Introduction
Aromatase inhibitor-resistant breast cancer cells
are modeled in vitro by long-term E2-deprived
breast cancer cell lines. The MCF-7:WS8 cell line represents a
clone of the estrogen receptor (ER)-positive cell line MCF-7 that
is highly sensitive to E2-stimulated growth (1). The MCF-7:5C and MCF-7:2A subclones
are derived from the parental MCF-7 cell line through long-term
E2 deprivation (1–4).
MCF-7:5C cells express wild-type ER at a higher level than the
parental line, and are progesterone receptor (PR)-negative
(3). These cells grow in the
absence of E2, and do not respond to 4-hydroxytamoxifen
(4-OHT) (2,3). MCF-7:2A cells can induce expression
of PR and express both wild-type (66 kDa) and mutant (77 kDa) ER
(4,5). The mutant ER contains a repeat of
exons 6 and 7 and cannot bind E2 nor anti-estrogens; it
is expressed 4- to 10-fold lower than the wild-type ER (6). The total ER level of MCF-7:2A cells
is higher than in parental MCF-7 cells, and they also grow in
E2-free media. 4-OHT and pure anti-E2 are
able to block their growth (4,5).
In addition to the different responses to
anti-E2 observed in MCF-7:5C versus MCF-7:2A cells, they
also have different apoptotic responses to E2. The
MCF-7:5C cells undergo apoptosis and die during the first week of
E2 treatment, whereas the MCF-7:2A cells die later,
after two weeks of E2 treatment (7). MCF-7:5C cell response to estrogens
and anti-estrogens has been extensively studied in our lab; the
data show that these cells undergo E2-induced apoptosis
through mechanisms associated with endoplasmic reticulum stress
(ERS) and oxidative stress (8,9).
Thus far, there has been less focus on the classification and
mechanisms of the MCF-7:2A response.
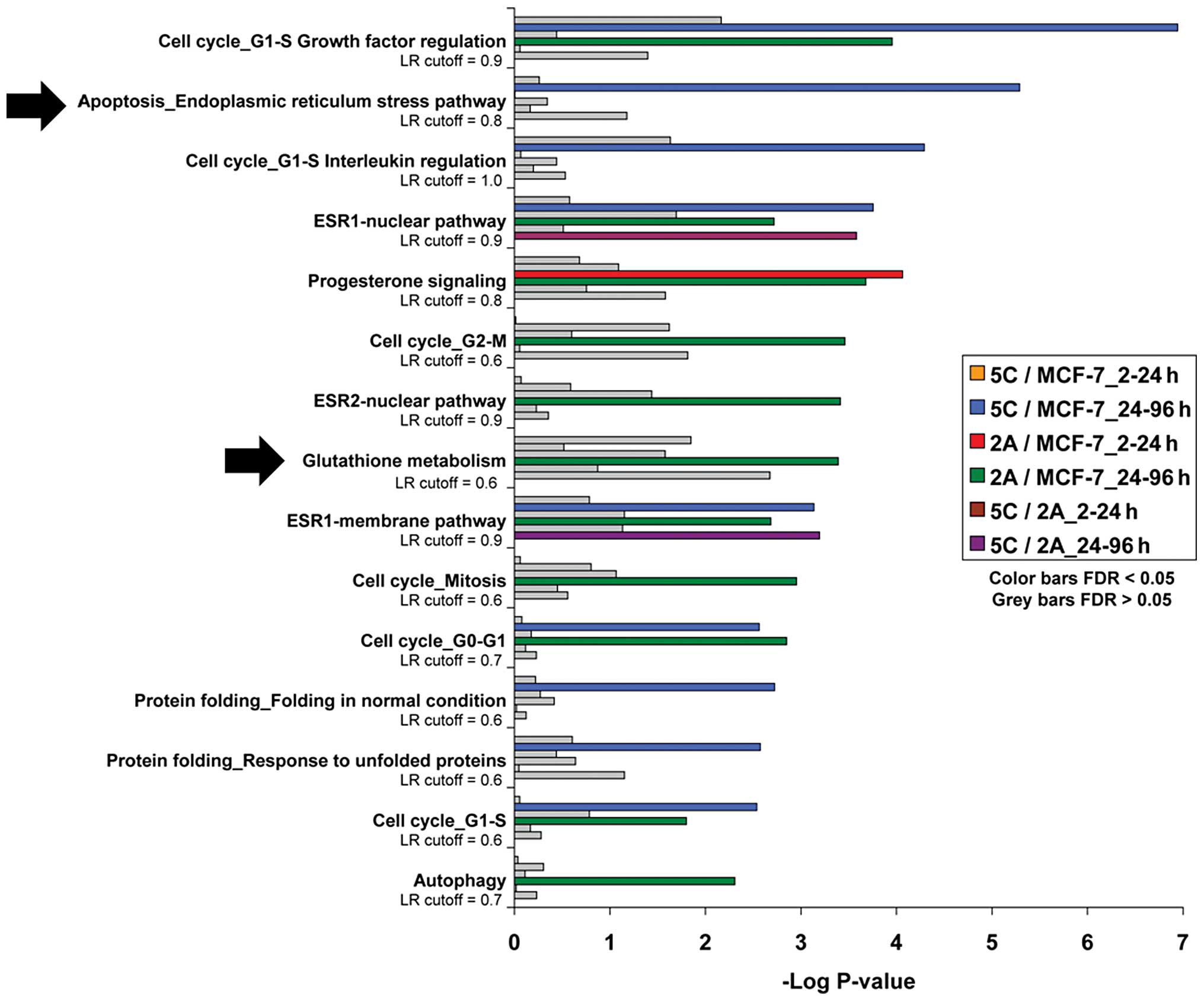
Network enrichment analyses done using gene arrays
in timecourse experiments show overexpression of apoptotic- and
stress-related pathways in the MCF-7:5C cells after 24–96 h of
E2 treatment; however, these analyses show the MCF-7:2A
cells expressing more genes associated with glutathione metabolism
during this time period of E2 exposure (Fig. 1). This suggests that the two cell
lines respond to E2 treatment using different signaling
pathways. The MCF-7:5C cells respond by quickly inducing apoptosis,
while the anti-oxidant pathway may be more relevant to the MCF-7:2A
cells. Experiments were designed to interrogate the apoptotic,
stress and antioxidant pathways in both cell lines to distinguish
signaling mechanisms in response to E2.
