
in the resting and differentiated status (Fig. 4).
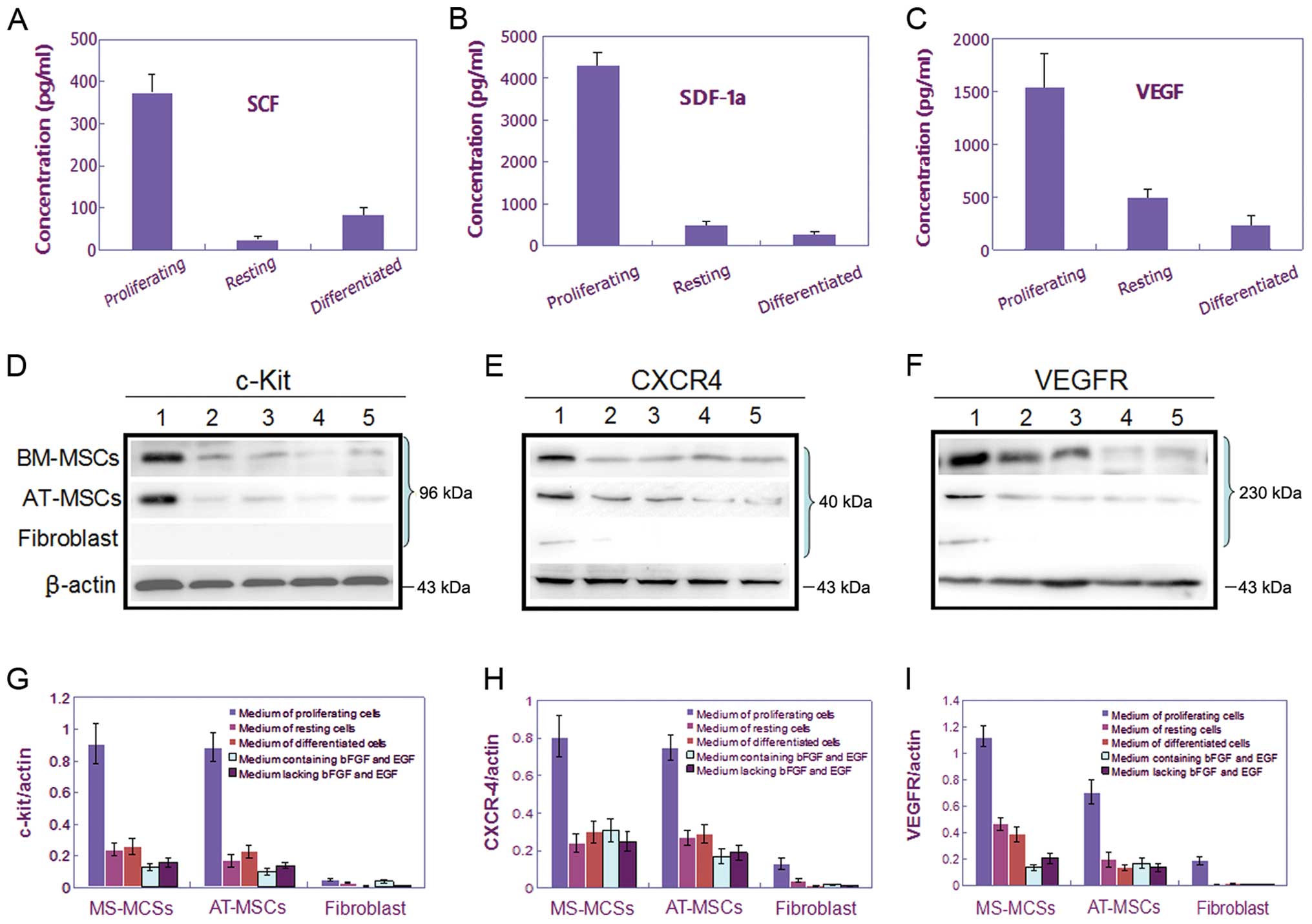 | Figure 3.Assay of the expression levels of
three cytokine/receptor pairs, SCF/c-Kit, SDF-1/CXCR4, and
VEGF/VEGFR, in media conditioned by CSGCs, MSCs, and fibroblast
cells. (A–C) The levels of three cytokines, SCF, SDF-1, and VEGF,
were measured by ELISA in CSGC medium when CSGCs were cultured
under different conditions. Only proliferating CSGCs expressed SCF,
SDF-1 and VEGF at high levels. (D–F) Western blotting was used to
measure the levels of three cytokine receptors, c-Kit, CXCR4, and
