Introduction
Breast cancer is the most commonly diagnosed type of
cancer and the second leading cause of cancer death in females
worldwide (1). Despite the fact
that many tumors initially respond to chemotherapy, breast cancer
cells can subsequently survive and gain resistance to the treatment
(2,3). It is therefore important to identify
novel agents with improved pharmacological and toxicological
profiles.
Cdks are a group of protein kinases first discovered
for their role in regulating cell cycle. Cdks are activated by the
formation of a complex with cyclin partners. Specific Cdks operate
in distinct phases of the cell cycle (4,5). In
addition to their cell cycle regulatory functions, Cdks, especially
Cdk7 and Cdk9, play important roles in the regulation of RNA Pol
II-mediated transcription. The general transcription factor II
(TFIIH) complex containing Cdk7 and cyclin H first phosphorylates
the serine-5 residue of carboxyl-terminal domain (CTD) of the large
subunit of RNA Pol II and facilitates the initiation of
transcription. Then the Cdk9-cyclin T complex forms the
transcription elongation factor b (p-TEFb) and phosphorylates the
5, 6-dichlorobenzimidazone-1-b-D-ribofuranoside (DRB)-sensitive
inducing factor and the negative elongation factor, followed by
serine-2 of the CTD to facilitate transcriptional elongation
(6–9).
Cdks play a critical role in cancer progression, and
misregulation of Cdks is one of the most frequent alterations in
human cancer (4,5,10,11).
For this reason, Cdks have been considered as very promising
therapeutic targets in human malignancies. Over the past decade,
the intensive search for pharmacological Cdk inhibitors has led to
several clinical candidates, and the focus on transcriptional Cdks
has underlined their antitumor activity (12). Flavopiridol, the first and
currently most promising Cdk inhibitor in preclinical and clinical
trials, has demonstrated marked antitumor activity in many types of
cancer (13,14). Flavopiridol acts largely through
inhibition of Cdk9. When RNA Pol II is repressed after Cdk9
inhibition, the result is a blockage of transcriptional elongation,
which in turn causes decreases in the cellular levels of
short-lived proteins including some antiapoptotic molecules such as
Mcl-1 and XIAP, and thus promotes the induction of apoptosis
(15,16).
SNS-032 is a novel Cdk inhibitor that has emerged in
clinical trials. Originally selected as an inhibitor of Cdk2,
SNS-032 was later found to be a potent inhibitor of Cdk9 and Cdk7
(17). SNS-032 is more selective
and less cytotoxic compared to flavopiridol, and in vitro
studies have shown more potent inhibition of RNA synthesis and cell
death induction by SNS-032 than by flavopiridol (18). In this study, we investigated the
efficacy of SNS-032 against breast cancer cells. SNS-032 suppressed
the proliferation of MCF-7 and MDA-MB-435 breast cancer cells as
well as the growth of MDA-MB-435 nude mouse xenografts. Such
effects occurred through the suppression of RNA synthesis of
antiapoptotic protein Mcl-1 and XIAP, which in turn led to the
reduction of the Mcl-1 and XIAP protein and the initiation of
apoptosis. Thus, we provide in vitro and in vivo
evidence for use of SNS-032 as a promising therapeutic agent for
the treatment of breast cancer.
Materials and methods
Chemicals
SNS-032 was obtained from Selleck Chemicals. LLC
(Houston, TX, USA), dissolved in dimethyl sulfoxide (DMSO) to give
a stock solution of 10 mM and stored at −20°C in small
aliquots.
Cell culture
Human breast cancer cell lines MCF-7 and MDA-MB-435
were cultured as previously described (19).
Cell viability analysis
Cells were seeded in 96-well flat-bottom plates at a
density of 1×104 cells per well and cultured in a
humidified incubator for 24 h, followed by exposure to various
concentrations of SNS-032 for additional 48 h. Cell viability was
measured by using the MTS
(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium)
assay to monitor cell proliferation, according to the
manufacturer’s recommendations. Briefly, 20 μl MTS solution
(CellTiter 96Aqueous One Solution reagent, Promega, Madison, WI,
USA) was added to each well and incubated for an additional 4 h at
37°C. The absorbance was measured on a 96-well plate reader at
wavelength 490 nm (Bio-Tek Synergy 2, Winooski, VT, USA). Cell
growth inhibition was determined using the following formula
according to a previously published method: growth inhibition (%) =
(1 - OD of treated cells/OD of control cells) × 100% (19). The half maximal inhibitory
concentration (IC50) was calculated with Bliss software
and the data were analyzed by SPSS. All experiments were performed
three times from which mean values were calculated.
