Introduction
The tumor suppressor protein p53 is a principal
modulator of various anti-carcinogenesis effects such as apoptosis,
cell cycle arrest, senescence, and DNA repair (1,2).
Regarding the induction of apoptosis, it is generally assumed that
the activation of the mitochondrial apoptotic pathway by
transcriptional target genes of p53 such as NOXA, PUMA, BAX and
APAF-1 is the major pathway (3).
In addition to transcription-dependent apoptosis by p53, p53 was
shown, more than a decade ago, to move to the mitochondria and,
when there, trigger intrinsic apoptosis (4). Mitochondrial p53 attenuates
anti-apoptotic activity of BCLXL through direct binding to it,
thereby leading to the oligomerization of BAK and the subsequent
formation of pores in the mitochondrial outer membranes, through
which cytochrome c, the pivotal inducer of intrinsic apoptosis,
exits to the cytosol (4,5). In skin epidermal cells, TPA directs
nuclear p53 to move to mitochondria where p53 interacts with
manganese superoxide dismutase (MnSOD), resulting in a decrease of
mitochondrial membrane potential which leads to the release of
cytochrome c (6). Based on
experimental findings demonstrating that the mitochondrial
translocation of p53 occurs earlier than the transcriptional
induction of p53 target genes, and artificial expression of p53
targeted to mitochondria which lacks transcriptional activity
induces apoptosis efficiently in cancer cells in vitro and
in vivo (7,8), it has been suggested that
mitochondrial p53 may have a more important role than nuclear p53
in the induction of apoptosis and may be a sole apoptosis-inducing
stimulus. In addition, a recent study reported that mitochondrial
p53 triggered mitochondrial permeability transition pore (MPTP)
opening by interacting with cyclophilin D (9). The binding of p53 to cyclophilin D
was shown to occur under conditions of oxidative stress and to
activate necrosis, instead of apoptosis, during
ischemia-reperfusion injury of the brain. Therefore, it can be
speculated that mitochondrial p53 could induce different forms of
cell death depending on the cellular contexts and the nature of the
stimuli, and its role as well as the underlying mechanism of
mitochondrial p53 in apoptosis induction should be clarified for a
complete understanding of p53-induced cancer cell death.
Nutlin-3, a cis-imidazoline analog which
upregulates the p53 protein by disrupting interactions between p53
and HDM2, is capable of inducing p53-dependent apoptosis in various
cancer cells including leukemia and multiple myeloma cells
(10,11). To be consistent with the
mitochondrial trafficking of p53 by anticancer therapeutics and
p53-overexpression plasmids, nutlin-3-upregulated p53 also moves to
mitochondria and triggers the intrinsic apoptotic pathway (12,13).
This mitochondrial p53 induced by nutlin-3 was found to be
sufficient to induce apoptosis, and moreover, the inhibition of
transcriptional activity of p53 potentiates nutlin-3-induced
apoptosis, suggesting that the mitochondrial translocation of p53
may be the primary and major initiator for nutlin-3-induced
apoptosis.
In a previous study, however, we reported that
mitochondrial p53 stimulates the activation of the MEK1/2-ERK1/2
pathway in cancer cells treated with nutlin-3 (14). This activation of MEK1/2 and ERK1/2
was attributed to an accumulation of mitochondrial ROS caused by
mitochondrial p53, and was found to suppress nutlin-3-induced
apoptosis, suggesting the possibility that mitochondrial p53 can
induce cell survival pathways, thus counteracting p53-induced
apoptosis, depending on the type of cancer cells. Although the
inhibition of ERK1/2 was shown to potentiate nutlin-3-induced
apoptosis, the mechanism how ERK1/2 suppresses apoptosis in
nutlin-3-treated cells remains unclear. To address this issue, we
report on attempts to identify a member of the anti-apoptotic BCL2
family which is expressed as a function of the level of ERK1/2
activity and inhibits nutlin-3-induced apoptosis as well.
Materials and methods
Reagents
Nutlin-3 and U0126 were purchased from Selleckchem
(Houston, TX, USA) and Tocris (Ellisville, MO, USA), respectively.
All the other chemicals were obtained from Sigma-Aldrich Inc. (St.
Louis, MO, USA), unless specified otherwise. The reagents were of
molecular biology or cell culture tested grade.
Cell culture
The human osteosarcoma cells U2OS and SAOS were
maintained in DMEM (Hyclone, Logan, UT, USA) containing 10%
heat-inactivated fetal bovine serum (Hyclone), 100 U/ml penicillin
(Hyclone) and glutamine (Invitrogen, Carlsbad, CA, USA) at 37°C in
a 5% CO2-humidified incubator.
Antibodies
Rabbit anti-BCL2A1 and other rabbit antibodies
against ERK1/2, phospho-CREB1, and phospho-MEK1/2 were purchased
from Abcam (Cambridge, UK) and Cell Signaling Technology (Boston,
MA, USA), respectively. All the other antibodies were also
commercially obtained from Santa Cruz Biotechnology (Santa Cruz,
CA, USA, mouse antibodies against phospho-ERK1/2, phospho-ELK1, and
p53), Epitomics (Burlingame, CA, USA, rabbit anti-MEK1/2 and
anti-CREB1), Merck (Billerica, MA, USA, chicken anti-GAPDH),
Sigma-Aldrich Inc. (HRP-conjugated anti-rabbit or -mouse IgG) and
KPL (Gaithersburg, MD, USA, HRP-conjugated anti-chicken IgG).
Transfection of small interfering RNAs
(siRNAs)
SiRNA against p53 was obtained from Santa Cruz
Biotechnology, and siRNAs against CREB1 and BCL2A1
were obtained from Sigma-Aldrich Inc. ELK1 siRNA was
purchased from Bioneer (Daejeon, Korea). Transfections of siRNAs
were carried out using Lipofectamine™ RNAiMAX (Invitrogen),
following the manufacturer’s instructions.