VEGFR, in MSCs and fibroblasts. (G–I) Bar charts showing the data
of (D–F). BM- and AT-MSCs expressed c-Kit, CXCR4, and VEGFR at high
levels only when cultured in medium conditioned by proliferating
CSGCs, thus not in medium conditioned by resting or differentiated
CSGCs. Fibroblasts displayed only slight tropism toward
proliferating CSGCs. |
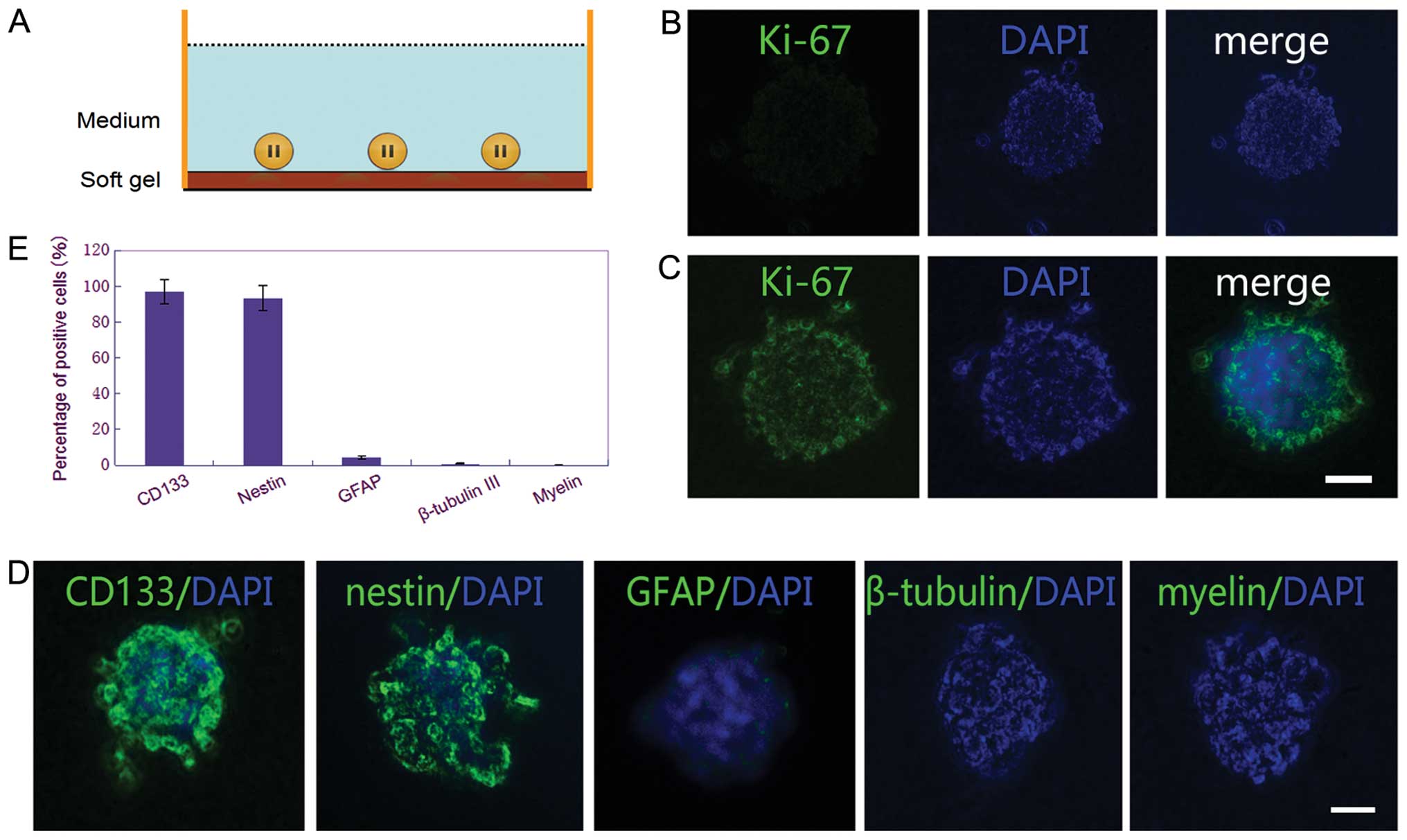
CSGCs in the resting status have
little tropism on MSCs
When cultured on surface of the soft gel culture
dishes with the same Neurobasal medium but lacking bFGF and EGF,
the CSGCs spheres showed growth arrest. Almost no cells were
positive for Ki-67 staining (Fig.