Annexin V-FITC (fluorescein
isothiocyanate)/propidium iodide (PI) staining assay
Apoptosis was determined by flow cytometry using
Annexin V-FITC Apoptosis Detection Kit II (BD Pharmingen, San
Diego, CA, USA) according to the manufacturer’s instructions. Cells
(2×105 cells/well) were seeded in 60-mm plates and
allowed to settle for 24 h before treatment with various
concentrations (0, 200 and 400 nM) of SNS-032 for an additional 8
h. The cells were harvested by trypsinization, washed twice with
cold phosphate-buffered saline (PBS), and then resuspended in 1×
binding buffer at a concentration of 1×106 cells/ml.
Following this, 100 μl of the sample solution was transferred to a
5-ml culture tube and incubated with 5 μl of FITC-conjugated
Annexin V and 10 μl of PI for 15 min at room temperature in the
dark. Subsequently, 400 μl of binding buffer was added to each
sample and the samples were analyzed using a flow cytometer
(FACSCanto II, Becton-Dickinson, San José, CA, USA).
TUNEL assay
The TUNEL assay for in situ detection of
apoptosis was performed by using the DeadEnd™ Fluorometric TUNEL
System assay kit (Promega) according to the manufacturer’s
instructions. Cells were plated in 24-well flat-bottom plates at a
density of 1×105 cells per well, treated with 400 nM
SNS-032 for 24 h. Following SNS-032 treatment, cells were fixed in
4% paraformaldehyde at 4°C for 25 min. Fixed cells were then
permeabilized in 0.1% Triton X-100 and labeled with
fluorescein-12-dUTP using terminal deoxynucleotidyl transferase.
After rinsing with PBS, nuclei were counterstained with PI (1
μg/ml) for 15 min. The localized green fluorescence of apoptotic
cells was detected by fluorescence microscopy (Zeiss Axiovert 100M,
Carl Zeiss, Germany).
Caspase activity assay
Activity of caspase-8 and -9 was measured using a
caspase colorimetric assay kit (Keygen Biotech, China), according
to the manufacturer’s protocol. Briefly, after treatment of SNS-032
at different concentrations (0, 200 and 400 nM) for 24 h, cells
were harvested, washed with PBS and then resuspended in chilled
lysis buffer. After incubation on ice for 40 min, cells were
centrifuged for 1 min at 10,000 × g. The supernatant was collected
in a fresh tube and protein concentration was determined by the
Bradford protein assay kit (Keygen Biotech), according to the
manufacturer’s protocol. Subsequently, 150 μg of each protein
sample was diluted with 50 μl lysis buffer and added to 50 ml of 2×
reaction buffer containing 10 mM dithiothreitol in a 96-well plate.
Then, 5 μl of a colorigenic substrate, IETD-pNA
(L-isoleucyl-L-glutamyl-L-Threonyl-L-aspartic-p-nitroanilide acid
amide) or LEHD-pNA
(L-leucine-L-glutamyl-L-histidyl-L-aspartic-p-nitroaniline acid
amide), was added to each well, and the plate was incubated at 37°C
in the dark for 4 h. ODs were determined at 405 nm using a
microplate reader (Bio-Tek Synergy 2).
Western blot analysis
After treatment with SNS-032 at different
concentrations (0, 200 and 400 nM) for 24 h, cells in each dish,
including dead cells floating in medium, were harvested and lysed
in 1× sampling buffer. Protein concentrations of the lysates were
determined using the bicinchoninic acid protein assay kit (Pierce
Biotech, Rockford, IL, USA). An aliquot of the denatured
supernatant containing 30 μg of protein was subjected to sodium
dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), and
then transferred to polyvinylidene fluoride (PVDF) membranes. After
blocking with blocking buffer (Tris-buffered saline, i.e. TBS,
containing 5% non-fat milk) for 1 h at room temperature, the
membranes were incubated overnight at 4°C with the following
specific primary antibodies: mouse anti-human caspase-8, mouse
anti-human caspase-9, rabbit anti-human PARP, rabbit anti-human
phospho-RNA Pol II (Ser2), rabbit anti-human phospho-RNA Pol II
(Ser5), rabbit anti-human RNA Pol II, rabbit anti-human Cdk7,
rabbit anti-human Cdk9, rabbit anti-human Mcl-1, rabbit anti-human
XIAP, rabbit anti-human Bcl-2 (Cell Signaling Technology, Beverly,
MA, USA); and mouse anti-human GAPDH (ProteinTech, Chicago, IL,
USA). Further incubation with appropriate horseradish
peroxidase-conjugated secondary antibodies, depending on the
primary antibody used, was performed for 1 h at room temperature.