The concept of E2-induced death is
important because of its clinical relevance. A clinical study
published in 2009 (10) compared
two doses of E2 for second-line treatment after breast
cancer patients had failed aromatase inhibitor therapy. The authors
showed that after long-term anti-hormone therapy, no response is
lost with the lower dose of E2; overall about 30% of
women responded to E2 treatment. The goal of this study
is to uncover the mechanisms preventing the other 70% of patients
from responding, and perhaps find ways to circumvent their
resistance. To this end, MCF-7:2A cells were used as a model for
E2-deprived breast tumors with the ability to evade
E2-induced apoptosis in the clinic.
Materials and methods
Cell culture
All cell lines were cultured in phenol red-free
RPMI-1640 media supplemented with 10% charcoal-stripped fetal
bovine serum (SFS). Media and treatments were replaced every three
days. Estradiol (E2) (Sigma-Aldrich, St. Louis, MO,
USA), buthionine sulfoximine (BSO) (Sigma-Aldrich), and
combinations were dissolved in ethanol and then in media. AG1024
(Calbiochem, San Diego, CA, USA) was dissolved in DMSO and then in
media.
DNA assays
MCF-7:WS8, MCF-7:5C and MCF-7:2A cells were
harvested after 7 or 14 days treatment with vehicle (0.1% ethanol),
E2 (10−9 mol/l, 1 nM), BSO (10−4
mol/l, 100 μM), or E2 (1 nM) + BSO (100
μM). DNA content was measured as previously described
(11).
Western blot analysis
Total MAPK (#9102), phosphorylated MAPK (#9101),
total AKT (#9272), phosphorylated AKT (#4051L), total eIF2α
(#9722S), phosphorylated eIF2α (#9721S), and IRE1α (#3294S)
antibodies were all purchased from Cell Signaling Technology
(Beverly, MA, USA). IGF-1Rβ antibody (sc-713) was purchased from
Santa Cruz Biotechnology (Santa Cruz, CA, USA). β-actin loading
control antibody (A5441) was purchased from Sigma-Aldrich. Proteins
were harvested from cells using cell lysis buffer (Cell Signaling
Technology) supplemented with Protease Inhibitor Cocktail Set I and
Phosphatase Inhibitor Cocktail Set II (Calbiochem). Bicinchoninic
acid (BCA) assay was used to quantify total protein content
(Rio-Rad Laboratories, Hercules, CA, USA). Protein (50 μg)
was probed and visualized as previously described (11).
Cell cycle analysis
MCF-7:2A cells were cultured in dishes and treated
with vehicle (0.1% ethanol) or E2 (10−9
mol/l, 1 nM). Cells were harvested after 24 h, fixed in 75% ethanol
on ice, stained with propidium iodide and sorted using FACS flow
cytometry (Becton Dickinson, San Jose, CA, USA). Results were
analyzed using CellQuest software.
RT-PCR
Cells were harvested using TRIzol, and RNA was
isolated using RNeasy mini kit (Qiagen, Valencia, CA, USA). RNA was
reverse transcribed to cDNA using a kit (Applied Biosystems, Foster
City, CA). SYBR-Green (Applied Biosystems) was used for
quantitative real-time polymerase chain reaction (RT-PCR) in a
7900HT Fast Real-Time PCR system (Applied Biosystems).
Glutathione assay
Cells were harvested and de-proteinized with 5%
5-sulfosalicylic acid solution (SSA) (Sigma-Aldrich). Total
glutathione [reduced glutathione (GSH) plus glutathione disulfide
(GSSG)] was measured spectroscopically at 412 nm using a
Glutathione Assay Kit (CS0260, Sigma-Aldrich) and the
manufacturer’s instructions.
ROS assay
MCF-7:2A cells were harvested, stained with
10−6 mol/l (1 μM) CM-H2DCFDA (Invitrogen, Eugene,
OR, USA), and analyzed for ROS fluorescence using flow
cytometry.
Statistical analysis
Values reported are means ± standard deviation (SD).
Significant differences were found by Student’s t-test. P-values
<0.05 were considered to indicate a statistically significant
difference.
Results
MCF-7:2A initial response to
E2
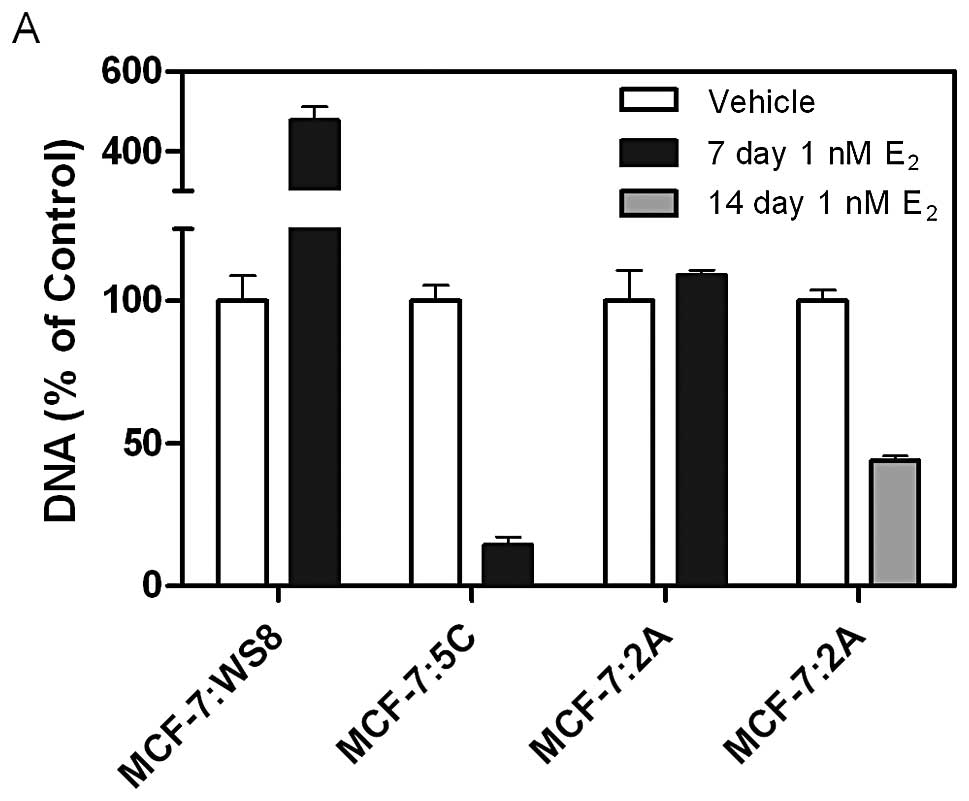
The MCF-7:WS8, MCF-7:5C and MCF-7:2A cell lines
respond differently to 10−9 mol/l (1 nM) E2.