5B). The results of immunocytochemical staining demonstrated
that ∼97% of cells were positive for CD133 and ∼90% were positive
for nestin, whereas <6% of cells were positive for GFAP, and
<2% were positive for β-tubulin III and myelin (Fig. 5D and E). These results confirmed
most of the CSGCs were in resting status. When re-adding bFGF and
EGF in medium, these CSGCs restored the rapid proliferation rate,
with the performance of expanding CSGC spheres and increasing
positive staining for Ki-67 (Fig.
5C).
The results of migration assay showed there was
scarcely any cell migration of BM- and AT-MSCs towards the culture
supernatants (Fig. 4), compared
with the homologous fibroblast cells. These results were consistent
with the findings of ELISA assay, that showed the expression of
SCF, SDF-1 and VEGF at very low level (Fig. 3A–C), and the results of western
blotting that displayed little protein express of c-Kit, CXCR4 and
VEGFR in both BM-MSCs and AT-MSCs (Fig. 3D–I).
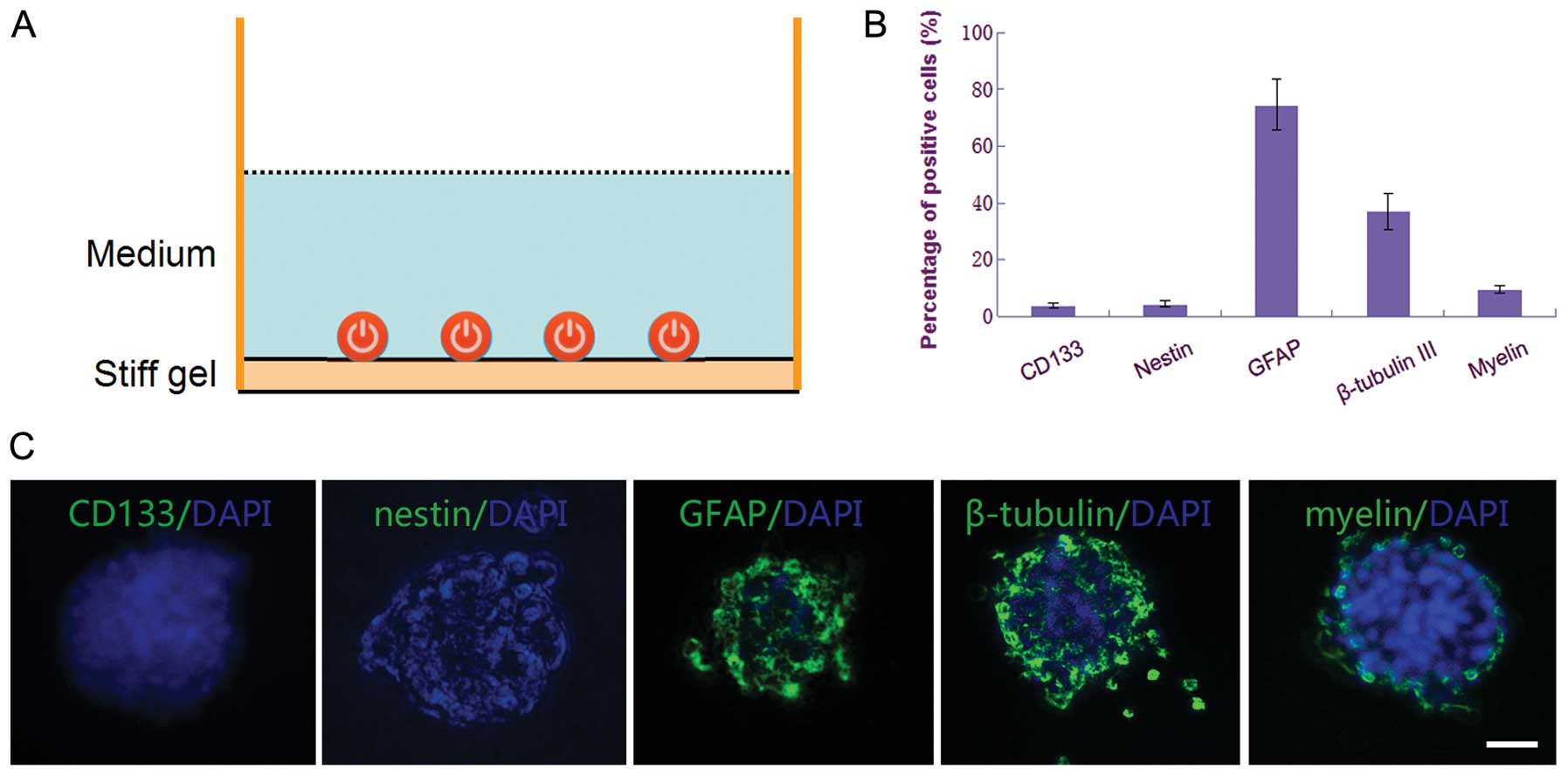
Differentiated CSGCs have little
tropism on MSCs
To induce differentiation of CSGCs into terminal
cells, the CSGC spheres were cultured on surface of the stiff gel
culture dishes with the same Neurobasal medium but lacking bFGF and
EGF. The result of telomeric repeat amplification protocol assay
showed obvious regression of telomerase activity (Fig. 2B), as well as the cell
differentiation of CSGCs into terminal cells tested by
immunocytochemistry. In contrast to the CSGCs that were in the
proliferation status, the differentiated cells of these spheroids
were negative for CD133 and nestin, with high level expression of
GFAP, β-tubulin III, and myelin (Fig.
6B and C).
Like the cells in the resting status, the
differentiated CSGCs have little tropism on either BM-MSCs or
AT-MSCs, with fewer migrating cells (Fig. 4), lower level expression of the
cytokine/receptor pairs, SCF/c-Kit, SDF-1/CXCR4 and VEGF/VEGFR,