Immunoblot analysis
Cells treated as described in the figure legends
were lysed in RIPA buffer supplemented with protease inhibitor
cocktail (Roche, Basel, Switzerland), and were subjected to
immunoblot analysis. Briefly, 20 μg aliquots of lysates were
separated on SDS-polyacrylamide gels and then, were transferred to
nitrocellulose membranes (Merck). After submerging in 5%
skim-milk/TTBS (Tris-buffered saline containing Tween-20 0.025%)
for 30 min, the membranes were incubated in 3% BSA/TTBS containing
primary antibodies, washed with TTBS and then incubated with
HRP-conjugated anti-IgG. The protein bands that reacted with
antibodies were then detected using enhanced chemiluminescence
reagents (ECL, GE Healthcare, Buckinghamshire, UK).
Quantitative real-time RT-PCR
(QRT-PCR)
Total RNA extracted using RNAiso Plus (Takara Bio
Inc., Shiga, Japan) was subjected to QRT-PCR. Briefly, cDNA was
generated using PrimeScript™ RT reagent kit (Takara Bio Inc.) and
QRT-PCR was performed using SYBR FAST qPCR kit (Kapabiosystems,
Woburn, MA, USA). All reactions were performed in triplicate by the
ABI 7300 Real-Time PCR system (Applied Biosystems, Carlsbad, CA,
USA). Relative changes in transcript level normalized by GAPDH mRNA
were calculated by the ΔΔCt method (15)
Apoptosis assay
Apoptosis was determined by ApoScan kit (BioBud,
Gyunggido, Korea) according to a previous report (16). Briefly, cells treated as indicated
in the figure legends were stained with Annexin V-Fluos at room
temperature for 15 min, washed and resuspended in binding buffer.
Propidium iodide (PI, 1 μg/ml) was added and stained cells were
then analyzed using a flow cytometer (FACSCalibur, BD Biosciences,
San Jose, CA, USA). For the measurement of hypo-diploidic cells,
cells were incubated in 70% ethanol solution for 2 h, and then
stained with propidium iodide for 15 min, followed by flow
cytometric analysis (17). Cell
cycle distribution of the PI-stained cells was analyzed by the
CellQuest and Modfit software following the manufacturer’s
instructions.
Results
Treatment of nutlin-3 induces the
expression of BCL2A1 gene by activating ERK1/2
We attempted to identify genes that are responsible
for the anti-apoptotic effect of nutlin-3-induced ERK1/2 activity.
To this end, we first compared changes in the expression levels of
anti-apoptotic BCL2 family members in nutlin-3-treated U2OS cells.
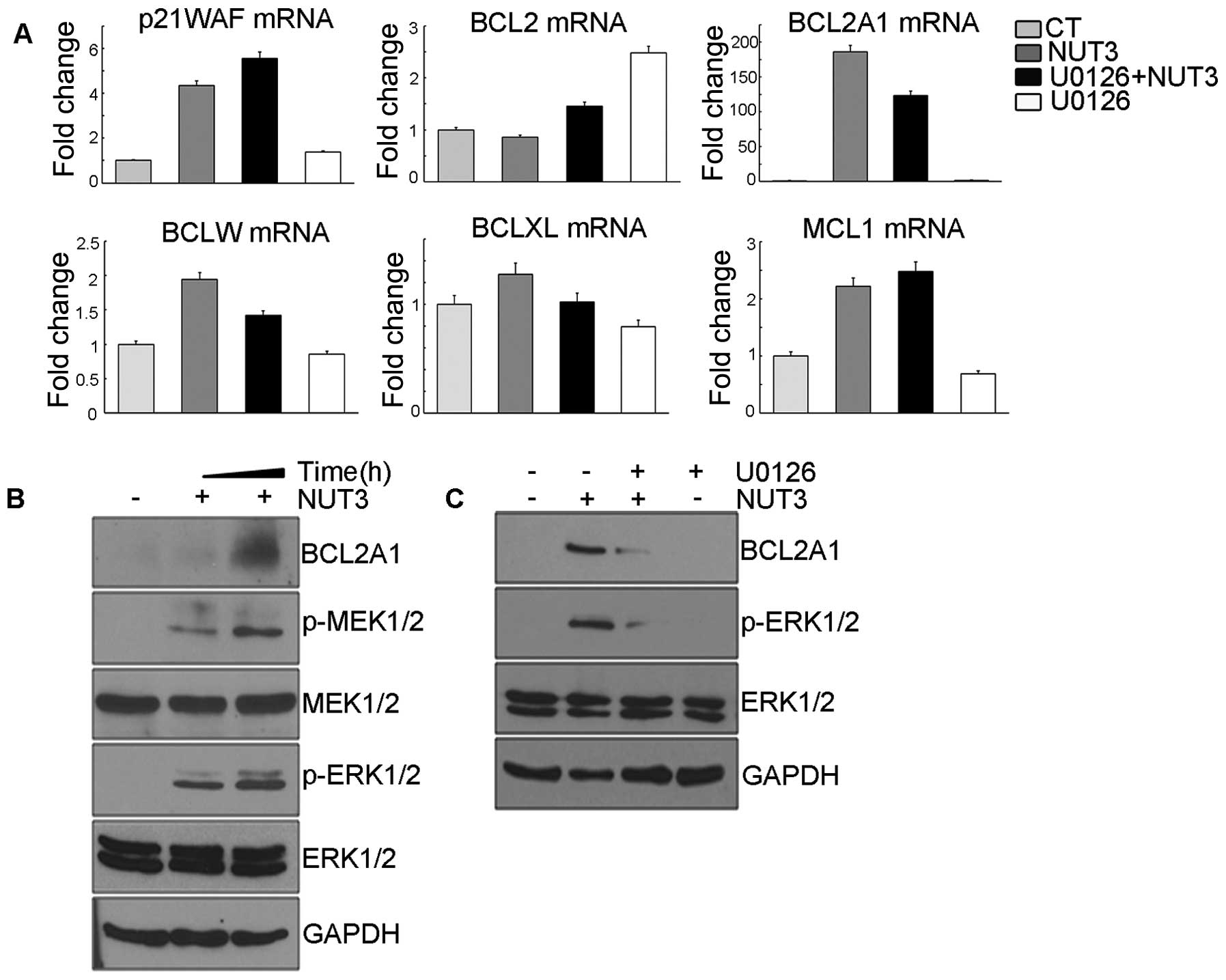
As shown in Fig. 1A, nutlin-3
increased the mRNA levels of BCL2A1, BCLXL and BCLW.