Detection of staining signals was performed by using the enhanced
chemiluminescence kit (Thermo Fisher Scientific, Rockford, IL, USA)
with Kodak film.
Real-time PCR
Total RNA was extracted using the RNeasy kit
(Qiagen, Crawley, UK). Each cDNA template was made from total RNA
with reverse transcriptase kit according to the manufacturer’s
instructions (Invitrogen). Amplification reactions were performed
using SYBRW Premix Ex Taq™ (Takara Shuzo, Kyoto, Japan) in a 25 μl
volume. The following cycling parameters were used: 30 sec at 95°C
for initial denaturing, 5 sec at 95°C for denaturing and 30 sec at
60°C for annealing and extension for the total of 40 cycles. The
fold change in mRNA was calculated by the 2−ΔΔCt method.
All samples were normalized to 18S ribosomal RNA, an RNA Pol I
transcript that is not modulated by inhibition of RNA Pol II. The
primer sequences used were: XIAP-up: 5′-CCATATACCCGAGGAA CCCT-3′;
XIAP-dn: 5′-TTTCCACCACAACAAAAGCA-3′; Mcl-1-up:
5′-AAAAGCAAGTGGCAAGAGGA-3′; Mcl-1-dn: 5′-TTAATGAATTCGGCGGGTAA-3′;
Bcl-2-up: 5′-AAG ATTGATGGGATCGTTGC-3′; Bcl-2-dn: 5′-TGTGCTTTGCA
TTCTTGGAC-3′; 18S rRNA-up: 5′-GTAACCCGTTGAACCCC ATT-3′; 18S
rRNA-dn: 5′-CCATCCAATCGGTAGTAGCG-3′.
Xenografted tumor model and antitumor
effect of SNS-032 in vivo
All animal care and experimental procedures were
approved by the Institutional Animal Care and Use Committee of
Guangzhou Medical University. Female BALB/c-nu mice (18–20 g) were
purchased from the Experimental Animal Center of Guangzhou
University of Chinese Medicine, and were housed in barrier
facilities on a 12-h light/dark cycle. On day 0, human breast
cancer MDA-MB-435 cells (5×106 cells in 0.1 ml per
mouse) were inoculated subcutaneously in the right mammary gland.
On day 6, the formed tumors were measured, and the mice were
randomly divided into a treatment group and a control group. The
treatment group received an intraperitoneal (i.p.) dosage of 200 μl
SNS-032 (15 mg/kg body weight) every 3 days, while animals in the
vehicle-control group received i.p. 200 μl 0.5% DMSO solution per
mouse. Tumors were measured every 3 days in blinded manner by
measuring perpendicular diameters with a digital caliper. The tumor
volumes (mm3) were calculated using the following
formula: volume = width × width × length × π/6. Data were presented
as the means ± standard deviation (SD) of six mice in each group.
At the end-point of the experiment, all the animals were
euthanized, and the tumors were dissected and weighed.
Statistical analysis
The data given in the text are expressed as means ±
SD. Statistical significances of the differences in the effects
between vehicle-treated mice and SNS-032-treated ones were analyzed
by Student’s t-test.
