Introduction
Primary liver cancer mainly refers to hepatocellular
carcinoma (HCC), cholangiocarcinoma and hepatic angiosarcoma. As
the third leading cause of cancer death, HCC accounts for 85–90% of
all primary liver cancers and ranks as the fifth most prevalent
malignancy worldwide (1). The high
mortality of HCC is due to late stage detection, and most of the
available therapies are not effective (2). The progression of hepatocellular
carcinogenesis is thought to involve the deregulation of genes that
are critical to cellular processes, such as cell cycle control,
cell growth, apoptosis, cell migration and spreading. In the past
few decades, studies have focused on investigating the genes and
proteins responsible for the development and progression of HCC
(3). Recently, an increasing
number of reports have implicated a new class of small regulatory
RNA molecules, termed microRNAs (miRNAs), in HCC progression.
Since their discovery in 1993, miRNAs have been
described in all multicellular organisms and are associated with a
vast breadth of biological functions, including cellular
proliferation, cellular differentiation and immunity, as well as
tissue remodeling and various human diseases, including cancer
(4). A recent demonstration of the
differential expression of miRNAs and their target mRNAs in cancer
and the discovery that some miRNAs can function as oncogenes or
tumor suppressors have sparked considerable interest in elucidating
their role in tumorigenesis (5,6).
Some specific miRNAs have been found to be frequently deregulated,
and this deregulation has been associated with clinicopathological
features of HCC, such as metastasis, recurrence and prognosis
(7–9). miRNAs are highly conserved, small,
non-coding RNAs that negatively regulate gene expression in
vertebrates through multiple mechanisms, such as complimentary base
pairing with the 3′-UTR of their target mRNAs, resulting in
translational repression, mRNA cleavage and mRNA decay initiated by
miRNA-guided rapid deadenylation (10). However, recent studies have
demonstrated that miRNAs can interact with the 5′-UTR of their
target mRNAs and the DNA methylation machinery, thereby affecting
chromatin status (11).
Epigenetic alternations in genomic DNA include
cytosine methylation in CpG islands, which usually extend
throughout the promoters and the first exons of genes. DNA
methylation, which is associated with gene silencing (12), is carried out by DNA
methyltransferases (DNMTs). Recent studies have established that,
similar to mutations, methylation-mediated silencing of tumor
suppressor genes plays a major role in tumorigenesis. However,
unlike mutations, methylation can be reversed by the inhibition of
DNA methyltransferase, resulting in restored expression of the
silenced tumor suppressor genes. The approval of drugs such as
Vidaza® (5-azacytidine) and Dacogen™
(5-aza-2′-deoxycytidine or decitabine) by the FDA (USA) as
anticancer agents underscores the usefulness of epigenetic therapy.
Similar to protein-coding genes, DNA sequences encoding miRNAs may
undergo aberrant DNA methylation, leading to miRNA upregulation
(through DNA hypomethylation) or downregulation (through DNA
hypermethylation) in human cancers. For example, previous studies
have shown that aberrant hypermethylation of the miR-148a coding
region occurs early in human pancreatic carcinogenesis and leads to
the downregulation of miR-148a expression (13).
In a previous study, miR-148a was found to be
silenced by DNA hypermethylation and to interact with DNMTs in
various cancers. Very recently, Gailhouste et al (14) found that miR-148a expression was
frequently downregulated in biopsy samples from HCC patients as
well as in mouse and human HCC cell lines; however, the mechanism
causing this downregulation has not yet been studied in detail.
Here, we examine i) whether DNA methylation is involved in the
miR-148a deregulation that occurs in HCC cell lines; ii) whether
there is a circular regulation loop between miR-148a and DNMT1; and
iii) the roles that miR-148a plays in the HCC cell cycle. Our study
provides new insight into the molecular mechanism of HCC
development and yield new strategies for HCC diagnostics and
treatment in the future.
Materials and methods
Cell culture and transient transfection
of miR-148a mimics and DNMT1 siRNA
The HCC cell lines HepG2, SMMC-7721 and HCCLM3 and
the normal liver cell line L-02 were obtained from Shanghai Fumeng
Gene Biological Corporation (Shanghai, China). The cells were
maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco,
Carlsbad, CA, USA) supplemented with 10% fetal calf serum (FCS),
100 U/ml penicillin, 100 mg/ml streptomycin, and 2 mM L-glutamine
and were incubated at 37°C in an atmosphere containing 5%
CO2. On the day of transfection, the HepG2 cells were
plated in DMEM supplemented with 10% FCS at a density of
2–3×105 cells/ml and were transfected with the miR-148a
mimics, DNMT1 siRNA or the non-specific (NS)-miRNA (all at 60 nM)
for 24 h using Lipofectamine 2000 (Invitrogen) according to the
manufacturer’s instructions. The culture medium was changed 6 h
after transfection.
The following oligonucleotide sequences were used:
miR-148a mimics, 5′-UCAGUGCACUACAGAACUUUGU-3′,
5′-AAAGUUCUGUAGUGCACUGAUU-3′; NS-miRNA,
5′-UUCUCCGAACGUGUCACGUTT-3′, 5′-ACGUGACACGU UCGGAGAATT-3′; DNMT1
siRNA, 5′-GAGGCCUAUAAU GCAAAGATT-3′,
5′-UCUUUGCAUUAUAGGCCUCTT-3′.
One-step quantitative real-time PCR
To confirm the expression of miR-148a, one-step
real-time qPCR was performed. Total RNA was extracted from HepG2
using TRIzol reagent (Invitrogen). miR-148a expression was measured
using the miScriptII RT kit (Qiagen, Frankfurt, Germany) and the
miScript SYBR-Green PCR kit (Qiagen) in an ABI Prism 7500 PCR
machine. PCR was performed at 95°C for 15 min, followed by 40
cycles of amplification at 94°C for 15 sec, 55°C for 30 sec, and
72°C for 30 sec. The melting curve was performed at 95°C for 30
sec, 60°C for 30 min, and 95°C for 30 sec. The relative miRNA
expression was calculated from three different experiments. The
fold change of the miRNA relative to the U6 RNA was determined
using the formula 2−ΔΔCt.
Quantitative real-time PCR and
semi-quantitative reverse transcription-polymerase chain reaction
(RT-PCR)
Total RNA was isolated from HepG2, SMMC-7721, HCCLM3
and L02 cells using TRIzol reagent (Invitrogen), and first-strand
cDNA was synthesized using the Thermoscript RT-PCR synthesis kit
(Fermentas, Pittsburgh, PA, USA) according to the manufacturer’s
instructions. Quantitative RT-PCR analyses for DNMT1 and GAPDH were
performed using the RT-PCR kit (Applied Biosystems, Foster City,
CA, USA). The mRNA level of GAPDH was used as an internal control.