In the presence of 1 nM E2, MCF-7:WS8 cells are
stimulated to proliferate over 7 days, whereas MCF-7:5C cells are
killed by this time point (Fig.
2A). MCF-7:2A cell growth is unaffected by the presence of
E2 after one week, but their DNA is reduced by 50% after
the second week of treatment (Fig.
2A). Interestingly, MCF-7:2A cells are initially stimulated to
proliferate in response to E2. After 24 h-treatment with
1 nM E2, both the mitogen-activated protein kinase
(MAPK) and serine/threonine protein kinase Akt (AKT) pathways are
activated, as shown by an increase in phosphorylated MAPK (p-MAPK)
and phosphorylated AKT (p-AKT) proteins, respectively (Fig. 2B). Further, MCF-7:2A cells treated
with E2 for 24 h show an increase in the percentage of
dividing cells compared with vehicle treatment (34.78 versus
20.17%), illustrated by S-phase in cell cycle analysis (Fig. 2C).
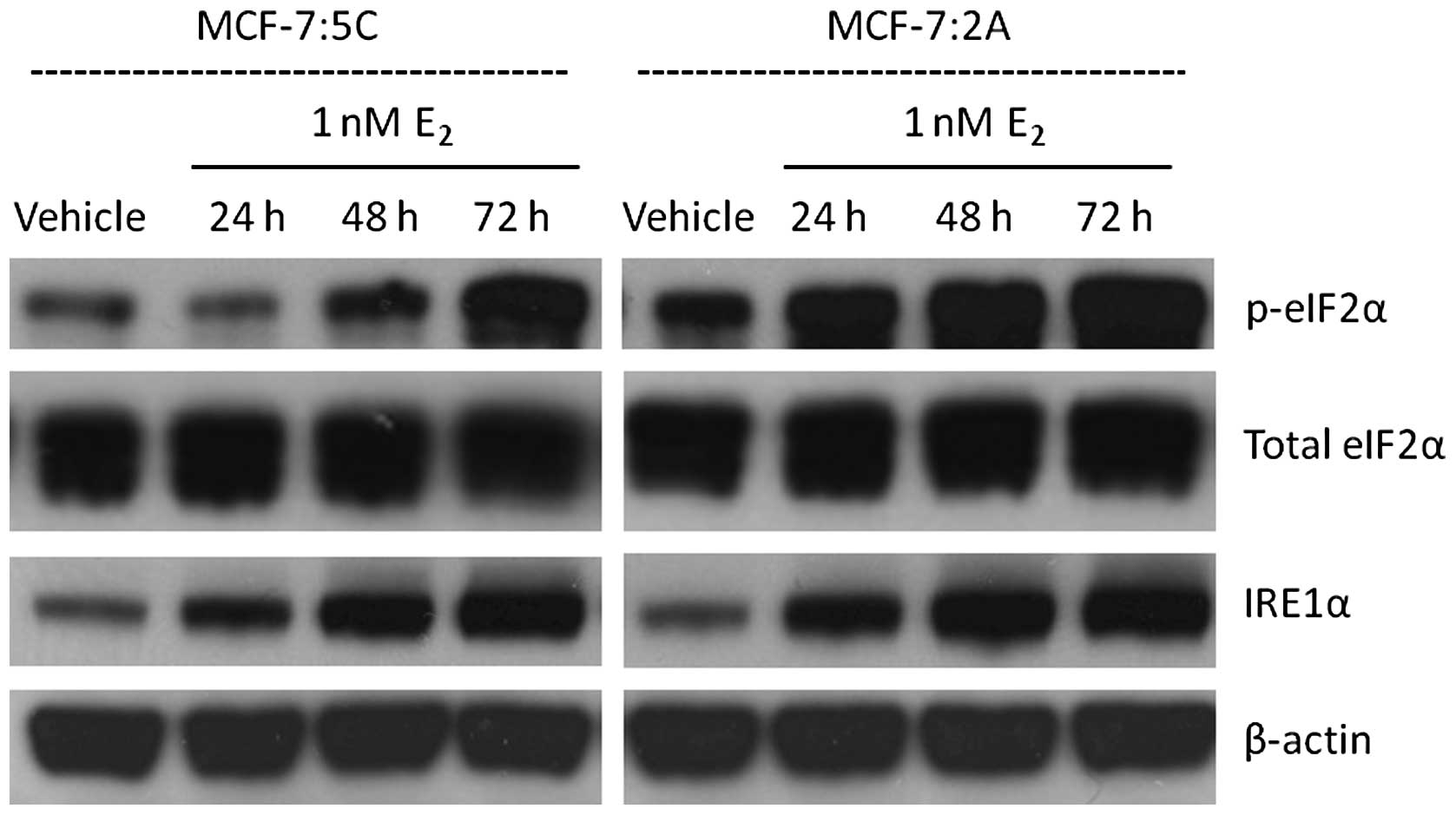
MCF-7:5C and MCF-7:2A UPR
To determine whether the different biological
effects observed in MCF-7:5C and MCF-7:2A cells is due to different
patterns of the unfolded protein response (UPR), proteins
associated with the UPR were measured over a 72 h timecourse. Two
markers of the UPR, phosphorylated eIF2α (p-eIF2α) and IRE1α, were
visualized by western blot analysis in MCF-7:5C and MCF-7:2A cells
in the presence of vehicle and 1 nM E2 (Fig. 3). p-eIF2α is directly downstream of
protein kinase RNA-like endoplasmic reticulum kinase (PERK), a
sensor which initiates UPR. Both cell lines show an increase in the
protein expression of p-eIF2α and IRE1α by 72 h of E2
treatment, indicating activated UPR. Though MCF-7:2A cells show a
slightly higher basal p-eIF2α level, no differences in UPR
activation can be seen between the two cell lines.