compared with the CSGCs in the proliferation status (Fig. 3).
CSGCs in the proliferation status show
slight tropism on fibroblast cells
In this study, fibroblast, as a negative control,
also showed slight tropism towards the proliferating CSGCs, but not
the resting and the differentiated CSGCs (Fig. 4), consistent with the
characteristic of weak expression of the receptors of CXCR4 and
VEGFR (Fig. 3D–I). This finding
seems to be contradictory to previous reports, which claimed
fibroblasts have hardly any tropism for tumor cells. We speculate
that fibroblasts may be involved in the cancer stem cell metastasis
as a helper cell component.
Discussion
MSCs can differentiate into a variety of adult
mesenchymal tissues including bone, cartilage, adipose, muscle,
ligament, and tendon, and seem to be a promising source for tissue
repair and gene therapy (29). In
the past decade, emerging studies show MSCs can be envisioned as
vehicles for both short-term and long-term gene therapy, as MSCs
are easily transduced by a variety of vectors with various
therapeutic drugs (30). In brain
tumor gene therapy, MSCs demonstrate acceptable therapeutic effect.
This is attributable to the fundamental ability of MSCs to migrate,
or home, to brain tumors irrespective of the blood-brain barrier
(BBB) and to be manipulated into expressing various therapeutic
molecules (31). However, many
challenges exist. The key issues include: i) Brain tumors exhibit
phenotypic heterogeneity, being composed of cells: a) resting stem
cells and b) proliferating cells with limited self-renewal, and c)
terminal cells: either resting differentiated cells or apoptotic
cells. It is unclear whether all the tumor cells may induce MSCs
migrating. ii) Most chemotherapeutic drugs are toxic preferentially
to proliferating cells. However, most tumor stem cells are resting,
that offers the possibility that cancer cells escape
chemotherapyand radiotherapy-induced cell death, and contribute to
local tumor recurrence (32).