RT-PCR was carried out according to the standard protocol using the
following primers: β-actin (forward, 5′-TGAGCTGCGTGTGGCCCCTGAG-3′;
reverse, 5′-GGGGCATCGGAACCGCTCATTG-3′), DNMT1 (forward,
5′-ACGAGGATGAGAGGGAGGAG-3′; reverse, 5′-GGCACTTTGGTGAGTTGAT-3′).
PCR was performed at 94°C for 5 min, followed by 30–35 cycles of
amplification at 94°C for 40 sec, 56°C for 40 sec and 72°C for 1
min using an ABI9700 system. The band intensities were measured
using a densitometer, and the results were normalized to the levels
of β-actin. The results were independently repeated at least three
times from three different pools of template, while each template
pool was extracted from at least eight batches of cells.
5-Aza-2prime;-deoxycytidine
treatment
HepG2 cells were seeded overnight in culture dishes,
and 5-aza-2′-deoxycytidine (5-azadC; Sigma-Aldrich, St. Louis, MO,
USA) was added. The medium was refreshed every 24 h until the 48-h
treatment was completed.
Methylation-specific PCR (MS-PCR)
The methylation status of the miR-148a promoter
region was determined by methylation-specific PCR (MSP) using
bisulfite-modified DNA. Genomic DNA was extracted using the QIAamp
DNA mini kit (Qiagen). Two primer sets were used to amplify the
promoter region of the miR-148a gene that contained several CpG
sites; one primer set was specific for the methylated sequence
(miR-148a-M: forward, 5′-TGATTCGTTTTATTA TCGGTC-3′; reverse,
5′-AACACTAACGACATCGACG-3′), and the other primer set was specific
for the unmethylated sequence (miR-148a-U: forward,
5′-TATGATTTGTTTTAT TATTGGTT-3′; reverse, 5′-AACACTAACAACATCAAC
AACC-3′). The primers used in the present study specifically detect
the promoter sequence of the PTEN gene rather than that of the PTEN
pseudogene. M and U are the PCR products of the methylated and
unmethylated alleles, respectively. The PCRs for miR-148a-M and
miR-148a-U were carried out in 50 μl volumes containing 1X
PCR buffer (15 mmol/l MgCl2), 2.5 mmol/l dNTP mixture,
10 pM each primer, 4 U HotStarTaq DNA polymerase (Qiagen), and
25–50 ng of bisulfite-modified DNA. Amplification was performed in
a thermocycler with the following conditions: 94°C for 2 min,
followed by 36 cycles of 94°C for 30 sec, 54°C or 50°C for 30 sec,
and 72°C for 45 sec, followed by an extension at 72°C for 7 min.
The methylation-specific PCRs were performed in duplicate.
Cell proliferation assay
Cell proliferation was determined using the standard
3-(4,5-dimethylthiazol-2-yl)-2,4-diphenyltetrazolium bromide (MTT)
assay. Briefly, the cells were seeded at a density of
5×103 cells per well in 96-well culture plates and
transfected with the miR-148a mimics and negative control as
described above. Cell proliferation was assessed after 24 h. After
culture, 5 mg/ml MTT was added and incubated at 37°C for an
additional 4 h; thereafter, the medium was replaced, and the
formazan crystals were dissolved in 150 μl of dimethyl
sulfoxide (DMSO). The optical density (OD) was determined using a
Thermomax microplate reader (Bio-Tek EL, Winooski, VT, USA) at a
wavelength of 570 nm. All experiments were performed in triplicate
and were repeated at least three times.
Cell cycle analysis
For the cell cycle analysis, we used the Cell Cycle
and Apoptosis Analysis kit. (Beyotime, Jiangsu, China). The cells
were washed three times with cold PBS and subsequently fixed in 70%
ethanol in PBS at −20°C for 12 h. After fixation, the cells were
washed with cold PBS and stained with 0.5 ml of propidium iodide
(PI) staining buffer, which contained 200 mg/ml RNase A and 50
μg/ml PI, at 37°C for 30 min in the dark. Analyses were
performed on a BD LSR flow cytometer (BD Biosciences, Franklin
Lakes, NJ, USA). The experiments were repeated three times.
Apoptosis analysis
For apoptosis analysis, the number of apoptotic
cells was quantified using the Annexin V-FITC Apoptosis Detection
Kit (BestBio, Shanghai, China) according to the manufacturer’s
instructions. Early apoptotic cells were defined as Annexin
V-positive, PI-negative cells. Analyses were performed on a BD LSR
flow cytometer (BD Biosciences). The experiments were repeated
three times.
Western blot analysis
The cells were lysed with RIPA lysis buffer
(Beyotime, Haimen, China). Whole extracts were prepared, and the
protein concentration was determined using a BCA protein assay kit
(Boster, Wuhan, China). Total protein (30 or 50 mg) from the
samples was separated by SDS-PAGE and blotted onto a PVDF membrane
(Millipore, Billerica, MA, USA). After blocking, the PVDF membrane
were incubated for 1 h with primary antibodies diluted in
TBS/Tween-20 (0.075%) containing 3% Marvel. A mouse monoclonal
antibody raised against DNMT1 (Santa Cruz Biotechnology, Santa
Cruz, CA, USA) and an anti-β-actin antibody (Santa Cruz
Biotechnology) were used at a dilution of 1:600. Horseradish
peroxidase-conjugated anti-mouse antibodies were used as the
corresponding secondary antibodies. After four washes in
TBS/Tween-20, the membranes were developed with distilled water,
and the proteins were detected using an enhanced chemiluminescence
system (ECL-plus kit, Thermo Scientific, Rockford, IL, USA).