MCF-7:5C and MCF-7:2A estrogen-induced
apoptosis
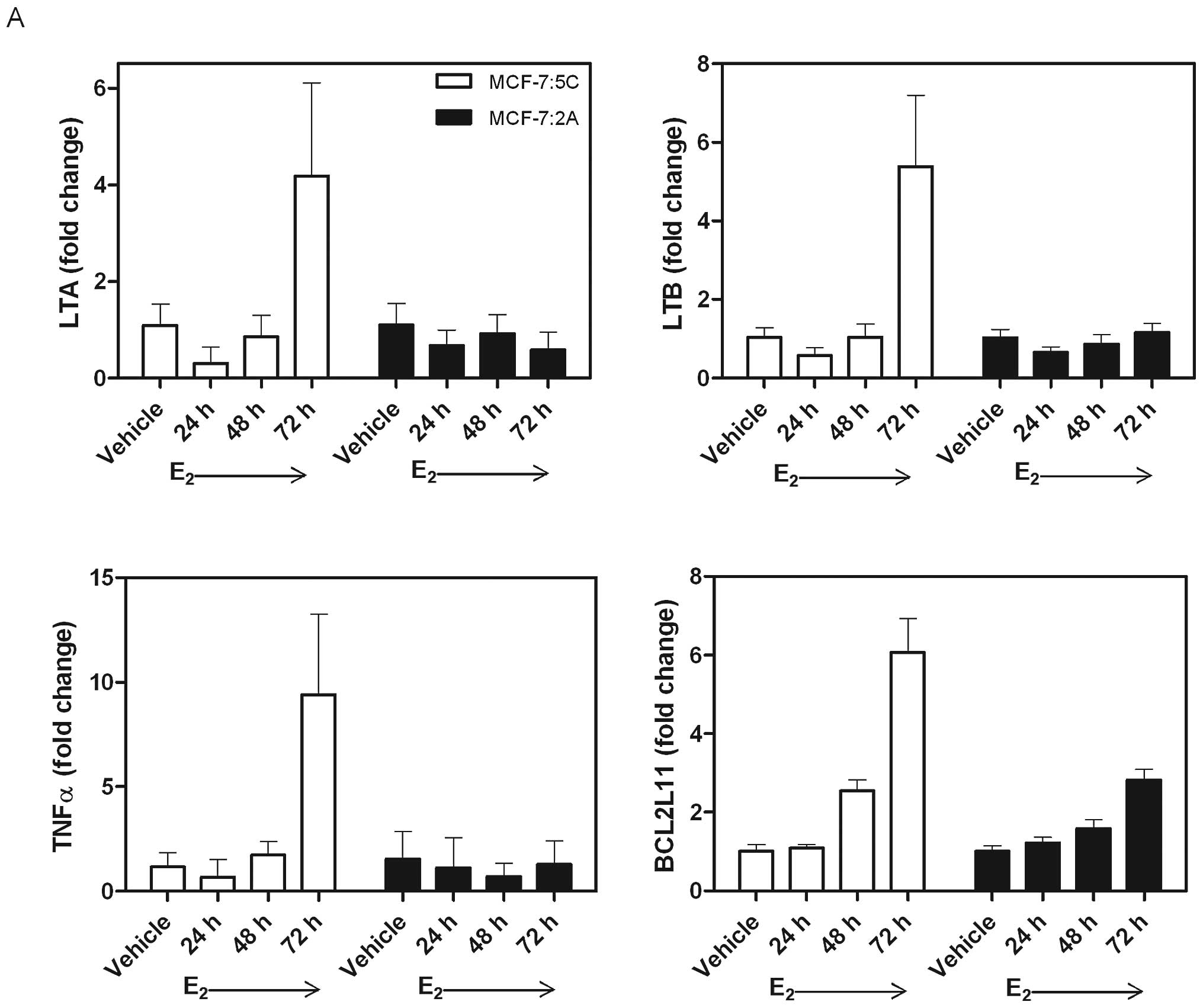
To determine whether MCF-7:2A cells experience
apoptosis through the same mechanism as MCF-7:5C cells, RT-PCR was
used to quantify mRNA levels of apoptosis-related genes. MCF-7:5C
cells noticeably upregulate LTA (4.19±1.92 fold change), LTB
(5.39±1.82), TNFα (9.40±3.86), and BCL2L11 (6.06±0.87) after 72 h
of E2 treatment, while MCF-7:2A cells show no major
changes during this time period (Fig.
4A). MCF-7:2A cells were then treated with E2 for a
longer time period to measure apoptosis-related genes during the
time when they appear to die. MCF-7:2A cells increase both TNFα
(33.55±12.09 fold change) and BCL2L11 (3.71±0.35 fold change) after
12 days of 1 nM E2 treatment (Fig. 4B). The upregulated
apoptosis-related genes correspond to the time when cell death is
most apparent in both cell lines, during week one in MCF-7:5C
cells, and during week two in MCF-7:2A cells.
MCF-7:5C and MCF-7:2A oxidative
stress
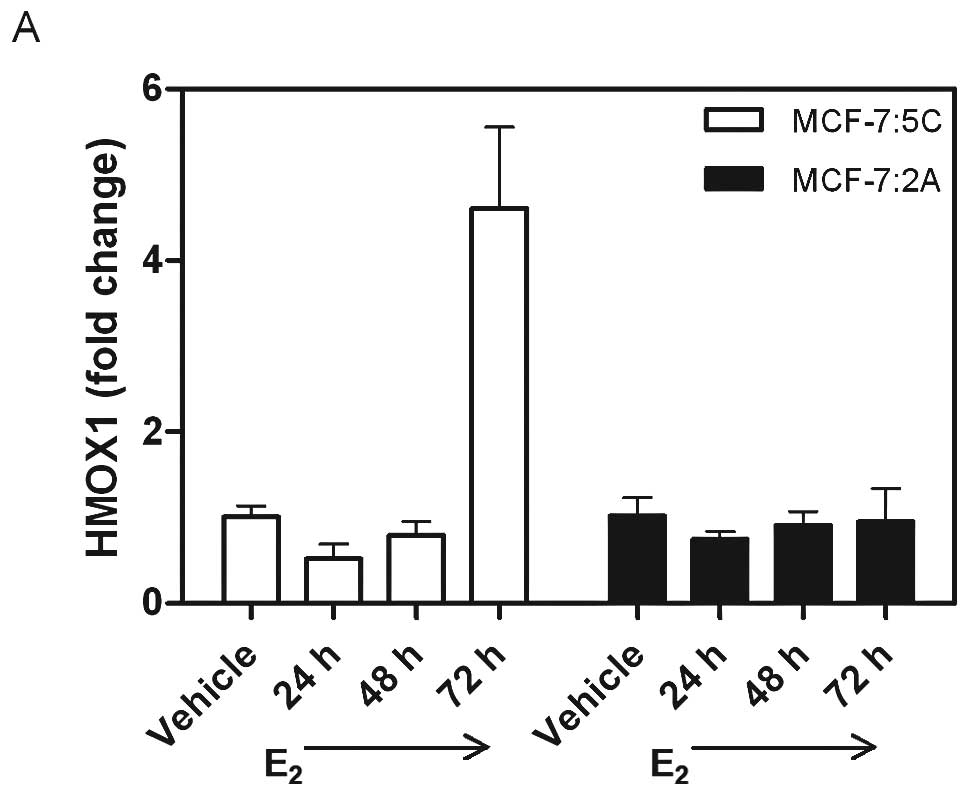
Heme oxygenase 1 (HMOX1) was used as an indicator to
illustrate when MCF-7:5C and MCF-7:2A cells experience oxidative
stress. After 72 h of 1 nM E2 treatment, HMOX1 mRNA was
increased 4.61-fold in MCF-7:5C cells (Fig. 5A), suggesting this cell line
undergoes oxidative stress at this time point. MCF-7:2A cells did
not generate an upregulation of HMOX1 mRNA until 12 days of 1 nM
E2 treatment when it increased 10.03-fold (Fig. 5B), suggesting an earlier protective
mechanism inherent in these cells to prevent oxidative stress
longer than MCF-7:5C cells.