Results
Growth inhibition of human breast cancer
cell lines induced by SNS-032
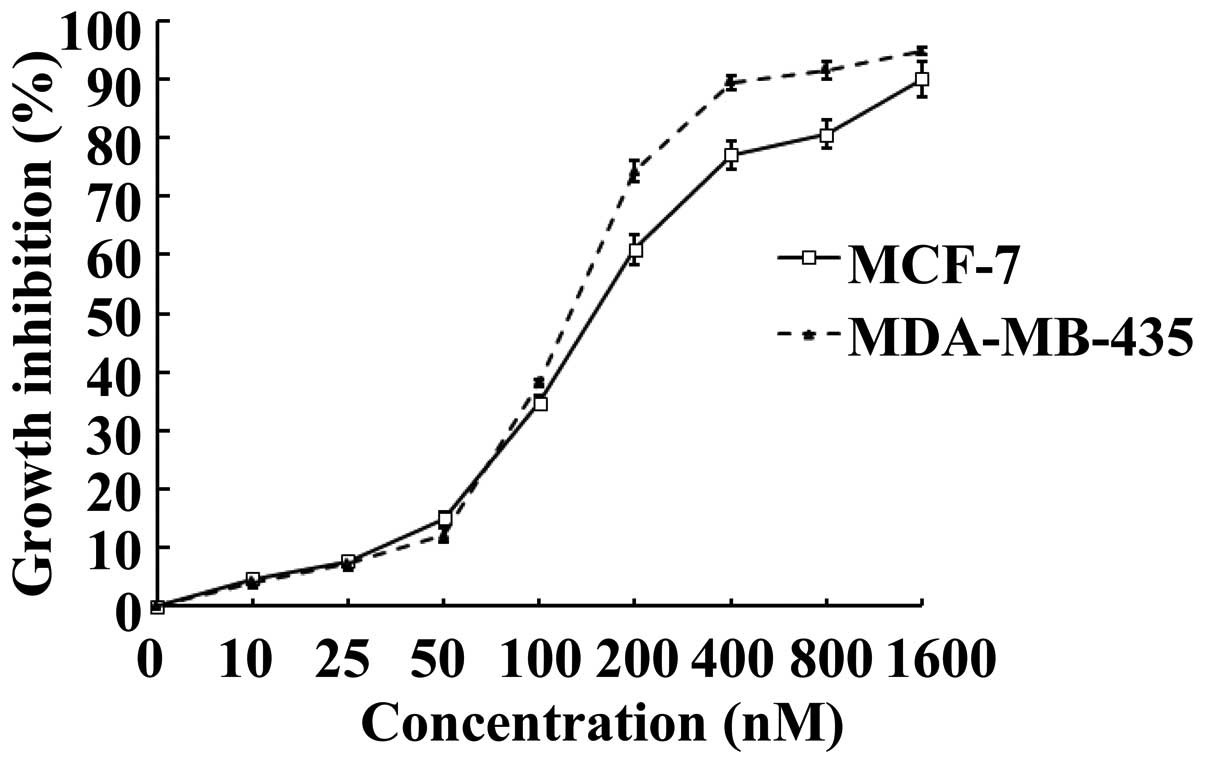
To investigate whether SNS-032 is able to inhibit
the growth of human breast cancer cells, its effects on MCF-7 and
MDA-MB-435 were examined using MTS assay. Upon treatment of SNS-032
for 48 h, cultured MCF-7 and MDA-MB-435 exhibited markedly
inhibited growth, as compared with vehicle-controlled cells in a
dose-dependent manner (Fig. 1).
Calculated IC50 values, i.e., concentrations of SNS-032
required for decreasing the growth rate of the cells by 50%, were
184.0 nM for MCF-7 and 133.6 nM for MDA-MB-435, respectively.
