Contents
Introduction
Expression, structure and characteristics of PTEN
and p53
Protein interaction and functional interplay between
PTEN and p53
Involvement of PTEN and p53 tumor suppressors in
hereditary cancer
Perspective
Introduction
PTEN (phosphatase and tensin homolog deleted
in chromosome 10) is a tumor suppressor gene that is deleted or
mutated in a variety of human cancers (1,2).
Germ line mutations of PTEN are also the cause of
PTEN hamartoma tumor syndromes (Cowden syndrome,
Bannayan-Riley-Ruvalcaba syndrome, PTEN-related Proteus
syndrome, Proteus-like syndrome) with increased risk for the
development of cancers (3). Cowden
syndrome is a rare, autosomal dominant, familial cancer syndrome
characterised by hamartomas, acral keratosis, multiple smooth
facial papules, and multiple oral papillomas (4). In contrast, Bannayan-Riley-Ruvalcaba
syndrome is characterized by lipomatosis, macrocephaly,
hemangiomatosis (5). Loss of
heterozygosity (LOH) studies suggest that PTEN may play its most
important role in advanced cancers of particular tissue (6). Alterations of PTEN in tumors
are associated with a poor prognosis (7). Germinal mutations of the p53
gene constitute an etiological genetic base of Li-Fraumeni
syndrome, which is a rare genetically and clinically heterogeneous
autosomal dominant inherited cancer disorder, characterized by a
specific range of tumors observed at an early age, in particular a
predominance of bone and soft tissue sarcomas and breast cancer
(8). Generally, cells with
dysfunctional p53 are predisposed to development of cancer
phenotype. Of importance, it is mutated frequently in the common
human malignancies of the breast and colon rectum and also in other
significant cancers such as glioblastoma with less frequency
(9).
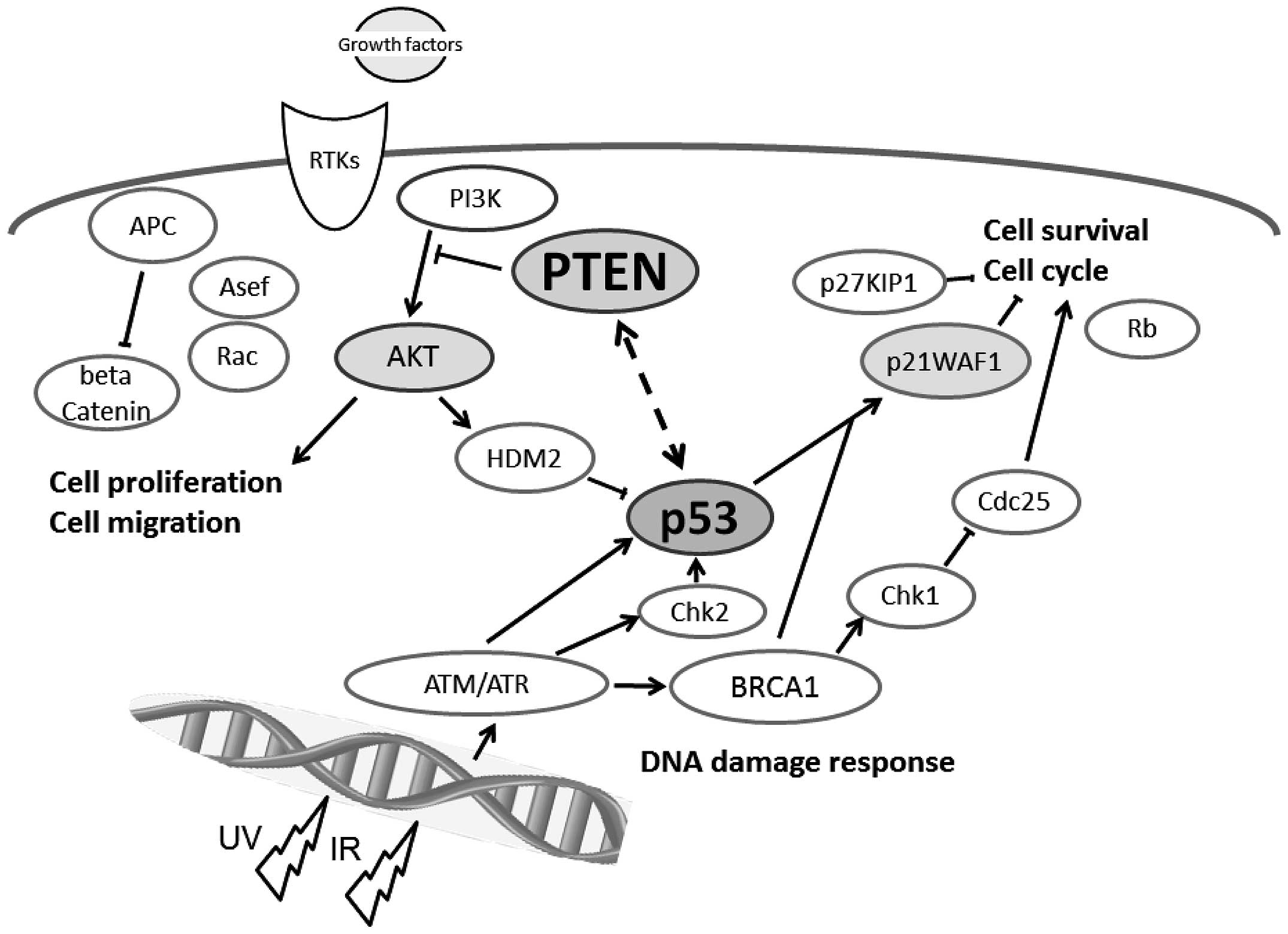
The PTEN and p53 tumor suppressors are
among the most commonly inactivated or mutated genes in human
cancer (10). The PTEN has been
shown to be involved in a complex network of interactions with p53
(Fig. 1). Although they are
functionally distinct, reciprocal cooperation has been proposed, as
PTEN is thought to regulate p53 stability, and p53 to enhance
PTEN transcription. Once PTEN is lost, however, the p53
pathway is strongly activated (11,12).