Luciferase reporter assay
We constructed 3′-UTR reporter plasmids for use in
the dual luciferase reporter assay. The 3′-UTR segments of the
DNMT1 gene containing the miR-148a binding sites were amplified by
PCR using KOD-Plus-DNA polymerase (Toyobo, Osaka, Japan) and were
cloned into the XhoI/NotI sites downstream of the luciferase
reporter gene in the psiCHECK-2-Report vector (Promega, Madison,
WI, USA); these constructs were named psiCHECK-2-TGF-β2 3′-UTR-wt
and psiCHECK-2-β-catenin 3′-UTR-wt, respectively. For the
luciferase assay, HepG2 cells (5×104 cells/well) were
cultured in 24-well plates. The cells were then co-transfected with
200 ng of the DNMT1 3′-UTR-wt plasmid or the empty vector plasmid
in the presence of 60 nmol of the miR-148a mimics (Gene Pharma,
Shanghai, China) using 2.5 μl of Lipofectamine 2000 and 100
μl of Opti-MEM reduced serum medium (Invitrogen, Carlsbad,
CA, USA). After 48 h, the luciferase activities were measured
consecutively using the Dual-Luciferase Reporter 1000 Assay System
(Promega). Renilla luciferase activity was used to normalize
the firefly luciferase activity. All the experiments were performed
in triplicate.
Statistical analysis
All results are expressed as the mean ± SE.
Statistical significance was determined using either Student’s
t-test for comparison between the means or a one-way analysis of
variance with a post hoc Dunnett’s test. P<0.05 was considered
to indicate a statistically significant difference.
Results
miR-148a is significantly downregulated
in hepatocellular carcinoma cell lines
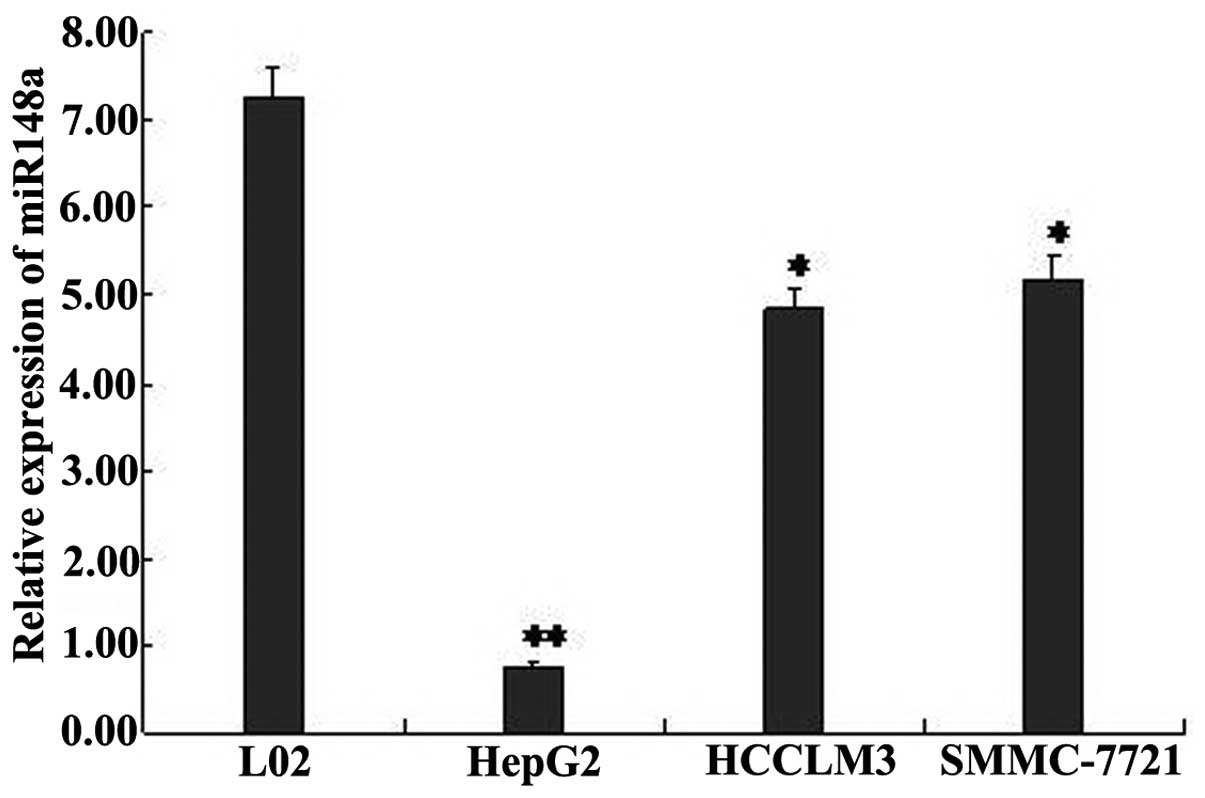
To determine whether miR-148a was silenced in HCC
cells, we examined the expression of miR-148a using real-time qPCR
in the HCC cell lines HepG2, SMMC-7721, and HCCLM3; the normal
liver cell line L-02 was used as a matched control. The results
show that miR-148a was significantly downregulated in the HCC cell
lines (Fig. 1), especially in the
HepG2 cells. Therefore, we chose to perform our subsequent studies
using the HepG2 cell line.