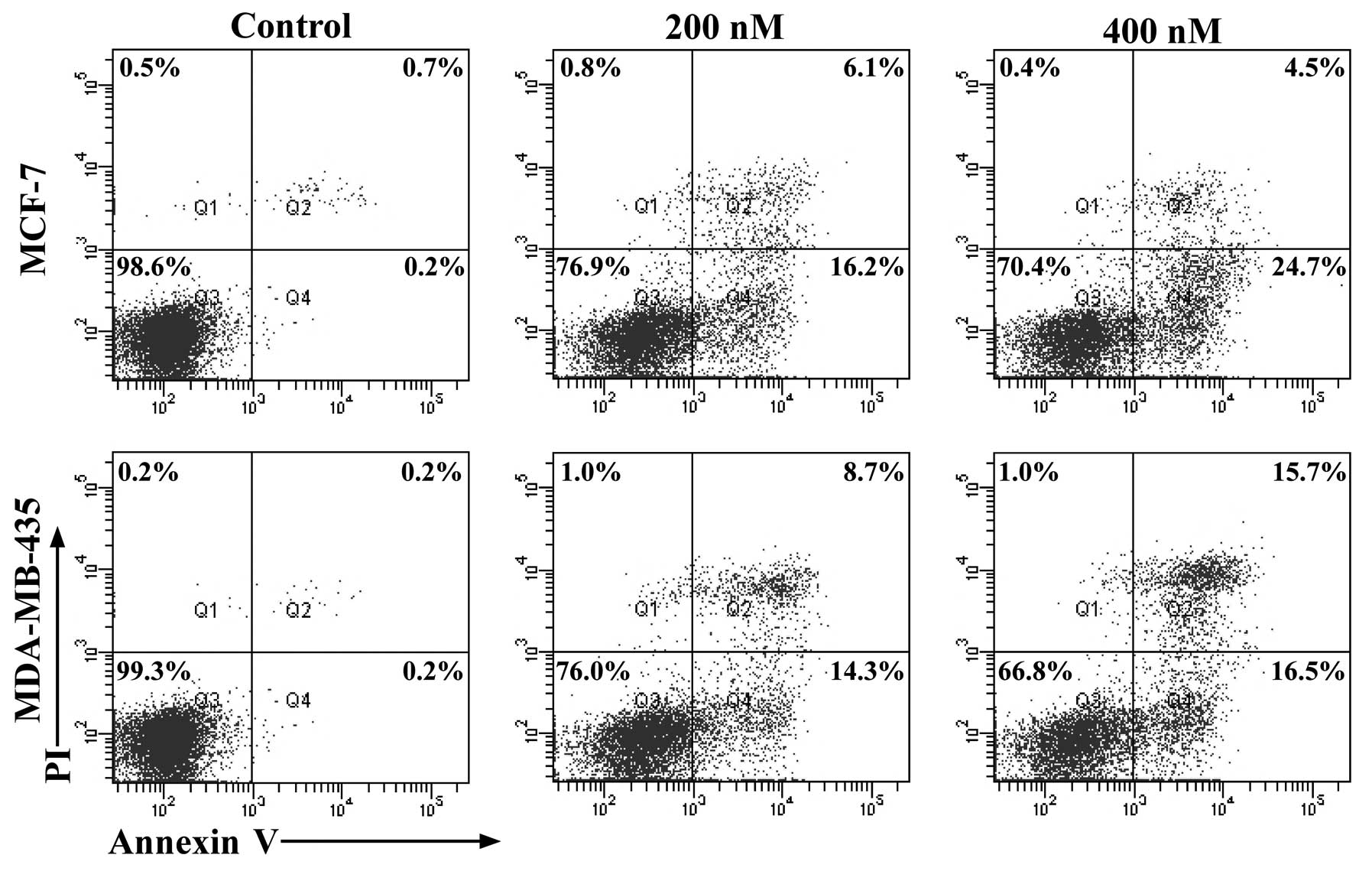
SNS-032 induces apoptosis in MCF-7 and
MDA-MB-435 cells
Apoptosis assays revealed that after treatment of
SNS-032 for 8 h at 200 and 400 nM, respectively, numbers of
apoptotic MCF-7 and MDA-MB-435 (Annexin
V+/PI−), as revealed by Annexin-V binding,
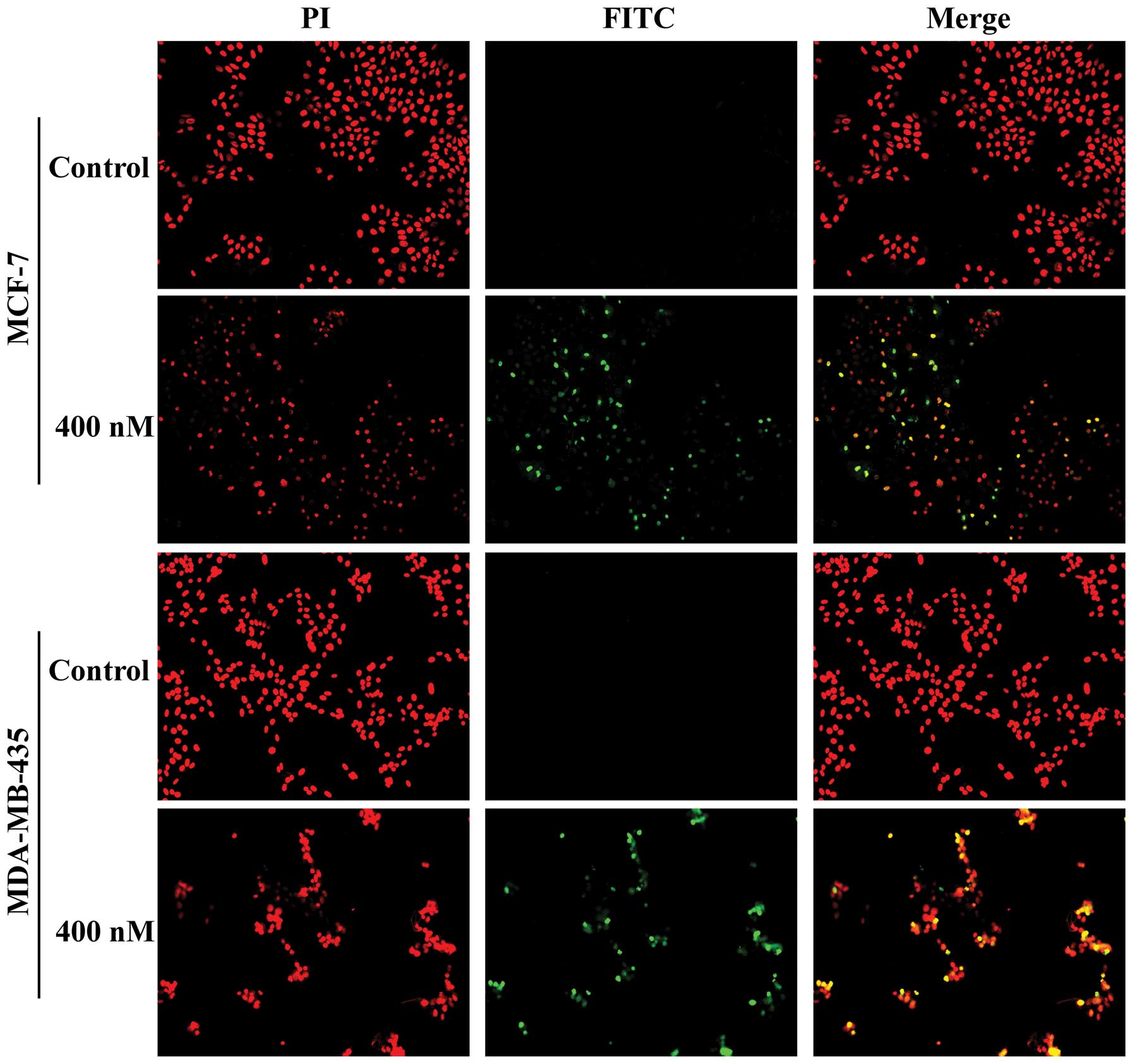
markedly increased in a dose-dependent manner (Fig. 2). When the TUNEL assay was
performed to assess DNA fragmentation as a late event in the
process of apoptosis of MCF-7 and MDA-MB-435 cells, a higher amount
of TUNEL-positive cells were visualized in MCF-7 and MDA-MB-435
cells treated for 24 h with SNS-032 at 400 nM, as compared to the
control (Fig. 3). The results of
two apoptosis assays, i.e., the Annexin V-binding and TUNEL assays,
strongly suggested a pro-apoptotic effect of SNS-032 on breast
cancer cells.