Furthermore, an absence of PTEN cooperates with an absence of p53
to promote cancer (13). The
inactivation of tumor suppression may be caused by lack of key
interaction partners. Recent studies have revealed a functional
ubiquitin ligase for tumor suppressors playing a pivotal role in
tumor cell survival (14,15). Mutations found in genes such as p53
and PTEN have emerged as high penetrance susceptibility genes and
are clinically relevant for determination of cancer risk. In this
review, we summarize the current research and our view of how and
when PTEN and p53 with their partners transduce signals downstream
and the implications for cancer-associated biology.
Expression, structure and characteristics of
PTEN and p53
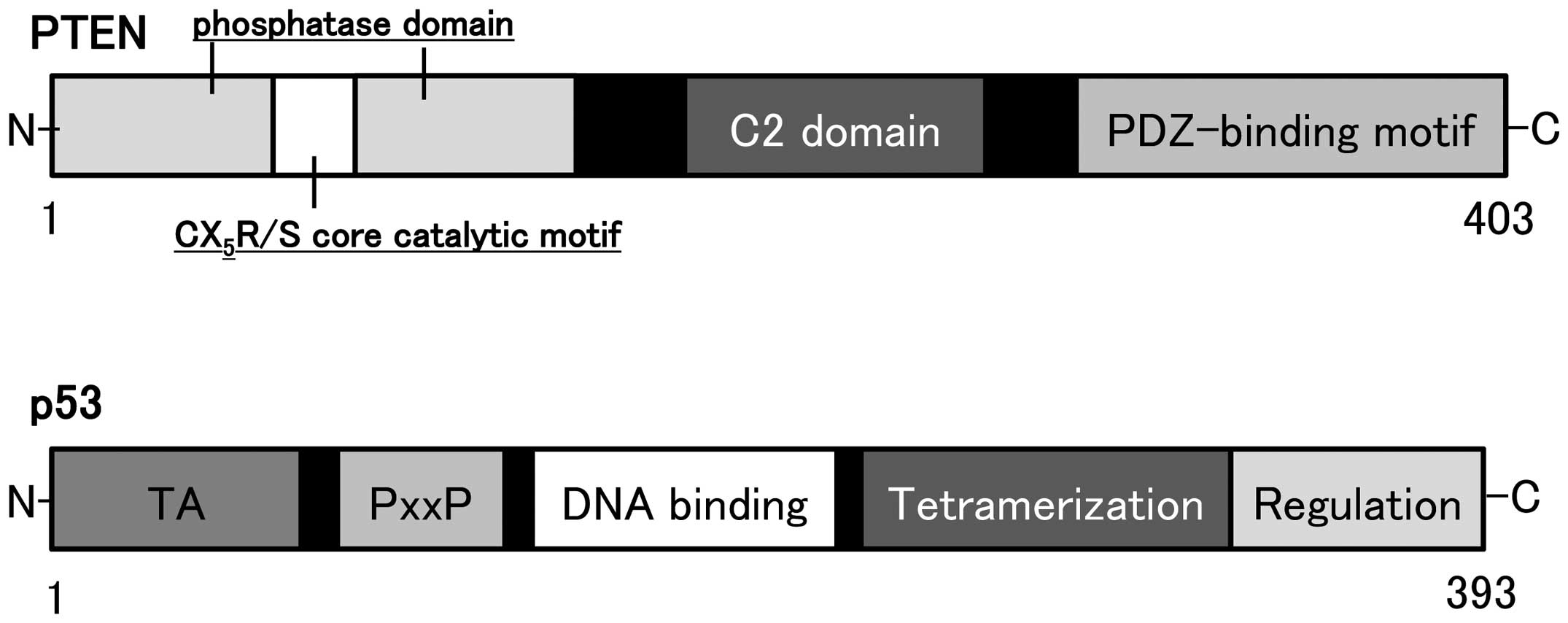
The human genomic PTEN locus consists of 9
exons on chromosome 10q23.3 encoding a 5.5 kb mRNA that specifies a
403 amino acid open reading frame (16,17).
The translation product is a 53 kDa protein with homology to tensin
and protein tyrosine phosphatases. PTEN is ubiquitously
expressed throughout early embryogenesis in mammals (18). PTEN gene can be upregulated
by early growth regulated transcription factor 1, peroxisome
proliferator activated receptor γ (PPARγ), p53, and activating
transcription factor 2 (ATF2) (19–22),
while transforming growth factor (TGF)-β, nuclear factor κB
(NF-κB), and Jun negatively regulate PTEN expression
(23–25). Of note, rosemary extract represses
PTEN expression in K562 leukemic culture cells (26). PTEN activity can be regulated by
posttranslational regulation including phosphorylation,
acetylation, and oxidation. Methylation of the PTEN promoter
can result in transcriptional silencing of the PTEN gene
(27). Schematic structure of the
predicted PTEN protein is shown in Fig. 2. PTEN negatively regulates the
activity of PI3K/AKT signaling through converting
phosphatidylinositol 3,4,5-triphosphate (PIP3) into
phosphatidylinositol 4,5- bisphosphate (PIP2). The PIP3 is the
principal second messenger of the PI3K pathway that mediates
receptor tyrosine kinase (RTK) signaling to the survival kinase
AKT. Activated AKT transfers a phosphate group to target proteins
involved in cell survival, cell cycling, proliferation, and
migration, which also are all critical for tumor development
(28,29).