Among these genes, the mRNA increase of BCL2A1 and
BCLW, but not BCLXL was suppressed by U0126
pretreatment. Although the expression of BCLW is also likely
to be regulated by ERK1/2, its induction level was <2-fold and
so, we analyzed the mechanism involved in the expression of
BCL2A1 and its effect on apoptosis in this model. The
expression of p21WAF1, a well-known transcriptional target
gene of p53, was also induced by nultin-3, but this induction was
not reduced by U0126 pretreatment, confirming that U0126 had no
effect on the transcriptional activity of p53 and thus,
nutlin-3-induced BCL2A1 expression is robustly regulated by
ERK1/2. Consistent with the induction of mRNA by nutlin-3, the
expression level of the BCL2A1 protein was increased by nutlin-3
along with the phosphorylation of MEK1/2 and ERK1/2 (Fig. 1B) and this increase was also
suppressed by U0126 pretreatment (Fig.
1C). Collectively, these data suggest that nutlin-3-induced
ERK1/2 activity should stimulate the expression of BCL2A1 at
both the mRNA and protein levels.
Mitochondrial p53 is critical in the
induction of BCL2A1 expression
Since nutlin-3 is an antagonist of HDM2 which
ubiquitylates p53 family proteins such as p63 and p73 as well as
p53, nutlin-3 can activate p73-dependent apoptosis in cancer cells
which have mutated p53 gene (18). Thus, we proceeded to determine the
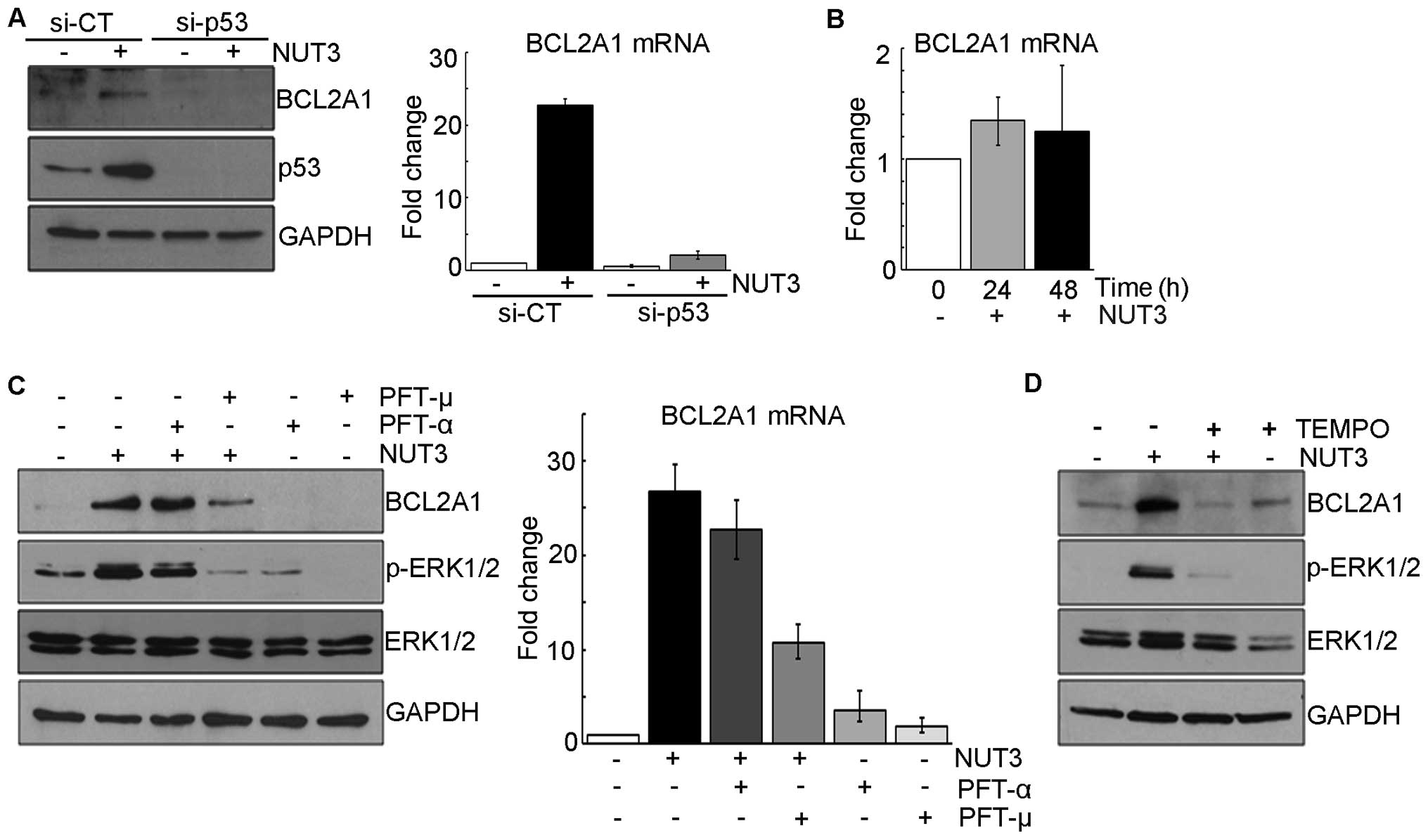
role of p53 in this nutlin-3-induced BCL2A1 expression. In
experiments using small interfering RNA against p53,
nutlin-3 did not increase either BCL2A1 mRNA or protein in
p53-knocked down U2OS cells (Fig.
2A) or in SAOS (human osteosarcoma) cells in which the
p53 gene was mutated (Fig.
2B), confirming the dependency of BCL2A1 induction on
the intact p53 protein. We previously showed that nutlin-3-induced
ERK1/2 activation was dependent on mitochondrial ROS generated by
mitochondrial p53 (14), which led
us to speculate that nutlin-3-induced BCL2A1 expression
would be also dependent on both the mitochondrial translocation of
p53 and ROS. As expected, the nutlin-3-induced expression of both
BCL2A1 mRNA and protein was suppressed by pretreatment with
PFT-μ and TEMPO, an inhibitor of the mitochondrial translocation of
p53 and a radical scavenger, respectively, which prevented the
phosphorylation of ERK1/2, but not in the case of pretreatment with
PFT-α, an inhibitor of the transcriptional activity of p53
(Fig. 2C and D and data not
shown). Accordingly, these data suggest that nutlin-3-induced
BCL2A1 expression is also dependent on the mitochondrial
translocation of p53 and ROS generation which are critical
regulators of ERK1/2 activation in nutlin-3-treated U2OS cells.