Downregulation of miR-148a is due to the
hypermethylation of the miR-148a gene promoter region in HCC
cells
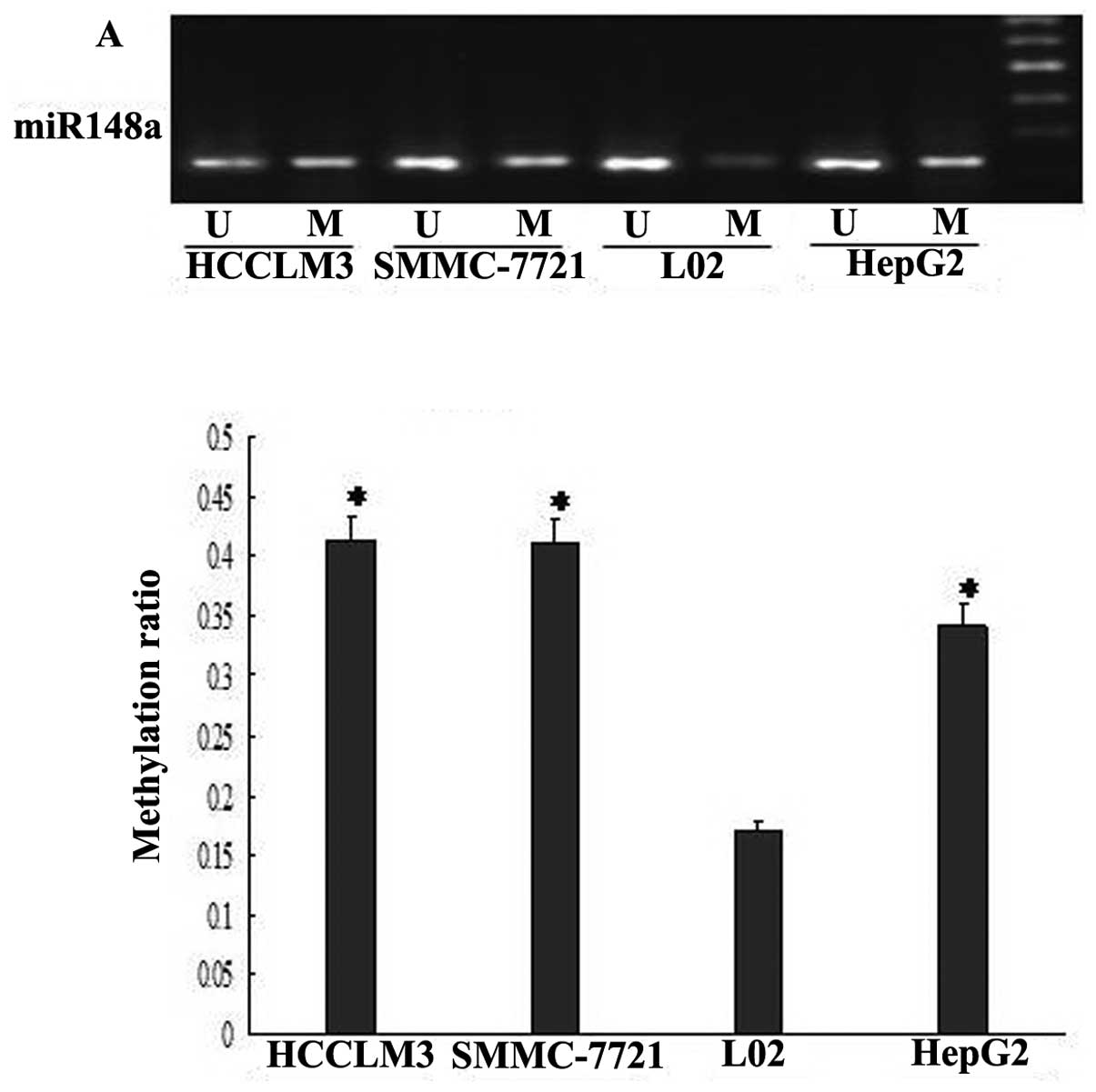
Methylation of gene promoters often occurs during
carcinogenesis, resulting in reduced expression or loss of
expression of the methylated gene (15). Previous studies have shown that the
genomic DNA sequence spanning the miR-148a gene contains a large
amount of CpG-rich regions (CpG islands) in the promoter (16). Thus, we hypothesized that DNA
methylation is responsible for the downregulation of miR-148a in
HCC. To verify this hypothesis, we performed MSP analysis to detect
the methylation status of the miR-148a promoter region; indeed,
hypermethylation of the CpG islands in the miR-148a promoter was
observed in the HepG2, SMMC 7721, and HCCLM3 cells compared to the
L-02 cells (Fig. 2A).
Additionally, to further support the functional relevance of the
DNA methylation, we found that demethylation by 5-aza-dC
dramatically restored miR-148a expression in HepG2 cells, and this
response was dose-dependent (Fig.
2C). Thus, the MSP results showed that 5-aza-dC treatment
caused the demethylation of the miR-148a promoter (Fig. 2B).
Overexpression of DNMT1 is responsible
for hypermethylation of the miR-148a gene promoter in
hepatocellular carcinoma cells
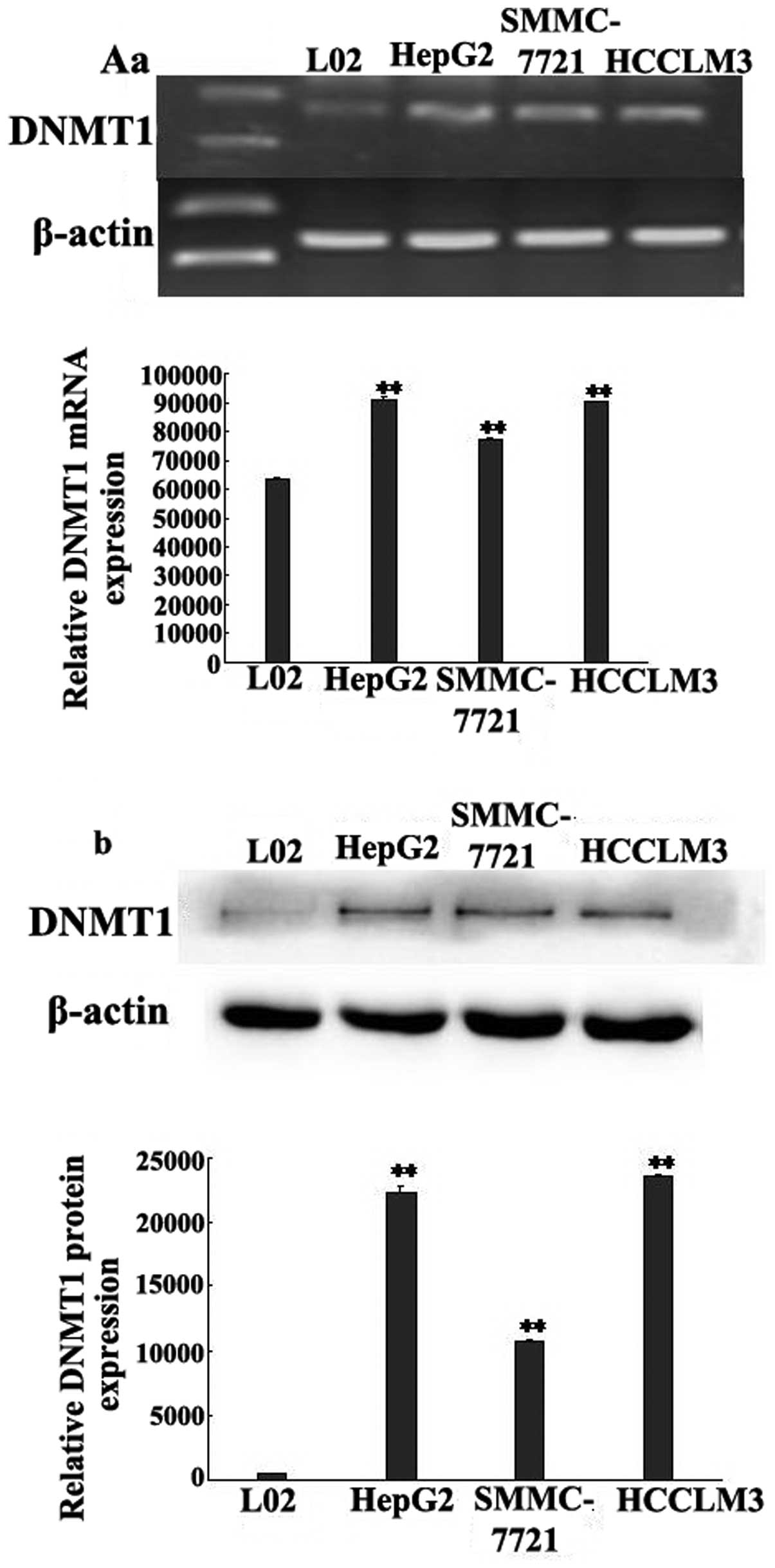
Previous studies have shown that DNMT1
overexpression contributes to gene promoter hypermethylation and is
associated with the malignant potential and poor prognosis of human
cancers (17,18). In our study, we found that DNMT1
expression was strongly increased in the HCC cell lines compared to
the L-02 cells (Fig. 3A). To
further explore the role of DNMT1 in the regulation of miR-148a
expression, we silenced de novo DNMT1 expression using a
siRNA targeted against DNMT1. DNMT1 knockdown abolished the
hypermethylation of the miR-148a gene (Fig. 3B) and resulted in the upregulation
of miR-148a expression (Fig. 3C).