PTEN acts as regulator of maintaining basal levels
of PIP3 below a threshold for the signaling activation. PTEN
protein consists of N-terminal phosphatase, and C-terminal C2, and
PDZ (PSD-95, DLG1, and ZO-1) binding domains. The PTEN CX5R(S/T)
motif resides within an active site that surrounds the catalytic
signature with three basic residues, which are critical for PTEN
lipid phosphatase activity. The structure endows PTEN with its
preference for acidic phospholipid substrates such as PIP3.
Overexpression of PTEN induces growth suppression by promoting cell
cycle arrest, which requires lipid phosphatase activity (30,31).
Overexpression of PTEN also correlates with decreased levels and
nuclear localization of cyclin D1 (32), a key cell cycle molecule regulated
by AKT. One mechanism by which PTEN induces cell cycle arrest is
the regulation of AKT activity such that levels of the cell cycle
inhibitor p27kip1 are increased (33). However, despite the main role of
PTEN as a negative regulator of the PI3K pathway, studies report
various tumor suppressive activities for PTEN that are exerted from
within the nucleus, where catalysis of PIP3 does not seem to
represent a dominant function of this enzyme (34). The nuclear PTEN activities may
include the regulation of genomic stability, cell cycle
progression, and gene expression.
The p53 gene, located on chromosome 17p 13.1
and encoding a nuclear 393-amino acid protein, acts to control cell
growth and apoptosis (Fig. 2). The
p53 protein is a transcription factor which is able to induce G1
arrest of the cell cycle by transactivating several downstream
genes. Inactivation of p53 gene is a common event in the
development of most types of cancer. The importance of p53 as an
inherited cancer susceptibility gene has been demonstrated in
Li-Fraumeni syndrome (35).
However, there are still a significant number of Li-Fraumeni
families for which no underlying genetic determinant has been
identified. It would be beneficial to understand the alterations of
genes or additional components involved in DNA damage recognition,
DNA repair, and/or cell cycle checkpoint pathways responsible for
the specific phenotype.
Protein interaction and functional interplay
between PTEN and p53
PTEN and p53 is known to interact and regulate each
other at the transcription as well as protein level, which could be
at the important control machinery for switching between survival
and death. This cross talk is frequently a combination of
reciprocally antagonistic pathways, which often involves another
tumor suppressor gene MDM2, and may serve as an added regulatory
effect on the expression of key genes involved in cancer. It has
also been revealed that PTEN regulates p53 stability and in turn
regulates its own transcriptional activity.
At transcription level
The PTEN and p53 complex enhances p53 DNA binding
and transcriptional activity (36). An important p53 function is to act
as a transcription factor by binding to the specific DNA consensus
sequence in responsive genes, which may increase the synthesis of
p21waf1 that is an important protein involved in cell cycle arrest
(37). In addition, one of
transcriptional targets of p53 is PTEN. One way by which p53
inhibits production of PIP3 indirectly is by inducing the
expression of PTEN (38). Under
hypoxic conditions PTEN and p53 form a complex in the nucleus and
induce strong expression of the tumor suppressor Maspin (39). Loss of PTEN attenuates the
induction of Maspin even in the presence of wild-type p53. The
integration of PTEN and p53 into a common pathway for the induction
of Maspin may constitute a tumor suppressor network (40).
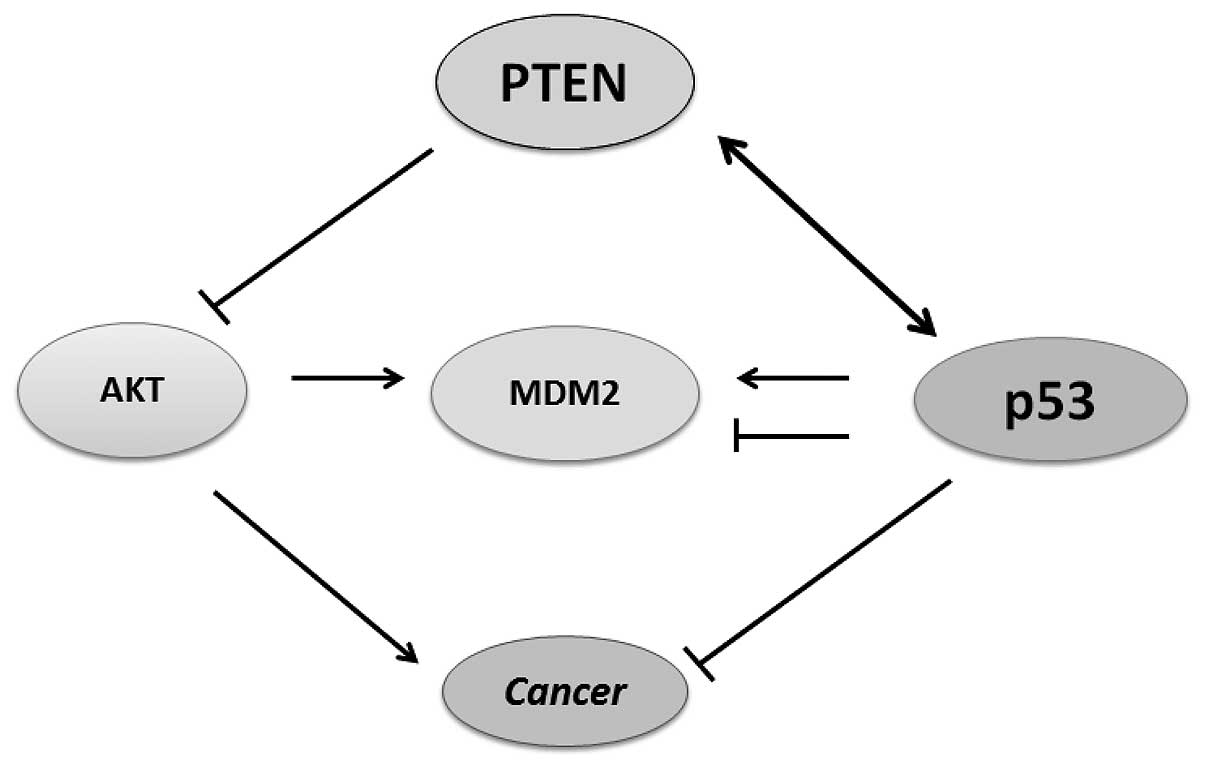
MDM2 is an oncoprotein that controls tumorigenesis,
its mRNA level is transcriptionally regulated by p53 in response to
DNA damage such as oxidative stress (41). The MDM2 protein and subcellular
localization are post-translationally modulated by AKT (42). PTEN inhibits PI3K/AKT signaling
that promotes translocation of MDM2 into the nucleus. In addition,
PTEN modulates MDM2 transcription and isoform selection by
negatively regulating its promoter (43). In PTEN-null cells, MDM2
promoter activity is upregulated, resulting in increased
MDM2 expression. Furthermore, PTEN controls MDM2
promoter activity through its lipid phosphatase activity,
independent of p53 (36). Although
another transcription factors such as AP-1 are able to modulate
MDM2 transcription, they have been characterized to work
through the p53 responsible promoter (44). MDM2 is a key regulator of p53. It
regulates the activity of p53 protein by blocking its
transcriptional activity, exporting nuclear p53 protein into the
cytoplasm, and/or by promoting the degradation of the p53 protein.