Induction of BCL2A1 expression is
mediated by ELK1
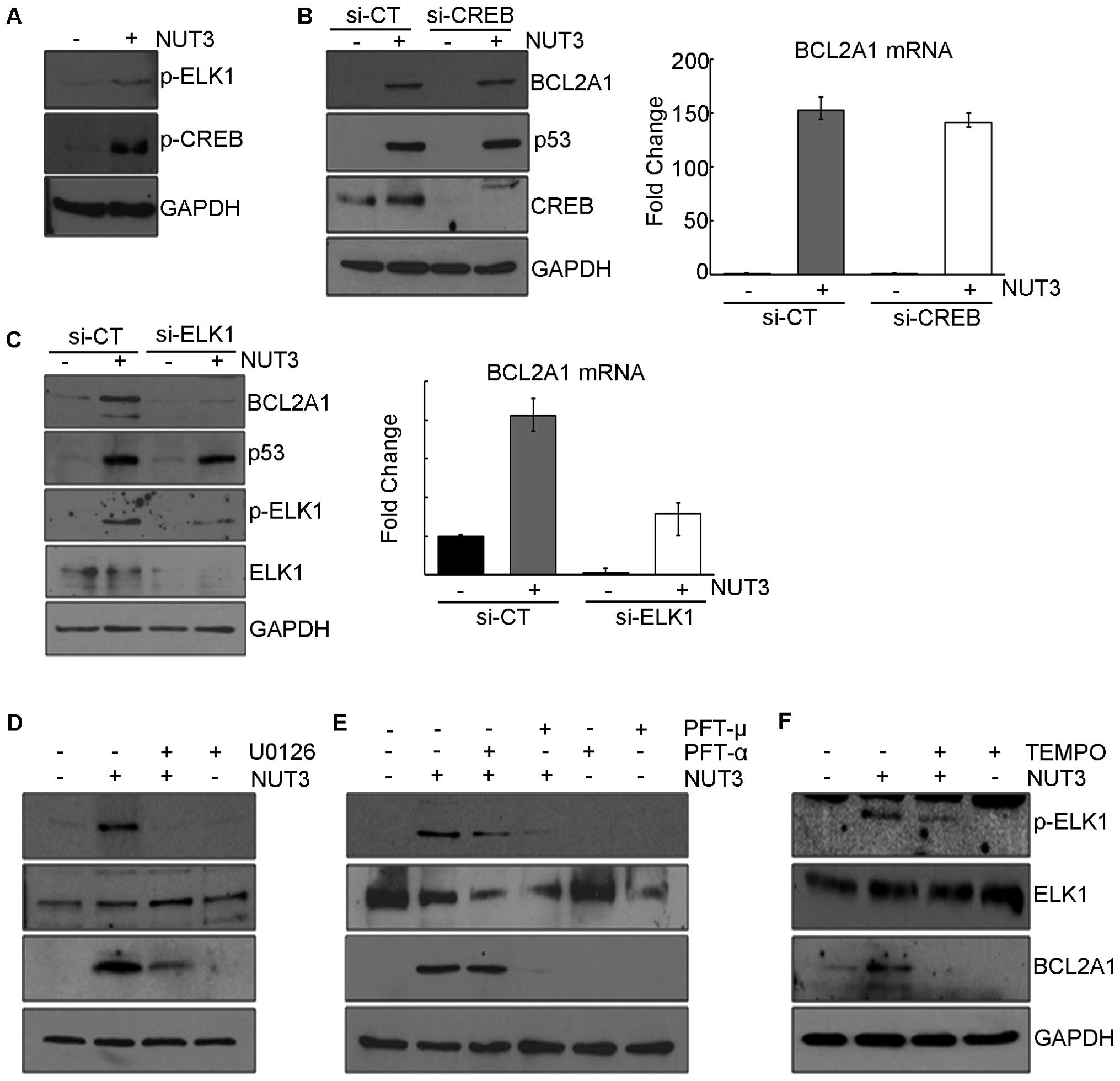
Next, we attempted to identify the signaling
molecules downstream of ERK1/2 that induce the expression of
BCL2A1. It is well known that ERK1/2 stimulates the activity
of several transcription factors, including ELK1 and CREB1 by
directly or indirectly phosphorylating them, which leads to the
transcriptional induction of a variety of genes (19,20).
In this model, nutlin-3 also increases the phosphorylation of CREB1
and ELK1, which prompted us to analyze the effect of these
transcriptional factors on BCL2A1 expression (Fig. 3A). In experiments using siRNAs to
knock down ELK1 and CREB1, nutlin-3-induced
BCL2A1 expression was suppressed by the knockdown of
ELK1 (Fig. 3C) but not
CREB1 (Fig. 3B), suggesting
that the ELK1 protein is required for the nutlin-3-induced
transcription of BCL2A1. Moreover, compounds such as U0126,
PFT-μ, and TEMPO which prevented both the nutlin-3-induced
phosphorylation of ERK1/2 and the expression of BCL2A1 also
inhibited the phosphorylation of ELK1 (Fig. 3D–F). In contrast, PFT-α, an
inhibitor of the transcriptional activity of p53, which did not
prevent either the nutlin-3-induced phosphorylation of ERK1/2 or
the expression of BCL2A1, also failed to inhibit the
phosphorylation of ELK1 (Fig. 3E).
These results collectively suggest that ROS generated by the
nutlin-3-induced mitochondrial translocation of p53 activates
ERK1/2, which in turn activates ELK1, finally leading to the
transcriptional induction of BCL2A1.