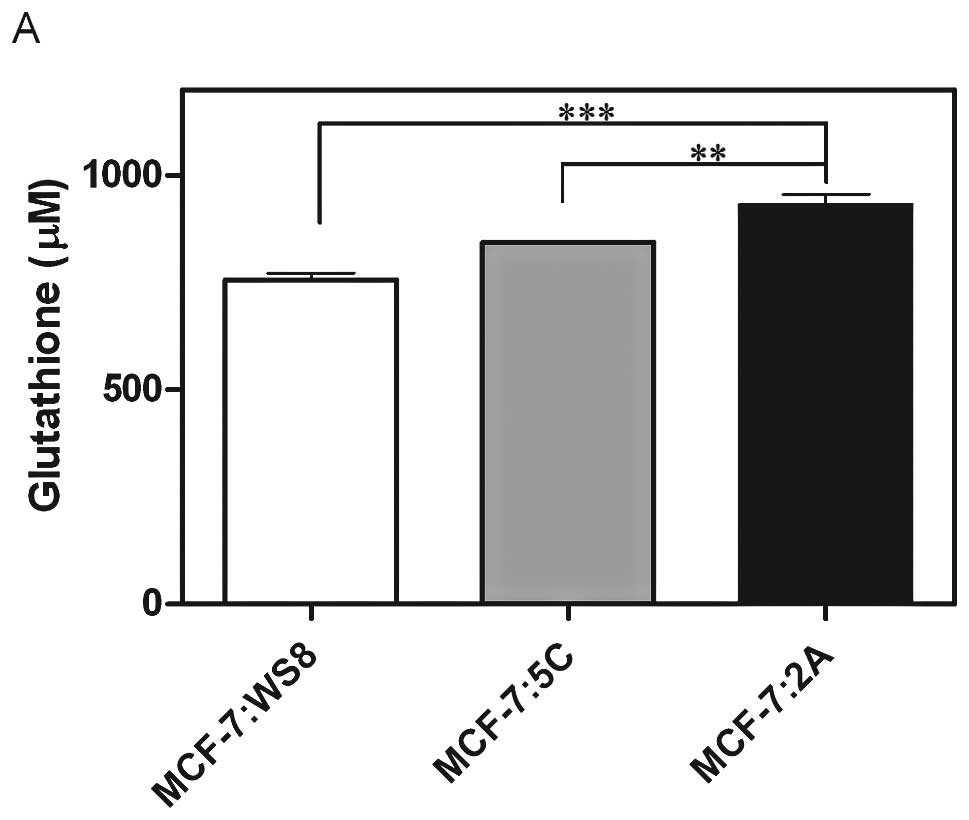
Glutathione is a potent antioxidant and was
quantified in MCF-7:5C and MCF-7:2A cells to illustrate a potential
protective mechanism in MCF-7:2A cells against oxidative stress
(Fig. 6A). In fact, MCF-7:2A cells
have significantly more basal glutathione than do MCF-7:WS8 and
MCF-7:5C cells (Fig. 6A).
Buthionine sulfoximine (BSO) is a synthetic amino acid that blocks
glutathione synthesis by inhibiting γ-glutamylcysteine synthetase.
BSO (100 μM) dramatically decreases glutathione levels in
both MCF-7:5C and MCF-7:2A cells (Fig.
6B). To ask the question of whether glutathione is protecting
MCF-7:2A cells from oxidative stress and E2-induced
apoptosis, HMOX1 was measured following treatment with vehicle, 1
nM E2 alone, 100 μM BSO alone, and 1 nM
E2 + 100 μM BSO after 24, 48 and 72 h (Fig. 6C). MCF-7:2A cells show increased
HMOX1 mRNA at 72 h after treatment with 100 μM BSO and 1 nM
E2 + 100 μM BSO (3.57±0.36 and 2.60±0.70 fold
change, respectively), suggesting a protective role of glutathione
in these cells. Reactive oxygen species (ROS) increased 634% over
vehicle in MCF-7:2A cells after 12 days of the combination
treatment (Fig. 6D). Furthermore,
1 nM E2 + 100 μM BSO treatment caused a
significant decrease in DNA after 14 days treatment (Fig. 6E), suggesting that oxidative stress
is a key factor in determining E2-induced MCF-7:2A cell
death.