PTEN upregulates the p53 level as well as its activity by
downregulating MDM2 transcription and p53 binding activity
(45). However, in the absence of
p53, PTEN may have a role inhibiting MDM2-mediated carcinogenesis
through regulation of MDM2 transcription as well as the
isoform selection. MDM2 is degraded, when restricted to the
cytoplasm. The ability of PTEN to inhibit the nuclear entry of MDM2
increases the cellular content and transactivation of the p53 to
promote the induction of responsive genes such as p21
(46).
Protein modification and interaction
p53 and AKT influence the process of apoptosis in
opposite ways. The AKT promotes cell survival by suppressing
pro-apoptotic proteins such as Bad through phosphorylation
(47). There is cross talk between
p53 and AKT involving gene transcription as well as
posttranslational protein modifications. p53 inhibits PIP3
production indirectly by repressing the catalytic subunit of PI3K.
A subsequent p53-induced expression of PTEN causes the p53-PTEN
interaction, which then suppresses the cell survival machinery of
AKT pathway. AKT phosphorylates MDM2 to translocate into the
nucleus (48). In addition, PTEN
physically associates with endogenous p53 and regulates the
transcriptional activity of p53 by modulating its DNA binding
(36). PTEN is required for the
maintenance of p53 acetylation, which is also required for target
gene transcription (49).
Growth factor-activated AKT signaling promotes
progression of cell cycles by acting on downstream factors involved
in controlling the G1/S and/or G2/M transitions. Several studies
have also implicated AKT in modulating DNA damage responses and
genome stability (50). AKT
therefore modifies downstream signaling in complex ways. In
addition, PTEN also plays a critical role in DNA damage repair and
DNA damage response through its interaction with p53 pathways in an
AKT-independent manner (51).
Furthermore, nuclear PTEN is sufficient to reduce tumor progression
in a p53-dependent manner. It has also been suggested that nuclear
PTEN plays a unique role to protect cells upon oxidative damage and
to regulate carcinogenesis (52),
thus, the PTEN-p53-MDM2-AKT loop becomes dominant (Fig. 3).
Protein degradation
One aspect of the PTEN tumor suppressor signaling is
achieved through stabilization of the p53 protein. PTEN has been
shown to physically interact with p53 and prevent its degradation
by excluding a portion of p53 protein from the p53 and MDM2
complex. Evidence indicates the existence of a link between PTEN
and p53 functions through the control of the phosphorylation state
of MDM2. AKT mediates MDM2 nuclear translocation by its
phosphorylation. In the nucleus, MDM2 negatively regulates p53 by
binding and signaling for destabilization (53). Therefore, attenuation of the AKT
pathway by PTEN protects p53 from MDM2 mediated degradation and
inactivation. The p53 and MDM2 complex is transported from the
nucleus into the cytoplasm where MDM2 serves as an E3 ubiquitin
ligase (54). Consequently, p53
and MDM2 form a regulatory feedback loop in which p53 positively
regulates MDM2 expression, whereas MDM2 negatively regulates the
level of p53 protein. Thus, PTEN may protect p53 from MDM2-mediated
degradation, whereas p53 can enhance the transcription of PTEN
(Fig. 3). Therefore, inactivation
of either gene result in lower protein levels of the other
gene.
The instability of PTEN correlated with its missense
mutations has been shown to involve protein interactions. PTEN may
be regulated by ubiquitin-mediated proteasomal degradation, a
common mechanism to control protein levels. In cells, a ubiquitin
ligase NEDD4-1 negatively regulates PTEN stability by catalyzing
PTEN ubiquitination (55). As
truncation or mutation of the C2 domain of PTEN makes the protein
unstable and accelerates protein degradation, the C2 domain seems
to regulate the phosphatase domain through maintaining the protein
stability (56). In addition to
the C2 domain, the C-terminus of PTEN contains two PEST (proline,
glutamic acid, serine and threonine) sequences involved in
ubiquitin protein degradation pathway. Treatment of cells with
proteasome inhibitors can cause an increase of PTEN protein level
(57,58). Several NEDD4-like E3 also regulate
p53. Interestingly, multiple NEDD4-like E3 show ligase independent
function. Furthermore, most of NEDD4-like E3 are frequently
regulated by phosphorylation, ubiquitination, translocation, and
transcription in cancer cells. NEDD4-like ubiquitin protein
ligase-1 (NEDL1) is a type E3 ubiquitin protein ligase. Functional
interaction of NEDL1 with p53 might contribute to the induction of
apoptosis in cancer cells bearing wild-type p53 (59,60).