BCL2A1 inhibits nutlin-3-induced
apoptosis
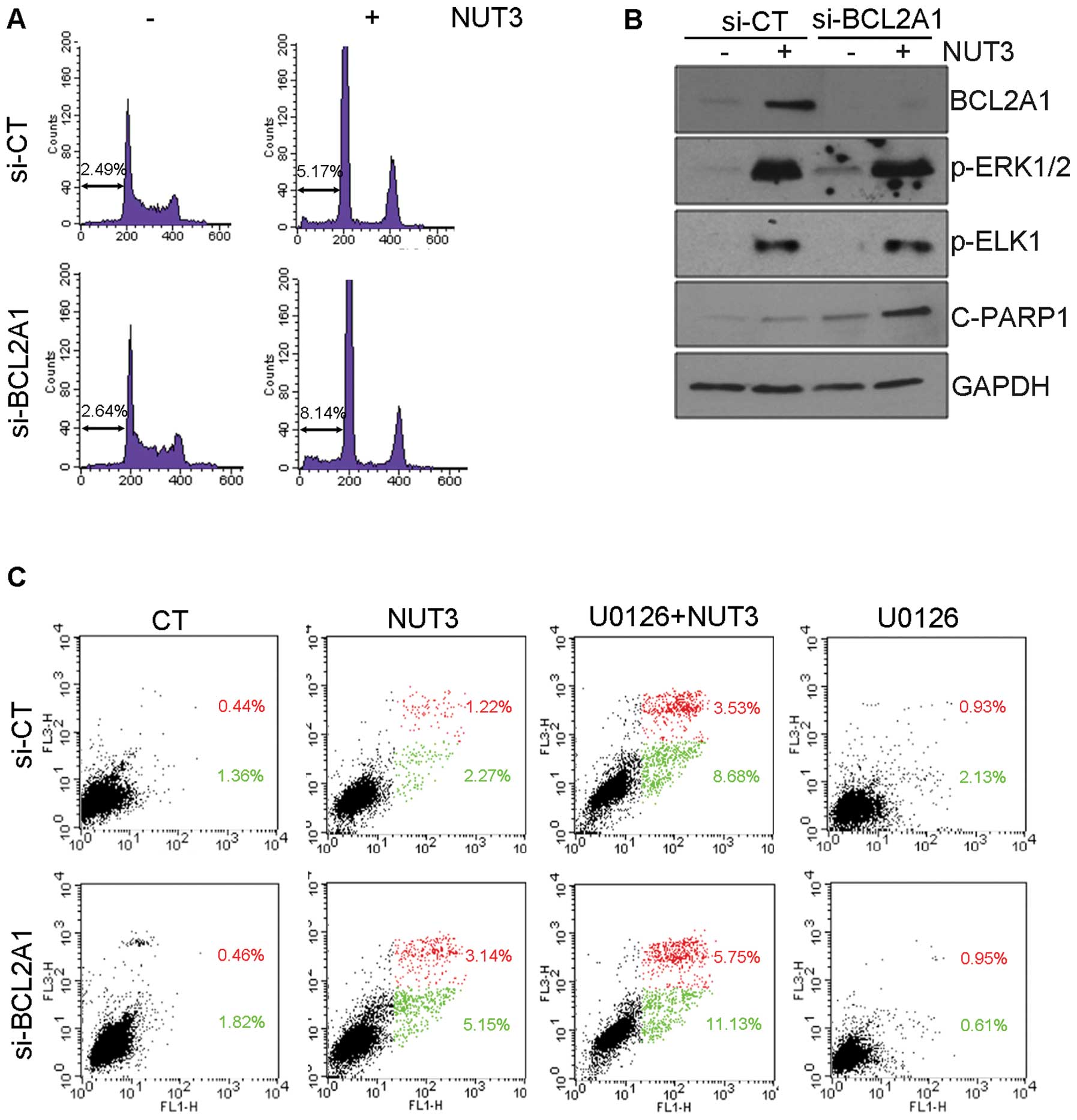
Finally, we analyzed the effect of BCL2A1 protein
expression on nutlin-3-induced apoptosis. As shown in Fig. 4A and C, the nutlin-3-induced
accumulation of both hypo-diploid cells in the sub-G1 phase and
Annexin V-positive cells was potentiated by BCL2A1
knockdown, suggesting that BCL2A1 has an inhibitory effect on
nutlin-3-induced apoptosis. Consistent with the assessment of
apoptosis, the knockdown of BCL2A1 augmented the expression
of cleaved poly(ADP-ribose) polymerase-1 (PARP-1) which is a
hallmark of apoptosis (Fig. 4B),
without affecting the phosphorylation level of ERK1/2 and ELK1. The
level of apoptosis induction by the combined treatment of nutlin-3
and siRNA against BCL2A1 appeared to be close to that for
the combined treatment of nutlin-3 and U0126 (Fig. 4C), implying that anti-apoptotic
effect of the BCL2A1 protein should be restricted to anti-apoptotic
functions of nutlin-3-activated ERK1/2, and that BCL2A1 might be a
gene responsible for the anti-apoptotic activity of ERK1/2.
Discussion
Contrary to reports that nutlin-3 induces apoptosis
via the mitochondrial translocation of p53 in cancer cells, our
previous study concluded that the nutlin-3-induced mitochondrial
translocation of p53 stimulated the activation of MAPK such as
ERK1/2, JNK and p38 MAPK via the generation of mitochondrial ROS
(14,21). This MAPK exerted anti-apoptotic
effect, suggesting that MAPK activated by mitochondrial p53 may
constitute a negative feedback loop of p53-induced apoptosis.
Whereas the JNK and p38 MAPK induced the expression of heme
oxygenase-1 (HO-1), an anti-apoptotic protein, we propose that
BCL2A1 may be a downstream gene of activated ERK1/2 and may
suppress nutlin-3-induced apoptosis.
BCL2A1 which belongs to the pro-survival BCL2
family has been reported to be expressed at the level of
transcription by inflammatory cytokines, CD40, and oxidative stress
in endothelial cells, B lymphocytes, and leukemic cells,
respectively (22–24). Elevated BCL2A1 protein prevents
apoptosis induced by various stimuli including TNF-α, TRAIL, Fas,
and chemotherapeutic agents by directly binding to tBid and BAK
(24–29). Regarding the transcriptional
induction of BCL2A1, it should be noted that NF-κB has been
invariably required for all the stimuli reported until now to
induce BCL2A1 expression, indicating that BCL2A1 is a
transcriptional target gene of NF-κB and a possible component of an
anti-apoptotic branch of the NF-κB-activated pathways. However, in
the present model, the expression of BCL2A1 was upregulated
by ELK1. The phosphorylation of ELK1 seemed to be induced by ERK1/2
which was activated by ROS generated by mitochondrial p53.