In this study we tested the tropism character of two
MSC cell lines, the BM- and AT-MSCs, toward the resting,
proliferating or differentiated CSGCs for the first time. In order
to define the three states of cells, we cultured the spheroids of
CSGCs on the surface of the substrate with different stiffness,
combined with or without bFGF and EGF in the medium. Study of Park
et al confirmed that stiffness of cell adhesion substrates
modulated stem cells proliferation and differentiation. Stem cells
on soft substrates had less spreading, fewer stress fibers and
lower proliferation rate than those on stiff substrates (33). Others reported glioblastoma
multiform-derived cell lines expanded slowly in the presence of the
only FGF2, switched to a faster growth mode when exposed to EGF
alone, and expanded even faster when exposed to both mitogens
simultaneously (34). Thus, we
tested the proliferation and differentiation status of CSGCs by
combining application of the substrate with different stiffness
with the culture medium containing or not bFGF and EGF. The
experimental results bear out our supposition. When cultured on
surface of the soft gel culture dishes with the Neurobasal medium
containing bFGF and EGF, the spheroids of CSGCs displayed highly
proliferative activity, with strong Ki-67 staining, high expression
of telomerase, and extremely low differentiation into terminal
nerve cell lines, such as neurons, astrocytes, and
oligodendrocytes. Whereas, when cultured on surface of the soft gel
culture dishes with the medium lacking bFGF and EGF, the spheroids
of CSGCs were still in resting status, with changes continuing till
the presence of FGF2 and EGF again. Lastly, when cultured on
surface of the stiff gel culture dishes with the medium lacking
bFGF and EGF, the spheroids of CSGCs differentiated into terminal
cell lines.
Furthermore, we tested the tropism of MSCs toward
the resting, proliferating or differentiated CSGCs by transwell
migration system. Taking into account that heterologous cells may
affect results of cell tropism, we obtained BM- and AT-MSCs,
fibroblast cells, and CSGCs from homologous tumor-bearing mice,
because homologous cells show the lowest immunogenicity. The
results of in vitro migration test showed that the
proliferating CSGCs displayed the strongest tropism on both BM- and
AT-MSCs, whereas, there were few MSC migrating towards CSGCs that
were in the resting and differentiated status.
It is known that the migration of MSCs in
vitro was stimulated by conditioned medium from cultured glioma
cells in a dose-dependent manner. This indicates that soluble
factors released from glioma cells could induce the migration of
MSCs. However, these brain tumor stem cells inducing migratory
properties require further elucidation. In this study, we detected,
for the first time, the level of three key cytokines, namely SCF,
SDF-1, and VEGF, secreted by the proliferating, resting, and
differentiated CSGCs. Concurrent with the results of in
vitro migration, the highest level expression of the three
cytokines was found in the proliferating CSGCs compared to that in
resting, and differentiated CSGCs. These cytokines further induced
expression of their respective receptors, c-Kit, CXCR4 and VEGF, on
both BM-MSCs and AT-MSCs. These findings showed that BM-and AT-MSCs
have little migration towards CSGCs that were in the resting and
differentiated status. Only when induced into proliferation
process, CSGCs secrete chemokines and induce migration of MSCs.
Moreover, we found, in the present study, that the
proliferating CSGCs showed slight tropism also on fibroblasts. This
finding seems to be contradictory with previous reports, which
claimed fibroblasts have hardly any tropism for tumor cells. We
speculate that fibroblasts may be involved in the cancer stem cells
metastasis as a helper cell component.
The essential significance of this study lies in
that it gives new insights into glioma treatment using MSCs as
vectors with various therapeutic drugs. The cancer stem cells, a
minority population within the tumor, play a key role in tumor
recurrence. A satisfactory clinical application is only achieved
when eliminating all cancer cells, especially the cancer stem
cells, by efficient targeting gene therapy strategy. In our study,
we found that CSGCs, when in resting status, have little tropism on
MSCs, limiting the application of MSCs as drug vectors.
Furthermore, most therapeutic drugs play a role in targeting the
tumor cells in the cell cycle. Thus, it is important to find an
alternative way to induce the resting cancer stem cells into
proliferating status, before or during using the tracking ability
of MSCs to kill all tumor cells.