Casein kinase II-mediated phosphorylation stabilizes
the PTEN protein by preventing its proteasomal degradation, while
keeping it in an inactive state. Inhibition of the PTEN
phosphorylation by the Casein kinase II results in increased PTEN
activity and a corresponding reduction in AKT activation (61). Importantly, inhibitors of Casein
kinase II also activate p53 function in wild-type, but not in p53
mutant cells. Activation of p53 function is involved in increased
DNA-binding ability, transcriptional activation, increased
expression of p53 target genes, associated with cell cycle
progression and apoptosis. In addition, inhibitors of Casein kinase
II increase senescence p53-dependently (62), thus, Casein kinase II may control
the PTEN and the p53 balance.
Involvement of PTEN and p53 tumor
suppressors in hereditary cancer
The PTEN gene is found in 80% of Cowden
syndrome patients (63,64). Mutations of the PTEN gene
are thus frequent in hereditary cancer syndromes, and are found in
all exons except 1, 4, and 9 in Cowden syndrome (63–65).
These mutations target the PTEN gene not only at its coding
regions, but also at exon-intron boundaries and promoter regions,
most of which have a major impact in the PTEN protein levels
of expression, being causative of PTEN functional deficiency and
considered as pathogenic (66,67).
It has been suggested that the differential expression of the
PTEN gene correlates with the different phenotypes.
Mutations targeting the PTEN coding region include
frame-shift and nonsense mutations, which also generate unstable
truncated PTEN proteins, as well as missense mutations that result
in individual amino acid changes. Functional analyses of these
missense mutations have revealed that the amino acid substitutions
generate PTEN proteins with impaired intrinsic catalytic activity
and/or protein stability (68–70).
Some examples have been described of missense PTEN
mutations that do not affect the intrinsic catalysis nor the
stability of the protein, but rather impair essential regulatory
PTEN properties, such as binding to membranes or nuclear entry
(71). PTEN hamartoma tumor
syndrome is the term used to describe Cowden syndrome (65). Mutations in PTEN together
with p53 make the tumor suppressor genes one of the most frequently
affected in human malignancies in solid tumors (72–74).
PTEN is considered haplo-insufficient to prevent certain
malignancies, suggesting that dosage is important for its function,
as it is influenced by a given point mutation in the catalytic
activity of the enzyme (75).
Tumors initiated by a subtle downregulation of a tumor suppressor
gene can progress in the absence of LOH of the wild-type allele
(76). These regulatory cues are
presumed to play a key role in tumorigenesis through the alteration
of the appropriate levels, localization, and activity of PTEN.
However, it has been shown that PTEN germ-line SNPs are
unlikely to have an important role in hereditary prostate
susceptibility (77). The lifetime
risk of breast cancer for Cowden syndrome patients is 81% (78), and bilateral risk-reducing
mastectomy with immediate reconstruction is performed to eliminate
further risk of breast cancer (79).
Perspective
Advances in the field of hereditary cancer genetics
have led to an improved understanding of detection and prevention
strategies. Germline genetic testing for mutations in PTEN and p53
allows for the identification of individuals at increased risk for
breast, ovarian and other cancers. PTEN and p53 may be regulated
and interact with each other at multiple levels including
transcription, protein modulation, and protein stability (80,81).
Understanding the regulation is crucial for the effective design of
novel cancer therapeutics. In addition, it is important to
investigate the functional linkage between PTEN, p53 and MDM2
isoforms in human cancer samples, and elucidation of
interaction-specific functions may provide insight into regulatory
aspects of these tumor suppressors as well as opportunities for
therapeutic intervention. Further mechanistic studies are needed in
order to understand the precise molecular mechanisms for the
effective treatment of both cancer and other diseases with their
alteration.
Abbreviations:
|
ATF2
|
activating transcription factor 2
|
|
NF-κB
|
nuclear factor κB
|
|
MDM2
|
murine double minute 2
|
|
PTEN
|
phosphatase and tensin homologue
deleted on chromosome 10
|
|
PIP3
|
phosphatidylinositol 3,4,5-tri
phosphate
|
|
PIP2
|
phosphatidylinositol
4,5-bisphosphate
|
|
PI3K
|
phosphoinositide-3 kinase
|
|
PTP
|
protein tyrosine phosphatase
|
|
PPAR
|
peroxisome proliferator-activated
receptor
|
|
TGF
|
transforming growth factor
|
Acknowledgements
This work was supported by
grants-in-aid from the Ministry of Education, Culture, Sports,
Science and Technology in Japan. In addition, this work was
supported in part by the grant from SHIN-EI Pharmaceutical Co.,
Ltd., and the grant from Nakagawa Masashichi Shoten Co., Ltd.
References
|
1.
|
Merritt MA and Cramer DW: Molecular
pathogenesis of endometrial and ovarian cancer. Cancer Biomark.
9:287–305. 2010.PubMed/NCBI
|
|
2.
|
Song MS, Salmena L and Pandolfi PP: The
functions and regulation of the PTEN tumour suppressor. Nat Rev Mol
Cell Biol. 13:283–296. 2012.PubMed/NCBI
|
|
3.
|
Hobert JA and Eng C: PTEN hamartoma tumor
syndrome: an overview. Genet Med. 11:687–694. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Conti S, Condò M, Posar A, et al:
Phosphatase and tensin homolog (PTEN) gene mutations and autism:
literature review and a case report of a patient with Cowden
syndrome, autistic disorder, and epilepsy. J Child Neurol.