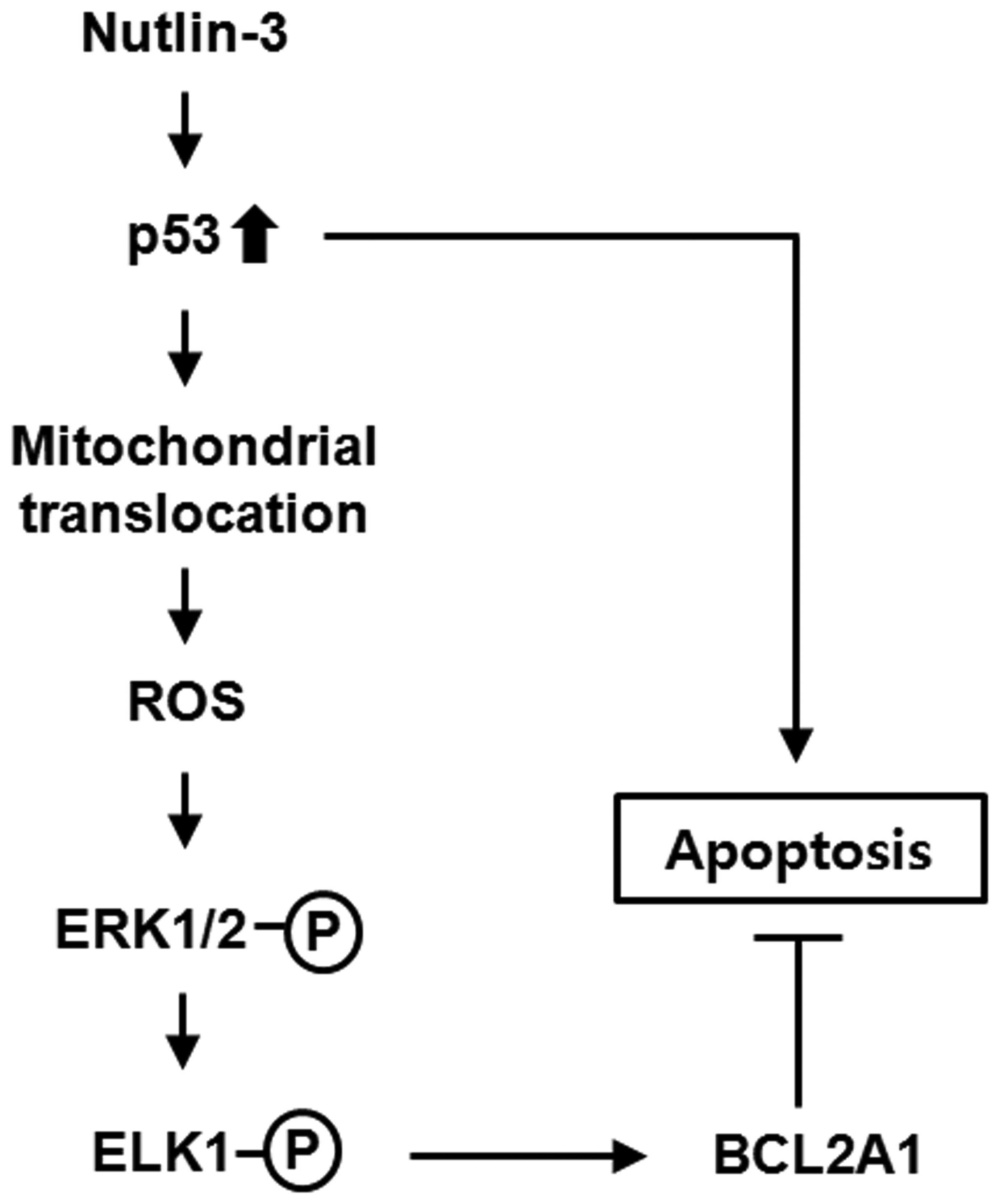
Therefore, the data presented herein can be summarized as shown in
Fig. 5, in which the proposed cell
survival pathway is comprised of the sequential induction of
mitochondrial p53, ROS generation, ERK1/2 activation, ELK1
activation, and expression of the BCL2A1 gene. This
induction of BCL2A1 protein expression suppressed the activation of
the nutlin-3-induced apoptosis program, implying that this pathway
might constitute a negative feedback loop of p53-induced
apoptosis.
ELK1 is a member of the ternary complex factor (TCF)
subfamily and is activated by mitogenic or growth factors such as
EGF. ELK1 functions as a transcription factor and regulates the
transcription of various genes that are involved in cell growth,
differentiation, and survival (30). Among the pro-survival BCL2
proteins, MCL1 was found to be a transcriptional target of
ELK1. For instance, it was reported that EGF and ovarian cancer
ascites cause an increase in MCL1 expression by activating ELK1 in
breast and ovarian cancer cells (27,31).
This ELK1-MCL1 pathway could be a potential target of cancer
therapy, as sorafenib, an inhibitor of multiple kinases, induces
apoptosis of endometrial carcinoma cells by interfering with
ELK1-dependent MCL1 transcription (32).
In the present model, although nutlin-3 treatment
induced ROS accumulation, NF-κB was not activated, and MCL1
expression was not induced, even in the presence of activated ELK1.
Therefore, it can be postulated that intracellular ROS may induce
BCL2A1 expression by directly activating NF-κB and/or by
activating ELK1 via the MEK1/2-ERK1/2 pathway. The mechanism
involved, regarding which of these two pathways are selected,
remains to be clarified.