These data strongly suggest that overexpression of DNMT1 is
responsible for the hypermethylation of the miR-148a gene promoter
in HCC cells.
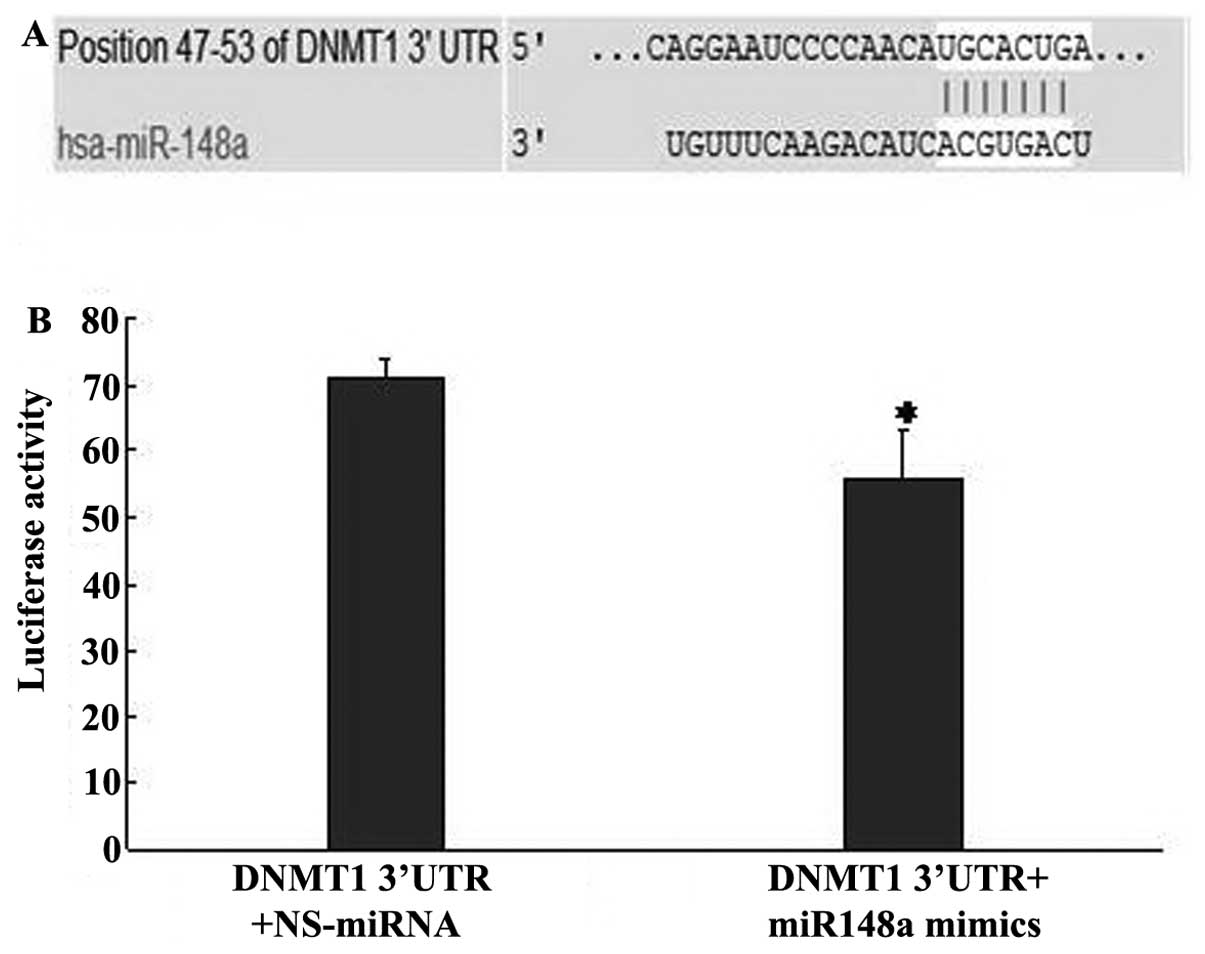
DNMT1 is a direct target of miR-148a
It has been reported that miR-148a directly targets
DNMT1 in lupus CD4+ T cells and gastric cancer (19,20).
The TargetScan 5.1 online software (http://www.targetscan.org/, Whitehead Institute for
Biomedical Research, Cambridge, MA, USA) was used to predict the
miR-148a target genes, and unsurprisingly, we found that the 3′-UTR
of the DNMT1 gene had 7 sequential bases that paired with the 5′
end of human miR-148a (Fig. 4A).
This finding indicates that DNMT1 may be a potential target of
miR-148a. Additionally, co-transfection of miR-148a mimics and the
DNMT1-wt construct caused a significant decrease in luciferase
activity compared to transfection with the NS-miRNA in HepG2 cells
(Fig. 4B). Furthermore, western
blot and real-time PCR analyses revealed that DNMT1 protein and
mRNA expression was significantly lower in the miR-148a-transfected
HepG2 cells compared to the control group (Fig. 4C). These results suggest that DNMT1
is a direct target of miR-148a in HCC cells.