27:392–397. 2012. View Article : Google Scholar
|
|
5.
|
Litzendorf M, Hoang K and Vaccaro P:
Recurrent and extensive vascular malformations in a patient with
Bannayan-Riley-Ruvalcaba syndrome. Ann Vasc Surg.
25:1138.e15-92011. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Morgan TM, Koreckij TD and Corey E:
Targeted therapy for advanced prostate cancer: inhibition of the
PI3K/Akt/mTOR pathway. Curr Cancer Drug Targets. 9:237–249. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Knobbe CB, Merlo A and Reifenberger G:
Pten signaling in gliomas. Neuro Oncol. 4:196–211. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Li FP, Fraumeni JF Jr, Mulvihill JJ, et
al: A cancer family syndrome in twenty-four kindreds. Cancer Res.
48:5358–5362. 1988.PubMed/NCBI
|
|
9.
|
Masui K, Cloughesy TF and Mischel PS:
Review: molecular pathology in adult high-grade gliomas: from
molecular diagnostics to target therapies. Neuropathol Appl
Neurobiol. 38:271–291. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Dong JT: Prevalent mutations in prostate
cancer. J Cell Biochem. 97:433–447. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Chen Z, Trotman LC, Shaffer D, et al:
Crucial role of p53-dependent cellular senescence in suppression of
Pten-deficient tumorigenesis. Nature. 436:725–730. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Kim J, Eltoum IE, Roh M, Wang J and
Abdulkadir SA: Interactions between cells with distinct mutations
in c-MYC and Pten in prostate cancer. PLoS Genet. 5:e10005422009.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Blanco-Aparicio C, Renner O, Leal JF and
Carnero A: PTEN, more than the AKT pathway. Carcinogenesis.
28:1379–1386. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Faesen AC, Dirac AM, Shanmugham A, Ovaa H,
Perrakis A and Sixma TK: Mechanism of USP7/HAUSP activation by its
C-terminal ubiquitin-like domain and allosteric regulation by
GMP-synthetase. Mol Cell. 44:147–159. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Sacco JJ, Coulson JM, Clague MJ and Urbé
S: Emerging roles of deubiquitinases in cancer-associated pathways.
IUBMB Life. 62:140–157. 2010.
|
|
16.
|
Okumura N, Yoshida H, Kitagishi Y,
Murakami M, Nishimura Y and Matsuda S: PI3K/AKT/PTEN signaling as a
molecular target in leukemia angiogenesis. Adv Hematol.
2012.8430852012.PubMed/NCBI
|
|
17.
|
Okumura N, Yoshida H, Kitagishi Y,
Nishimura Y and Matsuda S: Alternative splicings on p53, BRCA1 and
PTEN genes involved in breast cancer. Biochem Biophys Res Commun.
413:395–399. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Croushore JA, Blasiole B, Riddle RC, et
al: Pten a and pten b genes play distinct roles in zebrafish
embryogenesis. Dev Dyn. 234:911–921. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Teresi RE, Shaiu CW, Chen CS, Chatterjee
VK, Waite KA and Eng C: Increased PTEN expression due to
transcriptional activation of PPARgamma by Lovastatin and
Rosiglitazone. Int J Cancer. 118:2390–2398. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Shen YH, Zhang L, Gan Y, et al:
Up-regulation of PTEN (phosphatase and tensin homolog deleted on
chromosome ten) mediates p38 MAPK stress signal-induced inhibition
of insulin signaling. A cross-talk between stress signaling and
insulin signaling in resistin-treated human endothelial cells. J
Biol Chem. 281:7727–7736. 2006.
|
|
21.
|
Pan L, Lu J, Wang X, et al: Histone
deacetylase inhibitor trichostatin a potentiates
doxorubicin-induced apoptosis by up-regulating PTEN expression.
Cancer. 109:1676–1688. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Lee YR, Yu HN, Noh EM, et al: Peroxisome
proliferator-activated receptor gamma and retinoic acid receptor
synergistically up-regulate the tumor suppressor PTEN in human
promyeloid leukemia cells. Int J Hematol. 85:231–237. 2007.
View Article : Google Scholar
|
|
23.
|
Yang Y, Zhou F, Fang Z, et al:
Post-transcriptional and post-translational regulation of PTEN by
transforming growth factor-beta1. Cell Biochem. 106:1102–1112.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Han S, Ritzenthaler JD, Zheng Y and Roman
J: PPARbeta/delta agonist stimulates human lung carcinoma cell
growth through inhibition of PTEN expression: the involvement of
PI3K and NF-kappaB signals. Am J Physiol Lung Cell Mol Physiol.
294:L1238–L1249. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Vasudevan KM, Burikhanov R, Goswami A and
Rangnekar VM: Suppression of PTEN expression is essential for
antiapoptosis and cellular transformation by oncogenic Ras. Cancer
Res. 67:10343–10350. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Yoshida H, Okumura N, Kitagishi Y,
Nishimura Y and Matsuda S: Ethanol extract of rosemary repressed
PTEN expression in K562 culture cells. Int J Appl Boil Pharm
Technol. 2:316–322. 2011.
|
|
27.
|
Mueller S, Phillips J, Onar-Thomas A, et
al: PTEN promoter methylation and activation of the PI3K/Akt/mTOR
pathway in pediatric gliomas and influence on clinical outcome.
Neuro Oncol. 14:1146–1152. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Faurschou A, Gniadecki R, Calay D and Wulf
HC: TNF-alpha impairs the S-G2/M cell cycle checkpoint and
cyclobutane pyrimidine dimer repair in premalignant skin cells:
role of the PI3K-Akt pathway. J Invest Dermatol. 128:2069–2077.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Chen Y, Wang BC and Xiao Y: PI3K: a
potential therapeutic target for cancer. J Cell Physiol.