Acknowledgements
This study was supported by the China
National Natural Scientific Fund (81000901), Key Laboratory Project
of Tianjin Programs for Science and Technology (10SYSYJC28800), Key
Project of Chinese National Programs for Fundamental Research and
Development (973 Program, 2010CB529405), and Tianjin Health Bureau
Science and technology projects (2011KZ24 and 2011KZ26), Tianjin
Binhai New Area Health Bureau Science and technology projects
(2011BHKZ004).
References
|
1.
|
Raizer JJ, Grimm S, Chamberlain MC, et al:
A phase 2 trial of single-agent bevacizumab given in an
every-3-week schedule for patients with recurrent high-grade
gliomas. Cancer. 116:5297–5305. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Gilbert CA and Ross AH: Cancer stem cells:
cell culture, markers, and targets for new therapies. J Cell
Biochem. 108:1031–1038. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Bonavia R, Inda MM, Cavenee WK and Furnari
FB: Heterogeneity maintenance in glioblastoma: a social network.
Cancer Res. 71:4055–4060. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Masica DL and Karchin R: Correlation of
somatic mutation and expression identifies genes important in human
glioblastoma progression and survival. Cancer Res. 71:4550–4061.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Huang PH, Cavenee WK, Furnari FB and White
FM: Uncovering therapeutic targets for glioblastoma: a systems
biology approach. Cell Cycle. 6:2750–2754. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Hide T, Takezaki T, Nakatani Y, et al:
Combination of a ptgs2 inhibitor and an epidermal growth factor
receptor-signaling inhibitor prevents tumorigenesis of
oligodendrocyte lineage-derived glioma-initiating cells. Stem
Cells. 29:590–599. 2011. View
Article : Google Scholar
|
|
7.
|
Murphy AM and Rabkin SD: Current status of
gene therapy for brain tumors. Transl Res. 161:339–354. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Gong A and Huang S: FoxM1 and
Wnt/β-catenin signaling in glioma stem cells. Cancer Res.
72:5658–5662. 2012.
|
|
9.
|
Filatova A, Acker T and Garvalov BK: The
cancer stem cell niche(s): the crosstalk between glioma stem cells
and their microenvironment. Biochim Biophys Acta. 1830:2496–2508.
2013. View Article : Google Scholar
|
|
10.
|
Altaner C and Altanerova V: Stem cell
based glioblastoma gene therapy. Neoplasma. 59:756–760. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Patel M, Vogelbaum MA, Barnett GH, Jalali
R and Ahluwalia MS: Molecular targeted therapy in recurrent
glioblastoma: current challenges and future directions. Expert Opin
Investig Drugs. 21:1247–1266. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Serakinci N, Christensen R, Fahrioglu U,
Sorensen FB, Dagnæs-Hansen F, Hajek M, Jensen TH, Kolvraa S and
Keith NW: Mesenchymal stem cells as therapeutic delivery vehicles
targeting tumor stroma. Cancer Biother Radiopharm. 26:767–773.
2011. View Article : Google Scholar
|
|
13.
|
Bak XY, Lam DH, Yang J, Ye K, Wei EL, Lim
SK and Wang S: Human embryonic stem cell-derived mesenchymal stem
cells as cellular delivery vehicles for prodrug gene therapy of
glioblastoma. Hum Gene Ther. 22:1365–1377. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Dai LJ, Moniri MR, Zeng ZR, Zhou JX, Rayat
J and Warnock GL: Potential implications of mesenchymal stem cells
in cancer therapy. Cancer Lett. 305:8–20. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Hata N, Shinojima N, Gumin J, Yong R,
Marini F, Andreeff M and Lang FF: Platelet-derived growth factor BB
mediates the tropism of human mesenchymal stem cells for malignant
gliomas. Neurosurgery. 66:144–157. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Matuskova M, Baranovicova L, Kozovska Z,
Durinikova E, Pastorakova A, Hunakova L, Waczulikova I, Nencka R
and Kucerova L: Intrinsic properties of tumour cells have a key
impact on the bystander effect mediated by genetically engineered
mesenchymal stromal cells. J Gene Med. 14:776–787. 2012. View Article : Google Scholar
|
|
17.