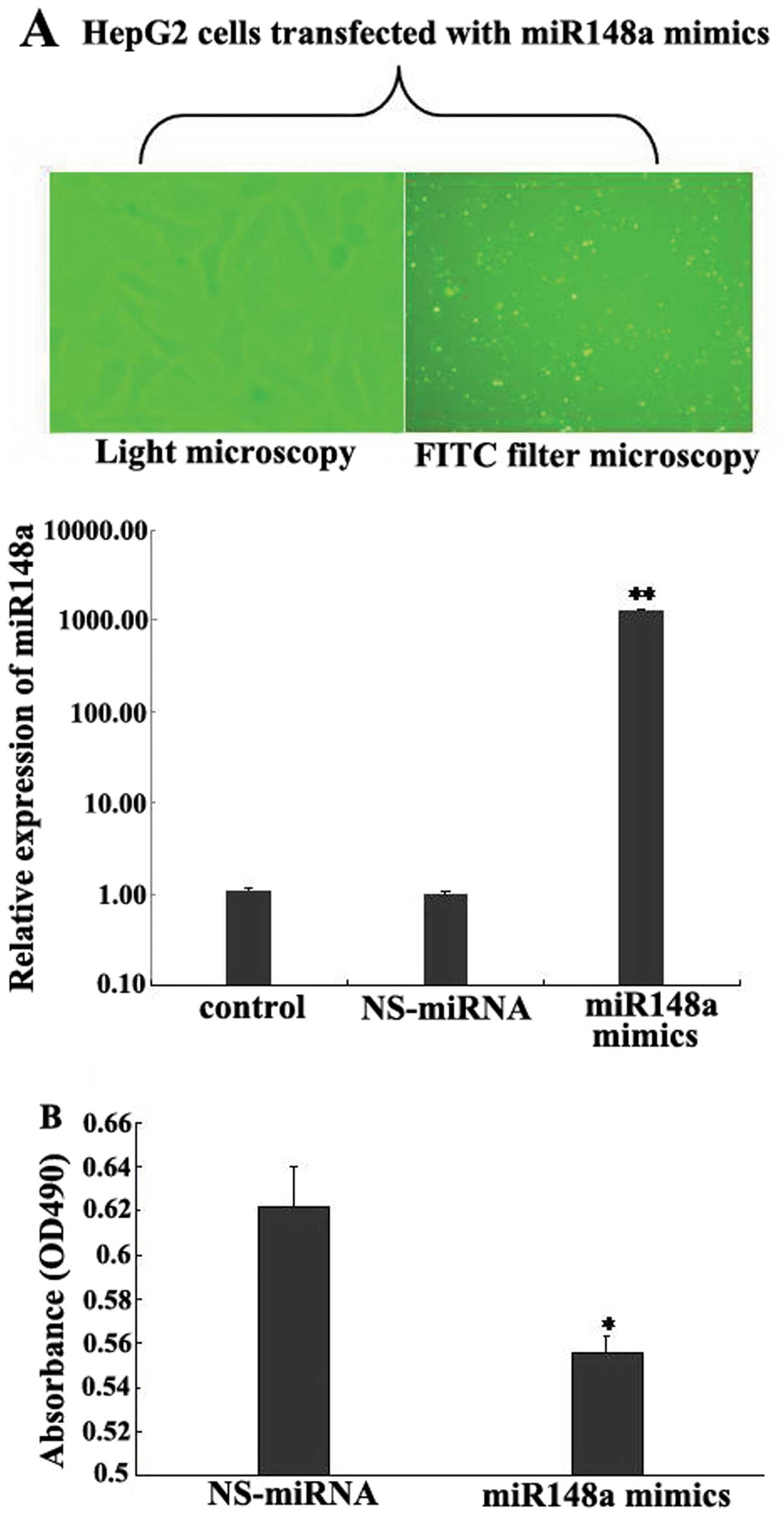
miR-148a inhibits cell proliferation
To investigate the roles of miR-148a in the
regulation of HCC cell proliferation and apoptosis, we tested the
effects of miR-148a on the proliferation of HepG2 cells. We found
that the cells transfected with the miR-148a mimics had
significantly increased expression of mature miR-148a (Fig. 5A). The MTT assay showed that the
introduction of miR-148a caused significant inhibition of HCC cell
proliferation (Fig. 5B). To
understand whether the reduced cell proliferation was due to cell
cycle arrest or apoptosis, we used FACS analysis to measure the
effect of miR-148a on cell cycle progression and apoptosis. We
found that overexpression of miR-148a had a striking effect on cell
cycle distribution, whereas the proportion of apoptotic cells
induced by the transfection of the miR-148a mimics was not
significantly different from that induced by transfection of
NS-miRNA (Fig. 5C). These results
indicated that overexpression of miR-148a inhibited HCC cell
proliferation, at least in part, through cell cycle arrest;
however, overexpression did not influence cell apoptosis.
Discussion
In recent years, there has been increasing interest
in the roles of epigenetic modifications in the etiology of human
diseases (13,21). For example, aberrant
hypermethylation of the CpG islands of tumor suppressor genes and
the resulting transcriptional silencing are associated with
malignant transformation in cancer (22). At the same time, a large number of
studies have revealed that microRNAs constitute effective
regulatory networks and can regulate approximately one-third of the
human protein coding genes at the post-transcriptional level. Some
miRNAs are known as gene silencers, and their expression profiles
have been negatively correlated with their target genes, including
oncogenes, during carcinogenesis. Interestingly, aberrant
expression of these miRNAs has been reported in most tumors; thus,
miRNAs may play a critical role in tumor formation and
development.
Hepatocellular carcinoma (HCC) has an extremely poor
prognosis and remains one of the most common and aggressive human
malignancies worldwide (23). The
effects of epigenetic changes on HCC, especially those related to
the biological functions of miRNAs, have been extensively reported
in recent years. Zhang et al (24) found that miR-148a suppresses the
epithelial-mesenchymal transition (EMT) and metastasis of hepatoma
cells by targeting the Met/Snail signaling pathway. Han et
al (25) indicated that Myc
induces HCC through a novel, microRNA-mediated feedback loop
composed of miR-148a-5p, miR-363-3p and ubiquitin-specific protease
28 (USP28). Moreover, Yan et al (26) found that miR-148a inhibits the
metastasis of HCC cells by blocking the EMT and CSC-like properties
through the Wnt signaling pathway. In this study, we confirmed the
downregulation of miR-148a in HCC cell lines using real-time qPCR
and demonstrated that the restoration of miR-148a expression in HCC
by transfection with miR-148a mimics could obviously inhibit cell
proliferation, suggesting that miR-148a plays a tumor suppressive
role in HCC. These findings also have potential therapeutic
implications. We found that the methylation level of the CpG
islands in the miR-148a promoter was higher in HCC cells than in
normal liver cells, and miR-148a was upregulated in HCC cell lines
upon treatment with the DNA hypomethylating agent 5-aza-2-dC. These
results indicate that the silencing of miR-148a was caused by the
hypermethylation of its promoter region in HCC.