227:2818–2821. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Choi Y, Zhang J, Murga C, et al: PTEN, but
not SHIP and SHIP2, suppresses the PI3K/Akt pathway and induces
growth inhibition and apoptosis of myeloma cells. Oncogene.
21:5289–5300. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Petrella BL and Brinckerhoff CE: PTEN
suppression of YY1 induces HIF-2 activity in von-Hippel-Lindau-null
renal-cell carcinoma. Cancer Biol Ther. 8:1389–1401. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Yamamoto M, Tamakawa S, Yoshie M, Yaginuma
Y and Ogawa K: Neoplastic hepatocyte growth associated with cyclin
D1 redistribution from the cytoplasm to the nucleus in mouse
hepatocarcinogenesis. Mol Carcinog. 45:901–913. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Andrés-Pons A, Gil A, Oliver MD, Sotelo NS
and Pulido R: Cytoplasmic p27Kip1 counteracts the pro-apoptotic
function of the open conformation of PTEN by retention and
destabilization of PTEN outside of the nucleus. Cell Signal.
24:577–587. 2012.PubMed/NCBI
|
|
34.
|
Planchon SM, Waite KA and Eng C: The
nuclear affairs of PTEN. J Cell Sci. 121:249–253. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Palmero EI, Achatz MI, Ashton-Prolla P,
Olivier M and Hainaut P: Tumor protein 53 mutations and inherited
cancer: beyond Li-Fraumeni syndrome. Curr Opin Oncol. 22:64–69.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Freeman DJ, Li AG, Wei G, et al: PTEN
tumor suppressor regulates p53 protein levels and activity through
phosphatase-dependent and -independent mechanisms. Cancer Cell.
3:117–130. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Lo PK, Lee JS and Sukumar S: The
p53-p21WAF1 checkpoint pathway plays a protective role in
preventing DNA rereplication induced by abrogation of FOXF1
function. Cell Signal. 24:316–324. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Puszyński K, Hat B and Lipniacki T:
Oscillations and bistability in the stochastic model of p53
regulation. J Theor Biol. 254:452–465. 2008.PubMed/NCBI
|
|
39.
|
Eitel JA, Bijangi-Vishehsaraei K,
Saadatzadeh MR, et al: EN and p53 are required for hypoxia induced
expression of maspin in glioblastoma cells. Cell Cycle. 8:896–901.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Zhang M: PTEN in action: coordinating with
p53 to regulate maspin gene expression. Cell Cycle. 8:1112–1113.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Wang X and Jiang X: Mdm2 and MdmX partner
to regulate p53. FEBS Lett. 586:1390–1396. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42.
|
Levav-Cohen Y, Haupt S and Haupt Y: Mdm2
in growth signaling and cancer. Growth Factors. 23:183–192. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Mayo LD and Donner DB: The PTEN, Mdm2, p53
tumor suppressor-oncoprotein network. Trends Biochem Sci.
27:462–467. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Kirch HC, Flaswinkel S, Rumpf H, Brockmann
D and Esche H: Expression of human p53 requires synergistic
activation of transcription from the p53 promoter by AP-1,
NF-kappaB and Myc/Max. Oncogene. 18:2728–2738. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45.
|
Zheng T, Meng X, Wang J, et al: PTEN- and
p53-mediated apoptosis and cell cycle arrest by FTY720 in gastric
cancer cells and nude mice. J Cell Biochem. 111:218–228. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Mayo LD, Dixon JE, Durden DL, Tonks NK and
Donner DB: PTEN protects p53 from Mdm2 and sensitizes cancer cells
to chemotherapy. J Biol Chem. 277:5484–5489. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Sen P, Mukherjee S, Ray D and Raha S:
Involvement of the Akt/PKB signaling pathway with disease
processes. Mol Cell Biochem. 253:241–246. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Zhou BP, Liao Y, Xia W, Zou Y, Spohn B and
Hung MC: HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2
phosphorylation. Nat Cell Biol. 3:973–982. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Li AG, Piluso LG, Cai X, Wei G, Sellers WR
and Liu X: Mechanistic insights into maintenance of high p53
acetylation by PTEN. Mol Cell. 23:575–587. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Quevedo C, Kaplan DR and Derry WB: AKT-1
regulates DNA-damage-induced germline apoptosis in C.
elegans. Curr Biol. 17:286–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51.
|
Ming M and He YY: PTEN in DNA damage
repair. Cancer Lett. 319:125–129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52.
|
Bonavida B and Baritaki S: The novel role
of Yin Yang 1 in the regulation of epithelial to mesenchymal
transition in cancer via the dysregulated NF-κB/Snail/YY1/RKIP/PTEN
circuitry. Crit Rev Oncog. 16:211–226. 2011.
|
|
53.
|
Vu BT and Vassilev L: Small-molecule
inhibitors of the p53-MDM2 interaction. Curr Top Microbiol Immunol.
348:151–172. 2011.PubMed/NCBI
|
|
54.
|
Bixby D, Kujawski L, Wang S and Malek SN:
The pre-clinical development of MDM2 inhibitors in chronic
lymphocytic leukemia uncovers a central role for p53 status in
sensitivity to MDM2 inhibitor-mediated apoptosis. Cell Cycle.
7:971–979. 2008. View Article : Google Scholar
|
|
55.
|
Amodio N, Scrima M, Palaia L, et al:
Oncogenic role of the E3 ubiquitin ligase NEDD4-1, a PTEN negative
regulator, in non-small-cell lung carcinomas. Am J Pathol.