MCF-7:5C and MCF-7:2A IGFR
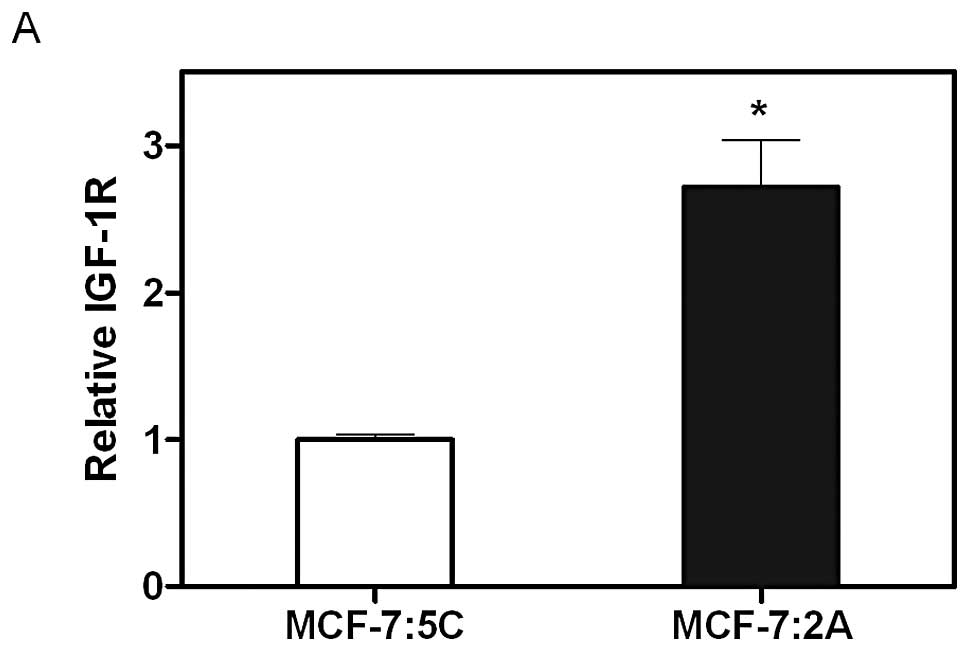
Insulin-like growth factor receptor β (IGF-1Rβ)
upregulation is another mechanism through which MCF-7:2A cells
could receive anti-apoptotic advantage over MCF-7:5C cells.
MCF-7:2A cells exhibit 2.71-fold
greater basal IGF-1Rβ mRNA than MCF-7:5C cells (Fig. 7A). This is consistent at the
protein level as shown by western blot analysis, where MCF-7:2A
cells exhibit more IGF-1Rβ protein expression than MCF-7:5C cells
(Fig. 7B). When treated with an
IGF-1Rβ inhibitor (10 μM AG1024) for 7 days, MCF-7:2A cells
show significantly decreased DNA content when compared to vehicle
and 1 nM E2 treatments (Fig. 7C). Combination treatment of 1 nM
E2 + 10 μM AG1024 decreased DNA content
significantly more than either treatment alone (Fig. 7C), suggesting an integral role of
IGF-1Rβ in MCF-7:2A cells evading E2-induced apoptosis.
To interrogate this further, growth pathway proteins were measured
in response to 10 μM AG1024 treatment. MAPK and AKT pathways
are both blocked by the IGF-1Rβ inhibitor after 72 h as shown by
decreased p-MAPK and p-AKT levels when compared to vehicle-treated
MCF-7:2A cells (Fig. 7D).
Discussion
This study investigated the mechanisms through which
MCF-7:2A cells evade E2-induced apoptosis in
vitro as a means to understand resistant breast cancer cells
after long-term anti-hormone therapy in the clinic. After failure
on an aromatase inhibitor, approximately 30% of breast cancer
patients will respond to treatment with E2 (10); their nascent or remaining breast
tumors will become cytostatic or disappear with physiological
levels of E2. Further, E2 replacement therapy
(ERT) has been shown to reduce the risk of breast cancer in
hysterectomized post-menopausal women (12), perhaps due to
E2-deprived breast cancer cells undergoing
E2-induced apoptosis before resulting in clinically
apparent disease. This study sought to discriminate between
E2-deprived breast tumors that will quickly respond to
treatment with E2 versus those that will respond more
slowly and less dramatically. We modeled these different scenarios
with MCF-7:5C and MCF-7:2A cell lines, respectively.
We have found that the UPR, associated with
endoplasmic reticulum stress (ERS), is a fundamental element in
E2-induced MCF-7:5C cell apoptosis (8). In this setting, E2
triggers UPR and rapidly causes apoptosis within one week of
treatment. Two main sensors of the UPR, IRE1α and PERK are
activated in both cell lines similarly. PERK activation is
confirmed by elevated p-eIF2α, since eIF2α is phosphorylated by
activated PERK. In MCF-7:2A cells, the same sensors are activated
as in MCF-7:5C cells (Fig. 3), but
significant cell death is not apparent at the same timepoint
(Fig. 2A). Despite similar
signaling patterns, the biological responses between the two cell
lines differ. Our data suggested that another mechanism was
preventing cell death after E2-induced UPR in MCF-7:2A
cells.
Oxidative stress is a critical pathway for MCF-7:2A
cells to undergo E2-induced apoptosis. MCF-7:2A cells
inherently exhibit stronger survival and antioxidant mechanisms
than MCF-7:5C cells (Figs.
4–6). This relationship is
consistent with previously published data showing that MCF-7 cells
with higher levels of glutathione peroxidase-1 (GSHPx-1) can
survive better under oxidative stress conditions, such as hydrogen
peroxide treatment (13), and that
MCF-7 cells can increase antioxidant enzymes (i.e. manganese
superoxide dismutase, MnSOD) to prevent TNF-mediated apoptosis
(14). Activation of
E2-induced apoptosis in MCF-7:2A cells also seems to
require TNF family member upregulation (Fig. 4A and B). Oxidative stress occurs
concurrently with upregulation of apoptosis-related genes in the
TNF family. Whether increased TNFα causes oxidative stress or
oxidative stress causes increased TNFα is not yet documented in
this setting.
Additionally, B cell lymphoma 2 (BCL2) plays a role
in preventing cell death caused by oxidative stress (15). In fact, MCF-7:2A cells exhibit
3.76-fold and 3.02-fold higher
basal BCL2 and B cell lymphoma extra large (BCL-xL, BCL2L1) mRNA
levels than MCF-7:5C cells, respectively (Table I), providing support for the idea
of a stronger survival signal. Other data from our lab shows that
MCF-7:2A cells exhibit 6.19-fold higher glutathione peroxidase 2
gene (GPX2) over MCF-7:5C cells (Table
II), illustrating more evidence in favor of increased
protection from E2-induced oxidative stress and
apoptosis in this context.
 | Table I.Basal apoptosis gene expression in
MCF-7:2A cells versus MCF-7:5C. |
Table I.