DNA methylation is carried out by the DNMTs, which
are ubiquitously expressed in normal human tissues (27). In cancer, they may be overexpressed
in various tumor types, such as leukemia, colorectal, ovarian,
prostate and breast cancer (28–31).
We assayed the expression of DNMT1 in the HCC cell lines compared
to the normal liver cells. Additionally, we examined the expression
of miR-148a and the methylation level of the miR-148a promoter
after siRNA-mediated DNMT1 depletion in HCC cell lines. The results
showed that the expression of DNMT1 was remarkably higher in the
HCC cells, whereas the methylation level of the miR-148a promoter
was significantly reduced, and miR-148a expression was
significantly upregulated after DNMT1 knockdown, suggesting that
DNMT1 was overexpressed and was responsible for the silencing of
miR-148a in HCC. This study is the first to reveal that DNMT1 plays
a critical role in regulating miR-148a expression by controlling
the methylation level of CpG islands in HCC. The regulation of
miR-148a by DNMT1 explains why miR-148a is upregulated after
treatment with 5-aza-2-dC or DNMT1 knockdown and suggests an
important functional link between DNMT1 and miR-148a.
DNMT1 is a methyltransferase that maintains
methylation patterns. Our previous research indicated that
DNMT1-mediated PTEN hypermethylation confers hepatic stellate cell
activation and liver fibrogenesis in rats (32). Other studies have shown that DNMT1
overexpression contributes to gene promoter hypermethylation and is
associated with the malignant potential and poor prognosis of human
cancers (17,18). Additionally, Huang et al
(33) found that the
downregulation of miRNA-152 could induce aberrant DNA methylation
in hepatitis B virus-related HCC by targeting DNMT1. In this study,
we found that the DNMT1 mRNA and protein levels were repressed
after the restoration of miR-148a expression, suggesting that DNMT1
might be one of the targets of miR-148a. This hypothesis was also
confirmed using the TargetScan program and luciferase reporter
assays. Recent studies in both human cholangiocarcinoma (34) and systemic lupus erythematosus
(SLE) (20) demonstrated that
DNMT1 was directly regulated by miR-148a. However, these studies
drew different conclusions regarding the way in which miR-148a
targets DNMT1; the recognition region was thought to be in either
the 3′-UTR (34) or the coding
region (20).
Taken together, the silencing of miR-148a and the
overexpression of DNMT1 may result in a regulatory feedback loop in
HCC. On the one hand, overexpression of DNMT1 leads to the
hypermethylation of the miR-148a promoter region, thus causing
miR-148a silencing; on the other hand, restoration of miR-148a
induces the downregulation of DNMT1. However, in hepatocellular
carcinogenesis, the silencing of miR-148a caused by
hypermethylation reduces its suppression of DNMT1, resulting in
higher DNMT1 expression and the hypermethylation of the miR-148a
gene. On the basis of the above results, we came to the following
conclusions. First, DNA methylation is involved in the deregulation
of miR-148a in HCC. Second, there is a regulatory feedback loop
between miR-148a and DNMT1. Third, miR-148a can inhibit cell
proliferation via cell cycle arrest but does not influence cell
apoptosis. Our study provides new insight into the molecular
mechanisms of HCC development and presents potential strategies for
HCC diagnostics and treatment in the future.
Very recently, Gailhouste et al (14) found that miR-148a expression was
frequently downregulated in biopsy samples from HCC patients as
well as in mouse and human HCC cell lines; however, these authors
did not focus on the relationship between miR-148a and systemic DNA
methylation. Therefore, the impact of this relationship on HCC
cells remained unknown until our study was performed. Recent
studies have shown that miR-148a suppressed the EMT by targeting
ROCK1 in non-small cell lung cancer cells (35) and regulated immune homeostasis by
targeting CaMKIIa (36). It is
likely that miR-148a may have different functional targets in
different types of cancers; this hypothesis requires further
investigation. However, a miR-148a knockout mouse has not yet been
generated, and the current knowledge of miR-148a functions in HCC
is still very limited. Further study using knockout and transgenic
animal models will aid in the identification of the in vivo
functions of miR-148a in HCC.
Due to the hypermethylation of its CpG island,
miR-148a undergoes methylation-mediated silencing in HCC cell
lines. Additionally, DNMT1 is aberrantly upregulated in HCC cell
lines, and its overexpression is responsible for hypermethylation
of the miR-148a promoter. Interestingly, the expression of DNMT1,
which is a target of miR-148a, is inversely correlated with the
expression of miR-148a in HCC cells. These results led us to
propose a negative feedback regulatory loop between miR-148a and
DNMT1 in HCC. Importantly, the overexpression of miR-148a
significantly inhibited HCC cell proliferation and cell cycle
progression. Our results suggest the existence of a novel
miR-148a-DNMT1 regulatory circuit and indicate that miR-148a may
act as a tumor suppressor in hepatocellular carcinogenesis.
Abbreviations:
|
HCC
|
hepatocellular carcinoma
|
|
miRNA
|
microRNA
|
|
DNMT1
|
DNA methyltransferase 1
|
|
RT-PCR
|
semi-quantitative reverse
transcription-polymerase chain reaction
|
|
RT-qPCR
|
quantitative real-time PCR
|
|
PBS
|
phosphate-buffered saline
|
|
SDS
|
sodium dodecyl sulfate
|
|
MSP
|
methylation-specific PCR
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,4-diphenyl-tetrazolium bromide
assay
|
|
DMSO
|
dimethyl sulfoxide
|
|
OD
|
optical density
|
Acknowledgements
This project was supported by the
National Science Foundation of China (nos. 81072686, 81273526 and
81202978).
References
|
1.
|
Farazi PA and DePinho RA: Hepatocellular
carcinoma pathogenesis: from genes to environment. Nat Rev Cancer.
6:674–687. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
2.
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Aravalli RN, Steer CJ and Cressman EN:
Molecular mechanisms of hepatocellular carcinoma. Hepatology.
48:2047–2063. 2008. View Article : Google Scholar
|
|
4.
|
Zhang B, Wang Q and Pan X: MicroRNAs and
their regulatory roles in animals and plants. J Cell Physiol.