Nutlin-3 may be a promising anticancer agent, since
it specifically activates p53-dependent anticancer programs without
genomic DNA damage, which exerts adverse effects on non-transformed
cells leading to cancer patients encountering difficulties in
adjusting to systemic and conventional anticancer treatments such
as radiotherapy and chemotherapy, and even in the case of secondary
tumor development (33,34). One of the features of nutlin-3 is
that it induces prominent cell cycle arrest with subtle apoptosis
in some solid cancer cells such as U2OS cells used in this study,
while it induces substantial apoptosis particularly in leukemia and
lymphoma cells (35). Although
cell cycle arrest can blunt the initiation and progression of
cancer, it can also diminish the therapeutic efficacy of anticancer
agents (36). The induction of
prominent cell cycle arrest by nutlin-3 was suggested to be due to
the dramatic expression of p21WAF1 and downregulation of HIPK2
(37,38). The negative feedback loop proposed
in this report could thus provide a novel explanation for
nutlin-3-induced subtle apoptosis, in part at least, because the
blockade of this loop enhanced apoptosis, while no interaction with
p21WAF1 and HIPK2 occurred.
A high level of BCL2A1 expression in cancer tissues
such as stomach, melanoma, and leukemia has been reported, and its
level of expression appears to be correlated with metastasis and
the poor prognosis of such types of cancers (39–41).
Accordingly, the knockdown of BCL2A1 protein expression was found
to sensitize B cell lymphoma cells to apoptosis induced by
anticancer chemicals (42).
Furthermore, it was recently reported that BCL2A1 confers melanoma
and lymphoma cells resistance to vemurafenib (PLX4032), an
inhibitor of BRAF, and ABT-737, a small molecule antagonist of
BCL2, respectively (43,44). Taking these findings into account,
the findings suggest that BCL2A1 might be expressed via
multiple signal transduction pathways including NF-κB stimulated by
molecular target therapeutics to cancer, and its expression would
diminish their therapeutic efficacy, resulting in the development
of an acquired resistance. Therefore, the pathway proposed in the
present study comprises the mechanism underlying the induced
expression of the BCL2A1 protein by a small molecule in cancer
cells and could contribute to the efficient use of small molecular
therapeutics against cancer including nutlin-3.
Acknowledgements
This study was supported by a grant
(2012R1A5A2047939) and a Basic Science Research Program through the
National Research Foundation of Korea (NRF-2010-0025420).
Abbreviations:
|
BCL2
|
B-cell CLL/lymphoma 2
|
|
BCL2A1
|
BCL2-related protein A1
|
|
BCLXL
|
BCL2-like 1
|
|
BCLW
|
BCL2-like 2
|
|
CREB1
|
cAMP response element binding protein
1
|
|
ELK1
|
member of ETS oncogene family
|
|
ERK
|
extracellular signal-regulated
kinases
|
|
HDM2
|
human double minute 2
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MCL1
|
myeloid cell leukemia sequence 1
|
|
MEK
|
MAPK/ERK kinase
|
|
P-CREB
|
phospho-CREB1 (S133)
|
|
P-ELK1
|
phospho-ELK1 (S383)
|
|
P-ERK1/2
|
phospho-ERK1/2 (T202/Y204)
|
|
P-MEK1/2
|
phospho-MEK1/2 (S217/221)
|
|
PFT
|
pifithrin
|
|
ROS
|
reactive oxygen species
|
|
PI
|
propidium iodide
|
|
TEMPO 2, 2, 6
|
6-tetramethyl-1-piperidinyloxy
|
References
|
1
|
Menendez D, Inga A and Resnick MA: The
expanding universe of p53 targets. Nat Rev Cancer. 9:724–737. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reinhardt HC and Schumacher B: The p53
network: cellular and systemic DNA damage responses in aging and
cancer. Trends Genet. 28:128–136. 2012. View Article : Google Scholar
|
|
3
|
Chipuk JE and Green DR: Dissecting
p53-dependent apoptosis. Cell Death Differ. 13:994–1002. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mihara M, Erster S, Zaika A, et al: p53
has a direct apoptogenic role at the mitochondria. Mol Cell.
11:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sot B, Freund SM and Fersht AR:
Comparative biophysical characterization of p53 with the
pro-apoptotic BAK and the anti-apoptotic BCL-xL. J Biol Chem.
282:29193–29200. 2007. View Article : Google Scholar
|
|
6
|
Zhao Y, Chaiswing L, Velez JM, et al: p53
translocation to mitochondria precedes its nuclear translocation
and targets mitochondrial oxidative defense protein-manganese
superoxide dismutase. Cancer Res. 65:3745–3750. 2005. View Article : Google Scholar
|
|
7
|
Erster S, Mihara M, Kim RH, Petrenko O and
Moll UM: In vivo mitochondrial p53 translocation triggers a rapid
first wave of cell death in response to DNA damage that can precede
p53 target gene activation. Mol Cell Biol. 24:6728–6741. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Palacios G and Moll UM: Mitochondrially
targeted wild-type p53 suppresses growth of mutant p53 lymphomas in
vivo. Oncogene. 25:6133–6139. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vaseva AV, Marchenko ND, Ji K, Tsirka SE,
Holzmann S and Moll UM: p53 opens the mitochondrial permeability
transition pore to trigger necrosis. Cell. 149:1536–1548. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Vassilev LT, Vu BT, Graves B, et al: In
vivo activation of the p53 pathway by small-molecule antagonists of
MDM2. Science. 303:844–848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Secchiero P, Bosco R, Celeghini C and
Zauli G: Recent advances in the therapeutic perspectives of
Nutlin-3. Curr Pharm Des. 17:569–577. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vaseva AV, Marchenko ND and Moll UM: The
transcription-independent mitochondrial p53 program is a major
contributor to nutlin-induced apoptosis in tumor cells. Cell Cycle.
8:1711–1719. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Saha MN, Jiang H and Chang H: Molecular
mechanisms of nutlin-induced apoptosis in multiple myeloma:
evidence for p53-transcription-dependent and -independent pathways.