Basal apoptosis gene expression in
MCF-7:2A cells versus MCF-7:5C.
| Gene symbol | Fold change |
|---|
| AIFM2 | 5.7601 |
| AKT1 | 2.5203 |
| ANXA1 | 57.2949 |
| ANXA4 | 2.7965 |
| APAF1 | 2.839 |
| ATF5 | 2.5303 |
| BAG1 | 2.7188 |
| BCL2 | 3.7598 |
| BCL2L1 | 3.0192 |
| BDNF | 5.8519 |
| BIK | 6.2803 |
| BIRC7 | 33.6437 |
| CARD9 | 2.7968 |
| CASP7 | 2.5278 |
| CD27 | 2.7439 |
| CD5 | 3.884 |
| CD70 | 8.1739 |
| CRYAB | 2.967 |
| CUL3 | 3.2377 |
| DAPK1 | 2.6145 |
| DAPK2 | 6.023 |
| EDAR | 5.7874 |
| ERCC3 | 2.7634 |
| ERN2 | 5.1671 |
| GRM4 | 6.4268 |
| HTT | 4.3186 |
| HIP1 | 5.7736 |
| HSPA1B | 2.5548 |
| HSPB1 | 7.5902 |
| IGF1R | 3.4421 |
| IL1A | 31.2667 |
| INHA | 2.5996 |
| LGALS1 | 430.9062 |
| MAL | 3.0587 |
| MALT1 | 3.2679 |
| NLRC4 | 2.84 |
| NOL3 | 2.9365 |
| PLAGL1 | 3.3963 |
| PLAGL2 | 3.0314 |
| PPP1R13B | 2.7465 |
| PPP2R1B | 4.5273 |
| PRKCA | 2.503 |
| PRODH | 3.8158 |
| PTH | 5.7472 |
| PYCARD | 3.1633 |
| RARG | 2.968 |
| SEMA4D | 2.9335 |
| SFN | 3.2245 |
| SIPA1 | 3.777 |
| SOCS2 | 4.3464 |
| STK17B | 3.8901 |
| TBX5 | 3.3289 |
| TNFRSF10D | 2.5864 |
| TNFRSF18 | 4.0067 |
| TNFRSF19 | 76.9083 |
| TNFRSF6B | 2.7982 |
| TNFRSF8 | 3.103 |
| TNFSF14 | 4.5599 |
| TP63 | 15.4118 |
| TRAF2 | 2.5655 |
| UNC13B | 3.0047 |
| VHL | 3.1063 |
| ZAK | 2.8369 |
| Table II.Top 10 overexpressed and
underexpressed oxidative stress-related genes in MCF-7:2A versus
MCF-7:5C. |
Table II.
Top 10 overexpressed and
underexpressed oxidative stress-related genes in MCF-7:2A versus
MCF-7:5C.
| Gene name | Gene symbol | Category | Fold change |
|---|
| Glutathione
peroxidase 2 |
GPX2 | Glutathione
peroxidases, oxidative stress responsive genes | 6.19 |
| Keratin 1 | KRT1 | Oxidative stress
responsive genes | 2.71 |
| Heme oxygenase
1 | HMOX1 | Oxidative stress
responsive genes | 2.66 |
| Thioredoxin
reductase 1 | TXNRD1 | Oxidative stress
responsive genes, other antioxidants | 2.24 |
| Peroxiredoxin
1 | PRDX1 | Peroxiredoxins
(TPx) | 2.22 |
|
24-Dehydrocholesterol reductase | DHCR24 | Oxidative stress
responsive genes | 2.21 |
| Aldehyde
oxidase | AOX1 | Other genes
involved in ROS metabolism | 2.20 |
| Forkhead box
M1 | FOXM1 | Oxidative stress
responsive genes | 1.83 |
| Thioredoxin | TXN | Oxidative stress
responsive genes | 1.71 |
|
Prostaglandin-endoperoxide synthase 1 | PTGS1 | Other
peroxidases | 1.71 |
| Copper chaperone
for superoxide dismutase | CCS | Other genes
involved in superoxide metabolism | −1.65 |
| Ring finger protein
7 | RNF7 | Oxidative stress
responsive genes | −1.65 |
| Neutrophil
cytosolic factor 2 | NCF2 | Other genes
involved in superoxide metabolism | −1.81 |
| NADPH oxidase,
EF-hand calcium binding domain 5 | NOX5 | Other genes
involved in superoxide metabolism | −1.83 |
| Scavenger receptor
class A, member 3 | SCARA3 | Oxidative stress
responsive genes | −1.98 |
| Superoxide
dismutase 3, extracellular | SOD3 | Superoxide
dismutases, other antioxidants | −2.55 |
| Cytochrome b-245,
beta polypeptide | CYBB | Other
peroxidases | −3.19 |
| Selenoprotein P,
plasma, 1 | SEPP1 | Oxidative stress
responsive genes | −4.94 |
| Apolipoprotein
E | APOE | Oxidative stress
responsive genes, other antioxidants | −8.55 |
| Chemokine (C-C
motif) ligand 5 | CCL5 | Oxidative stress
responsive genes | −50.23 |
Increased IGFR promotes anti-hormone resistance in
breast cancer, likely through growth factor receptor crosstalk and
aberrant ER, MAPK, and AKT signal transduction pathway activation
(16–18). Our data correlate with these
findings in that higher IGF-1Rβ mRNA and protein expression confer
a growth advantage and apoptotic resistance in MCF-7:2A cells
despite treatment with E2 (Fig. 7). This suggests an IGF-1Rβ
signaling pathway that can circumvent normal ER signaling in
long-term estrogen-deprived breast cancer cells. Studies using
hepato-cellular carcinoma cells (HCC) have demonstrated that IGF-1R
overexpression can potentially cause increased glutathione
transferase (GST) and protection from oxidative stress (19). Although this mechanism is shown in
liver cancer cells, it may apply to our models of breast cancer as
well. Perhaps the higher level of IGF-1Rβ in MCF-7:2A cells
generates the increased glutathione levels necessary to escape cell
death in the presence of E2.
The evidence thus far shows that TNF family member
gene expression, protection against oxidative stress, and growth
factor signaling are major mechanisms underlying the different
biological responses to E2 seen in MCF-7:2A cells versus
MCF-7:5C cells. Despite similar UPR signaling patterns, MCF-7:2A
cells resist ERS-induced death longer and stronger than MCF-7:5C
cells. Additional studies may provide further insight into the
connection between IGF-1Rβ and glutathione in MCF-7:2A cells, and
how this relationship functions in the presence and absence of a
stressor such as E2. In order to effectively treat
breast cancer patients who have undergone exhaustive anti-hormone
treatment, and to explain why ERT can prevent breast cancer in some
post-menopausal women, the examination of breast cancer cell models
of E2 deprivation is proving invaluable. By
understanding mechanisms that prevent apoptosis in these breast
cancer cells, we can translate key findings into clinical
practice.
Acknowledgements
This study was supported by the
Department of Defense Breast Program (award number
W81XWH-06-1-0590) Center of Excellence, the Susan G. Komen for the
Cure Foundation (award number SAC100009), and the Lombardi
Comprehensive Cancer Center Support Grant (core grant NIH P30
CA051008). The views and opinions of the authors do not reflect
those of the US Army or the Department of Defense. We would like to
acknowledge Heather Cunliffe, PhD at Translational Genomics
(Phoenix, AZ) for her work on the network enrichment analysis in
Fig. 1.