210:279–289. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Calin GA and Croce CM: Chromosomal
rearrangements and microRNAs: a new cancer link with clinical
implications. J Clin Invest. 117:2059–2066. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Braconi C and Patel T: MicroRNA expression
profiling: a molecular tool for defining the phenotype of
hepatocellular tumors. Hepatology. 47:1807–1809. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Ladeiro Y, Couchy G, Balabaud C, et al:
MicroRNA profiling in hepatocellular tumors is associated with
clinical features and oncogene/tumor suppressor gene mutations.
Hepatology. 47:1955–1963. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Mott JL: MicroRNAs involved in tumor
suppressor and oncogene pathways: implications for hepatobiliary
neoplasia. Hepatology. 50:630–637. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Filipowicz W, Bhattacharyya SN and
Sonenberg N: Mechanisms of post-transcriptional regulation by
microRNAs: are the answers in sight? Nat Rev Genet. 9:102–114.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Qiu L, Fan H, Jin W, et al:
miR-122-induced down-regulation of HO-1 negatively affects
miR-122-mediated suppression of HBV. Biochem Biophys Res Commun.
398:771–777. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Wolffe AP and Matzke MA: Epigenetics:
regulation through repression. Science. 286:481–486. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Hanoun N, Delpu Y, Suriawinata AA, et al:
The silencing of microRNA 148a production by DNA hypermethylation
is an early event in pancreatic carcinogenesis. Clin Chem.
56:1107–1118. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Gailhouste L, Gomez-Santos L, Hagiwara K,
et al: miR-148a plays a pivotal role in the liver by promoting the
hepatospecific phenotype and suppressing the invasiveness of
transformed cells. Hepatology. 58:1153–1165. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Wilson AS, Power BE and Molloy PL: DNA
hypomethylation and human diseases. Biochim Biophys Acta.
1775:138–162. 2007.PubMed/NCBI
|
|
16.
|
Xu Q, Jiang Y, Yin Y, et al: A regulatory
circuit of miR-148a/152 and DNMT1 in modulating cell transformation
and tumor angiogenesis through IGF-IR and IRS1. J Mol Cell Biol.
5:3–13. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Bernardino J, Roux C, Almeida A, et al:
DNA hypomethylation in breast cancer: an independent parameter of
tumor progression? Cancer Genet Cytogenet. 97:83–89. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Soares J, Pinto AE, Cunha CV, et al:
Global DNA hypomethylation in breast carcinoma: correlation with
prognostic factors and tumor progression. Cancer. 85:112–118. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Zhu A, Xia J, Zuo J, et al: MicroRNA-148a
is silenced by hypermethylation and interacts with DNA
methyltransferase 1 in gastric cancer. Med Oncol. 29:2701–2709.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Pan W, Zhu S, Yuan M, et al: MicroRNA-21
and microRNA-148a contribute to DNA hypomethylation in lupus
CD4+ T cells by directly and indirectly targeting DNA
methyltransferase 1. J Immunol. 184:6773–6781. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Egger G, Liang G, Aparicio A and Jones PA:
Epigenetics in human disease and prospects for epigenetic therapy.
Nature. 429:457–463. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Balaguer F, Link A, Lozano JJ, et al:
Epigenetic silencing of miR-137 is an early event in colorectal
carcinogenesis. Cancer Res. 70:6609–6618. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Thorgeirsson SS and Grisham JW: Molecular
pathogenesis of human hepatocellular carcinoma. Nat Genet.
31:339–346. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Zhang JP, Zeng C, Xu L, Gong J, Fang JH
and Zhuang SM: MicroRNA-148a suppresses the epithelial-mesenchymal
transition and metastasis of hepatoma cells by targeting Met/Snail
signaling. Oncogene. Sep 9–2013.(Epub ahead of print).
|
|
25.
|
Han H, Sun D, Li W, et al: A
c-Myc-MicroRNA functional feedback loop affects
hepatocarcinogenesis. Hepatology. 57:2378–2389. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Yan H, Dong X, Zhong X, et al: Inhibitions
of epithelial to mesenchymal transition and cancer stem cells-like
properties are involved in miR-148a-mediated anti-metastasis of
hepatocellular carcinoma. Mol Carcinog. Jul 17–2013.(Epub ahead of
print).
|
|
27.
|
Robertson KD, Uzvolgyi E, Liang G, et al:
The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate
mRNA expression in normal tissues and overexpression in tumors.
Nucleic Acids Res. 27:2291–2298. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Ahluwalia A, Hurteau JA, Bigsby RM and
Nephew KP: DNA methylation in ovarian cancer. II Expression of DNA
methyltransferases in ovarian cancer cell lines and normal ovarian
epithelial cells. Gynecol Oncol. 82:299–304. 2001.PubMed/NCBI
|
|
29.
|
Karpf AR and Matsui S: Genetic disruption
of cytosine DNA methyltransferase enzymes induces chromosomal
instability in human cancer cells. Cancer Res. 65:8635–8639. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Mizuno S, Chijiwa T, Okamura T, et al:
Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal
hematopoiesis and in acute and chronic myelogenous leukemia. Blood.
97:1172–1179. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Roll JD, Rivenbark AG, Jones WD and
Coleman WB: DNMT3b overexpression contributes to a hypermethylator
phenotype in human breast cancer cell lines. Mol Cancer. 7:152008.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Bian EB, Huang C, Ma TT, et al:
DNMT1-mediated PTEN hypermethylation confers hepatic stellate cell
activation and liver fibrogenesis in rats. Toxicol Appl Pharmacol.
264:13–22. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Huang J, Wang Y, Guo Y and Sun S:
Down-regulated microRNA-152 induces aberrant DNA methylation in
hepatitis B virus-related hepatocellular carcinoma by targeting DNA
methyltransferase 1. Hepatology. 52:60–70. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Braconi C, Huang N and Patel T:
MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor
suppressor gene expression by interleukin-6 in human malignant
cholangiocytes. Hepatology. 51:881–890. 2010.PubMed/NCBI
|
|
35.
|
Li J, Song Y, Wang Y, Luo J and Yu W:
MicroRNA-148a suppresses epithelial-to-mesenchymal transition by
targeting ROCK1 in non-small cell lung cancer cells. Mol Cell
Biochem. 380:277–282. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Liu X, Zhan Z, Xu L, et al:
MicroRNA-148/152 impair innate response and antigen presentation of
TLR-triggered dendritic cells by targeting CaMKIIalpha. J Immunol.
185:7244–7251. 2010. View Article : Google Scholar : PubMed/NCBI
|