|
Keung EZ, Nelson PJ and Conrad C: Concise
review: genetically engineered stem cell therapy targeting
angiogenesis and tumor stroma in gastrointestinal malignancy. Stem
Cells. 31:227–235. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Kosaka H, Ichikawa T, Kurozumi K, Kambara
H, Inoue S, Maruo T, Nakamura K, Hamada H and Date I: Therapeutic
effect of suicide gene-transferred mesenchymal stem cells in a rat
model of glioma. Cancer Gene Ther. 19:572–578. 2012. View Article : Google Scholar
|
|
19.
|
Doucette T, Rao G, Yang Y, Gumin J,
Shinojima N, Bekele BN, Qiao W, Zhang W and Lang FF: Mesenchymal
stem cells display tumor-specific tropism in an RCAS/Ntv-a glioma
model. Neoplasia. 13:716–725. 2011.PubMed/NCBI
|
|
20.
|
Liu Y, Wang L, Fatahi R, Kronenberg M,
Kalajzic I, Rowe D, Li Y and Maye P: Isolation of murine bone
marrow derived mesenchymal stem cells using Twist2 Cre transgenic
mice. Bone. 47:916–925. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Kisiel AH, McDuffee LA, Masaoud E, Bailey
TR, Esparza Gonzalez BP and Nino-Fong R: Isolation,
characterization, and in vitro proliferation of canine mesenchymal
stem cells derived from bone marrow, adipose tissue, muscle, and
periosteum. Am J Vet Res. 73:1305–1317. 2012. View Article : Google Scholar
|
|
22.
|
Dembo M and Wang YL: Stresses at the
cell-to-substrate interface during locomotion of fibroblasts.
Biophys J. 76:2307–2316. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Qin L, Huang J, Xiong C, Zhang Y and Fang
J: Dynamical stress characterization and energy evaluation of
single cardiac myocyte actuating on flexible substrate. Biochem
Biophys Res Commun. 360:352–356. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Huang J, Peng X, Xiong C and Fang J:
Influence of substrate rigidity on primary nucleation of cell
adhesion: a thermal fluctuation model. J Colloid Interface Sci.
366:200–208. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Lee J, Elkahloun AG, Messina SA, Ferrari
N, Xi D, Smith CL, Cooper R Jr, Albert PS and Fine HA: Cellular and
genetic characterization of human adult bone marrow-derived neural
stem-like cells: a potential antiglioma cellular vector. Cancer
Res. 63:8877–8889. 2003.
|
|
26.
|
Paroo Z, Ye X, Chen S and Liu Q:
Phosphorylation of the human microRNA-generating complex mediates
MAPK/Erk signaling. Cell. 139:112–122. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Liu N, Landreh M, Cao K, et al: The
microRNA miR-34 modulates ageing and neurodegeneration in
Drosophila. Nature. 482:519–523. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Bischoff DS, Makhijani NS and Yamaguchi
DT: Constitutive expression of human telomerase enhances the
proliferation potential of human mesenchymal stem cells. Biores
Open Access. 1:273–279. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Sensebé L, Bourin P and Tarte K: Good
manufacturing practices production of mesenchymal stem/stromal
cells. Hum Gene Ther. 22:19–26. 2011.PubMed/NCBI
|
|
30.
|
Boeuf S and Richter W: Chondrogenesis of
mesenchymal stem cells: role of tissue source and inducing factors.
Stem Cell Res Ther. 1:312010.PubMed/NCBI
|
|
31.
|
Uccelli A, Morando S, Bonanno S, Bonanni
I, Leonardi A and Mancardi G: Mesenchymal stem cells for multiple
sclerosis: does neural differentiation really matter? Curr Stem
Cell Res Ther. 6:69–72. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Augello A and De Bari C: The regulation of
differentiation in mesenchymal stem cells. Hum Gene Ther.
21:1226–1238. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Park JS, Chu JS, Tsou AD, Diop R, Tang Z,
Wang A and Li S: The effect of matrix stiffness on the
differentiation of mesenchymal stem cells in response to TGF-β.
Biomaterials. 32:3921–3930. 2011.PubMed/NCBI
|
|
34.
|
Galli R, Binda E, Orfanelli U, Cipelletti
B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F and Vescovi
A: Isolation and characterization of tumorigenic, stem-like neural
precursors from human glioblastoma. Cancer Res. 64:7011–7021.
2004.PubMed/NCBI
|