References
|
1.
|
Sweeney EE, McDaniel RE, Maximov PY, Fan P
and Jordan VC: Models and mechanisms of acquired resistance in
breast cancer: Significant clinical progress despite limitations.
Hom Mol Biol Clin Investig. 9:143–163. 2012.PubMed/NCBI
|
|
2.
|
Jiang SY, Wolf DM, Yingling JM, Chang C
and Jordan VC: An estrogen receptor positive MCF-7 clone that is
resistant to anti-estrogens and estradiol. Mol Cell Endocrinol.
90:77–86. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Lewis JS, Osipo C, Meeke K and Jordan VC:
Estrogen-induced apoptosis in a breast cancer model resistant to
long-term estrogen withdrawal. J Steroid Biochem Mol Biol.
94:131–141. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Pink JJ, Jiang SY, Fritsch M and Jordan
VC: An estrogen-independent MCF-7 breast cancer cell line which
contains a novel 80-kilodalton estrogen receptor-related protein.
Cancer Res. 55:2583–2590. 1995.
|
|
5.
|
Pink JJ and Jordan VC: Models of estrogen
receptor regulation by estrogens and antiestrogens in breast cancer
cell lines. Cancer Res. 56:2321–2330. 1996.PubMed/NCBI
|
|
6.
|
Pink JJ, Wu SQ, Wolf DM, Bilimoria MM and
Jordan VC: A novel 80 kDa human estrogen receptor containing a
duplication of exons 6 and 7. Nucleic Acids Res. 24:962–969. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Lewis JS, Meeke K, Osipo C, Ross EA,
Kidawi N, Li T, Bell E, Changel NS and Jordan VC: Intrinsic
mechanism of estradiol-induced apoptosis in breast cancer cells
resistant to estrogen deprivation. J Natl Cancer Inst.
97:1746–1759. 2005. View Article : Google Scholar
|
|
8.
|
Ariazi EA, Cunliffe HE, Lewis-Wambi JS,
Slif ker MJ, Willis AL, Ramos P, Tapia C, Kim HR, Yerrum S, Sharma
CG, Nicolas E, Balagurunathan Y, Ross EA and Jordan VC: Estrogen
induces apoptosis in estrogen deprivation-resistant breast cancer
through stress responses as identified by global gene expression
across time. Proc Natl Acad Sci USA. 108:18879–18886. 2011.
View Article : Google Scholar
|
|
9.
|
Fan P, Griffith OL, Agboke FA, Anur P, Zou
X, McDaniel RE, Creswell K, Kim SH, Katzenellenbogen JA, Gray JW
and Jordan VC: c-Src modulates estrogen-induced stress and
apoptosis in estrogen-deprived breast cancer cells. Cancer Res.
73:4510–4520. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Ellis MJ, Gao F, Dehdashti F, Jeffe DB,
Marcom PK, Carey LA, Dickler MN, Silverman P, Fleming GF,
Kommareddy A, Jamalabadi-Majidi S, Crowder R and Siegel BA:
Lower-dose vs high-dose oral estradiol therapy of hormone receptor
positive, aromatase inhibitor resistant advanced breast cancer: A
Phase 2 randomized study. JAMA. 302:774–780. 2009. View Article : Google Scholar
|
|
11.
|
Fan P, McDaniel RM, Kim HR, Clagett D,
Haddad B and Jordan VC: Modulating therapeutic effects of the c-Src
inhibitor via oestrogen receptor and human epidermal growth factor
receptor 2 in breast cancer cell lines. Eur J Cancer. 48:3488–3498.
2012. View Article : Google Scholar
|
|
12.
|
Anderson GL, Limacher M, Assaf AR,
Bassford T, Beresford SA, Black H, Bonds D, Brunner R, Brzyski R,
Caan B, Chlebowski R, Curb D, Gass M, Hays J, Heiss G, Hendrix S,
Howard BV, Hsia J, Hubbell A, Jackson R, Johnson KC, Judd H,
Kotchen JM, Kuller L, LaCroix AZ, Lane D, Langer RD, Lasser N,
Lewis CE, Manson J, Margolis K, Ockene J, O’Sullivan MJ, Phillips
L, Prentice RL, Ritenbaugh C, Robbins J, Rossouw JE, Sarto G,
Stefanick ML, Van Horn L, Wactawski-Wende J, Wallace R and
Wassertheil-Smoller S; Women’s Health Initiative Steering
Committee: Effects of conjugated equine estrogen in post-menopausal
women with hysterectomy: the Women’s Health Initiative randomized
controlled trial. J Am Med Assoc. 291:1701–1712. 2004.
|
|
13.
|
Doroshow JH: Glutathione peroxidase and
oxidative stress. Toxicol Lett. 82–83:395–398. 1995.
|
|
14.
|
Siemankowski LM, Morreale J and Briehl MM:
Antioxidant defenses in the TNF-treated MCF-7 cells: selective
increase in MnSOD. Free Radic Biol Med. 26:919–924. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Lee YJ, Chen JC, Amoscato AA, Bennouna J,
Spitz DR, Suntharalingam M and Rhee JG: Protective role of Bcl2 in
metabolic oxidative stress-induced cell death. J Cell Sci.
114:677–684. 2001.PubMed/NCBI
|
|
16.
|
Brodie A, Macedo L and Sabnis G: Aromatase
resistance mechanisms in model systems in vivo. J Steroid Biochem
Mol Biol. 118:283–287. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Fox EM, Miller TW, Balko JM, Kuba MG,
Sanchez V, Smith RA, Liu S, Gonzalez-Angulo AM, Mills GB, Ye F,
Shyr Y, Manning HC, Buck E and Arteaga CL: A kinome-wide screen
identifies the insulin/IGF-I receptor pathway as a mechanism of
escape from hormone dependence in breast cancer. Cancer Res.
71:6773–6784. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Osborne CK and Schiff R: Mechanisms of
endocrine resistance in breast cancer. Annu Rev Med. 62:233–247.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Lee JY, Han CY, Yang JW, Smith C, Kim SK,
Lee EY, Kim SG and Kang KW: Induction of glutathione transferase in
insulin-like growth factor type I receptor-overexpressed hepatoma
cells. Mol Pharmacol. 72:1082–1093. 2007. View Article : Google Scholar : PubMed/NCBI
|