177:2622–2634. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56.
|
Valiente M, Andrés-Pons A, Gomar B, et al:
Binding of PTEN to specific PDZ domains contributes to PTEN protein
stability and phosphorylation by microtubule-associated
serine/threonine kinases. J Biol Chem. 280:28936–28943. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Torres J and Pulido R: The tumor
suppressor PTEN is phosphorylated by the protein kinase CK2 at its
C terminus. Implications for PTEN stability to proteasome-mediated
degradation. J Biol Chem. 276:993–998. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
58.
|
Wu W, Wang X, Zhang W, et al: Zinc-induced
PTEN protein degradation through the proteasome pathway in human
airway epithelial cells. J Biol Chem. 278:28258–28263. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
59.
|
Li Y, Ozaki T, Kikuchi H, Yamamoto H,
Ohira M and Nakagawara A: A novel HECT-type E3 ubiquitin protein
ligase NEDL1 enhances the p53-mediated apoptotic cell death in its
catalytic activity-independent manner. Oncogene. 27:3700–3709.
2008. View Article : Google Scholar
|
|
60.
|
Shinada K, Tsukiyama T, Sho T, Okumura F,
Asaka M and Hatakeyama S: RNF43 interacts with NEDL1 and regulates
p53-mediated transcription. Biochem Biophys Res Commun.
404:143–147. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
61.
|
Barata JT: The impact of PTEN regulation
by CK2 on PI3K-dependent signaling and leukemia cell survival. Adv
Enzyme Regul. 51:37–49. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Kang JY, Kim JJ, Jang SY and Bae YS: The
p53-p21(Cip1/WAF1) pathway is necessary for cellular senescence
induced by the inhibition of protein kinase CKII in human colon
cancer cells. Mol Cells. 28:489–494. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63.
|
Marsh DJ, Kum JB, Lunetta KL, et al: PTEN
mutation spectrum and genotype-phenotype correlations in
Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with
Cowden syndrome. Hum Mol Genet. 8:1461–1472. 1999. View Article : Google Scholar
|
|
64.
|
Marsh DJ, Coulon V, Lunetta KL, et al:
Mutation spectrum and genotype-phenotype analyses in Cowden disease
and Bannayan-Zonana syndrome, two hamartoma syndromes with germline
PTEN mutation. Hum Mol Genet. 7:507–515. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
65.
|
Zbuk KM and Eng C: Hamartomatous polyposis
syndromes. Nat Clin Pract Gastroenterol Hepatol. 4:492–502. 2007.
View Article : Google Scholar
|
|
66.
|
Agrawal S, Pilarski R and Eng C: Different
splicing defects lead to differential effects downstream of the
lipid and protein phosphatase activities of PTEN. Hum Mol Genet.
14:2459–2468. 2005. View Article : Google Scholar
|
|
67.
|
Teresi RE, Zbuk KM, Pezzolesi MG, Waite KA
and Eng C: Cowden syndrome-affected patients with PTEN promoter
mutations demonstrate abnormal protein translation. Am J Hum Genet.
81:756–767. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
68.
|
Han SY, Kato H, Kato S, et al: Functional
evaluation of PTEN missense mutations using in vitro
phosphoinositide phosphatase assay. Cancer Res. 60:3147–3151.
2000.PubMed/NCBI
|
|
69.
|
Kato H, Kato S, Kumabe T, et al:
Functional evaluation of p53 and PTEN gene mutations in gliomas.
Clin Cancer Res. 6:3937–3943. 2000.PubMed/NCBI
|
|
70.
|
Georgescu MM, Kirsch KH, Kaloudis P, Yang
H, Pavletich NP and Hanafusa H: Stabilization and productive
positioning roles of the C2 domain of PTEN tumor suppressor. Cancer
Res. 60:7033–7038. 2000.PubMed/NCBI
|
|
71.
|
Trotman LC, Wang X, Alimonti A, et al:
Ubiquitination regulates PTEN nuclear import and tumor suppression.
Cell. 128:141–156. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
72.
|
Bonneau D and Longy M: Mutations of the
human PTEN gene. Hum Mutat. 16:109–122. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
73.
|
Keniry M and Parsons R: The role of PTEN
signaling perturbations in cancer and in targeted therapy.
Oncogene. 27:5477–5485. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
74.
|
Simpson L and Parsons R: PTEN: life as a
tumor suppressor. Exp Cell Res. 264:29–41. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
75.
|
Carracedo A, Alimonti A and Pandolfi PP:
PTEN level in tumor suppression: how much is too little? Cancer
Res. 71:629–633. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
76.
|
Alimonti A, Carracedo A, Clohessy JG, et
al: Subtle variations in Pten dose determine cancer susceptibility.
Nat Genet. 42:454–458. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
77.
|
Xie CC, Lu L, Sun J, et al: Germ-line
sequence variants of PTEN do not have an important role in
hereditary and non-hereditary prostate cancer susceptibility. J Hum
Genet. 56:496–502. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
78.
|
Riegert-Johnson DL, Gleeson FC, Roberts M,
Tholen K, Youngborg L, Bullock M and Boardman LA: Cancer and
Lhermitte-Duclos disease are common in Cowden syndrome patients.
Hered Cancer Clin Pract. 8:62010. View Article : Google Scholar : PubMed/NCBI
|
|
79.
|
Ali E, Athanasopoulos PG, Forouhi P and
Malata CM: Cowden syndrome and reconstructive breast surgery: case
reports and review of the literature. J Plast Reconstr Aesthet
Surg. 64:545–549. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
80.
|
Zhang G, Yang P, Guo P, Miele L, Sarkar
FH, Wang Z and Zhou Q: Unraveling the mystery of cancer metabolism
in the genesis of tumor-initiating cells and development of cancer.
Biochim Biophys Acta. 1836:49–59. 2013.PubMed/NCBI
|
|
81.
|
Dean JL and Knudsen KE: The role of tumor
suppressor dysregulation in prostate cancer progression. Curr Drug
Targets. 14:460–471. 2013. View Article : Google Scholar : PubMed/NCBI
|