Cancer Biol Ther. 10:567–578. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee SY, Shin SJ and Kim HS: ERK1/2
activation mediated by the nutlin-3-induced mitochondrial
translocation of p53. Int J Oncol. 42:1027–1035. 2013.PubMed/NCBI
|
|
15
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jang JY, Kim MK, Jeon YK, Joung YK, Park
KD and Kim CW: Adenovirus adenine nucleotide translocator-2 shRNA
effectively induces apoptosis and enhances chemosensitivity by the
down-regulation of ABCG2 in breast cancer stem-like cells. Exp Mol
Med. 44:251–259. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee K, Lee MH, Kang YW, Rhee KJ, Kim TU
and Kim YS: Parkin induces apoptotic cell death in
TNF-alpha-treated cervical cancer cells. BMB Rep. 45:526–531. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lau LM, Nugent JK, Zhao X and Irwin MS:
HDM2 antagonist Nutlin-3 disrupts p73-HDM2 binding and enhances p73
function. Oncogene. 27:997–1003. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Johannessen M, Delghandi MP and Moens U:
What turns CREB on? Cell Signal. 16:1211–1227. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Choe YJ, Lee SY, Ko KW, Shin SJ and Kim
HS: Nutlin-3 induces HO-1 expression by activating JNK in a
transcription-independent manner of p53. Int J Oncol. 44:761–768.
2014.PubMed/NCBI
|
|
22
|
Karsan A, Yee E, Kaushansky K and Harlan
JM: Cloning of human Bcl-2 homologue: inflammatory cytokines induce
human A1 in cultured endothelial cells. Blood. 87:3089–3096.
1996.PubMed/NCBI
|
|
23
|
Lee HH, Dadgostar H, Cheng Q, Shu J and
Cheng G: NF-kappaB-mediated up-regulation of Bcl-x and Bfl-1/A1 is
required for CD40 survival signaling in B lymphocytes. Proc Natl
Acad Sci USA. 96:9136–9141. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim H, Kim YN, Kim H and Kim CW: Oxidative
stress attenuates Fas-mediated apoptosis in Jurkat T cell line
through Bfl-1 induction. Oncogene. 24:1252–1261. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Karsan A, Yee E and Harlan JM: Endothelial
cell death induced by tumor necrosis factor-alpha is inhibited by
the Bcl-2 family member, A1. J Biol Chem. 271:27201–27204. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim HR, Heo YM, Jeong KI, et al: FGF-2
inhibits TNF-alpha mediated apoptosis through upregulation of
Bcl2-A1 and Bcl-xL in ATDC5 cells. BMB Rep. 45:287–292. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Goncharenko-Khaider N, Matte I, Lane D,
Rancourt C and Piche A: Ovarian cancer ascites increase Mcl-1
expression in tumor cells through ERK1/2-Elk-1 signaling to
attenuate TRAIL-induced apoptosis. Mol Cancer. 11:842012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang CY, Guttridge DC, Mayo MW and Baldwin
AS Jr: NF-kappaB induces expression of the Bcl-2 homologue A1/Bfl-1
to preferentially suppress chemotherapy-induced apoptosis. Mol Cell
Biol. 19:5923–5929. 1999.PubMed/NCBI
|
|
29
|
Simmons MJ, Fan G, Zong WX, Degenhardt K,
White E and Gelinas C: Bfl-1/A1 functions, similar to Mcl-1, as a
selective tBid and Bak antagonist. Oncogene. 27:1421–1428. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kasza A: Signal-dependent Elk-1 target
genes involved in transcript processing and cell migration. Biochim
Biophys Acta. 1829:1026–1033. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Booy EP, Henson ES and Gibson SB:
Epidermal growth factor regulates Mcl-1 expression through the
MAPK-Elk-1 signalling pathway contributing to cell survival in
breast cancer. Oncogene. 30:2367–2378. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun NK, Huang SL, Chang TC and Chao CC:
Sorafenib induces endometrial carcinoma apoptosis by inhibiting
Elk-1-dependent Mcl-1 transcription and inducing
Akt/GSK3beta-dependent protein degradation. J Cell Biochem.
114:1819–1831. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shangary S, Qin D, McEachern D, et al:
Temporal activation of p53 by a specific MDM2 inhibitor is
selectively toxic to tumors and leads to complete tumor growth
inhibition. Proc Natl Acad Sci USA. 105:3933–3938. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Travis LB, Ng AK, Allan JM, et al: Second
malignant neoplasms and cardiovascular disease following
radiotherapy. Health Phys. 106:229–246. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tovar C, Rosinski J, Filipovic Z, et al:
Small-molecule MDM2 antagonists reveal aberrant p53 signaling in
cancer: implications for therapy. Proc Natl Acad Sci USA.
103:1888–1893. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Moreno CS, Matyunina L, Dickerson EB, et
al: Evidence that p53-mediated cell-cycle-arrest inhibits
chemotherapeutic treatment of ovarian carcinomas. PLoS One.
2:e4412007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rinaldo C, Prodosmo A, Siepi F, et al:
HIPK2 regulation by MDM2 determines tumor cell response to the
p53-reactivating drugs nutlin-3 and RITA. Cancer Res. 69:6241–6248.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Enge M, Bao W, Hedstrom E, Jackson SP,
Moumen A and Selivanova G: MDM2-dependent downregulation of p21 and
hnRNP K provides a switch between apoptosis and growth arrest
induced by pharmacologically activated p53. Cancer Cell.
15:171–183. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Choi SS, Park IC, Yun JW, Sung YC, Hong SI
and Shin HS: A novel Bcl-2 related gene, Bfl-1, is overexpressed in
stomach cancer and preferentially expressed in bone marrow.
Oncogene. 11:1693–1698. 1995.PubMed/NCBI
|
|
40
|
Riker AI, Enkemann SA, Fodstad O, et al:
The gene expression profiles of primary and metastatic melanoma
yields a transition point of tumor progression and metastasis. BMC
Med Genomics. 1:132008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Simpson LA, Burwell EA, Thompson KA,
Shahnaz S, Chen AR and Loeb DM: The antiapoptotic gene A1/BFL1 is a
WT1 target gene that mediates granulocytic differentiation and
resistance to chemotherapy. Blood. 107:4695–4702. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Brien G, Trescol-Biemont MC and
Bonnefoy-Berard N: Downregulation of Bfl-1 protein expression
sensitizes malignant B cells to apoptosis. Oncogene. 26:5828–5832.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Vogler M, Butterworth M, Majid A, et al:
Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately
1000-fold resistance to ABT-737 in chronic lymphocytic leukemia.
Blood. 113:4403–4413. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Haq R, Yokoyama S, Hawryluk EB, et al:
BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that
confers resistance to BRAF inhibition. Proc Natl Acad Sci USA.
110:4321–4326. 2013. View Article : Google Scholar : PubMed/NCBI
|