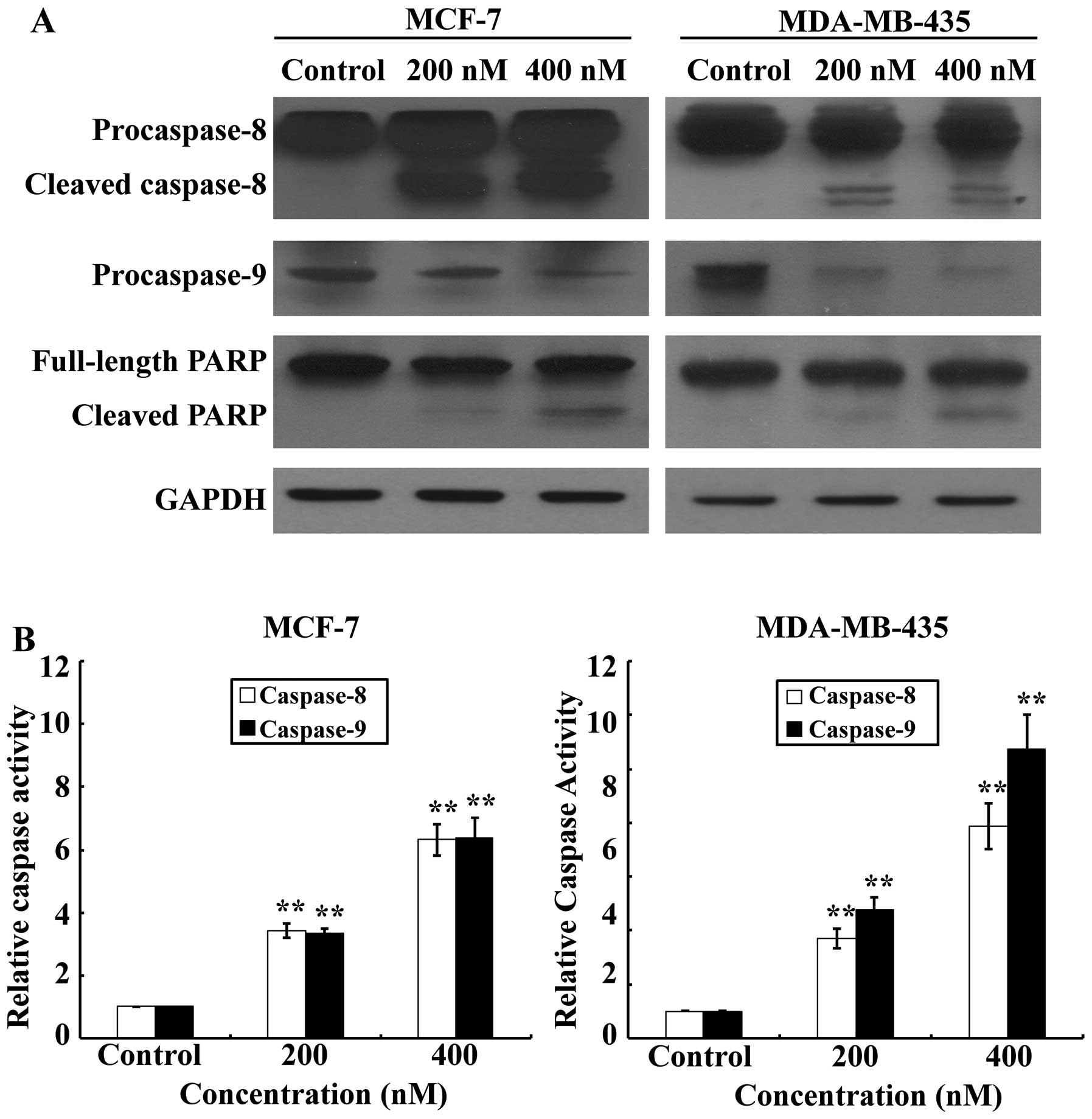
Activation of both the extrinsic and
intrinsic apoptotic pathways by SNS-032
Activation of effector caspases plays a central role
in the execution of apoptosis. To further characterize the cell
apoptotic process in MCF-7 and MDA-MB-435 cells, pro-apoptotic
caspases, i.e., caspase-9 and -8, and the effector molecule PARP,
were examined on SNS-032 treated or untreated MCF-7 and MDA-MB-435
cells, comparatively. Our results (Fig. 4A) showed that treatments with
SNS-032 for 24 h dramatically increased activating cleavage of
caspase-8 dose-dependently in MCF-7 and MDA-MB-435 cells.
Consistent with the results of western blot analysis, the enzymatic
activity of caspase-8 showed a dose-dependent increase with SNS-032
treatment (Fig. 4B). Concurrently,
cleavage of the caspase-9 precursor and increased caspase-9
activity were also detected (Fig.
4). Cleavage of PARP from 116 to 85 kDa was clearly
demonstrated after SNS-032 treatment in MCF-7 and MDA-MB-435 cells
(Fig. 4A). Taken together, these
data suggest that apoptosis induced by SNS-032 in breast cancer
cells may involve both intrinsic and extrinsic apoptotic
pathways.
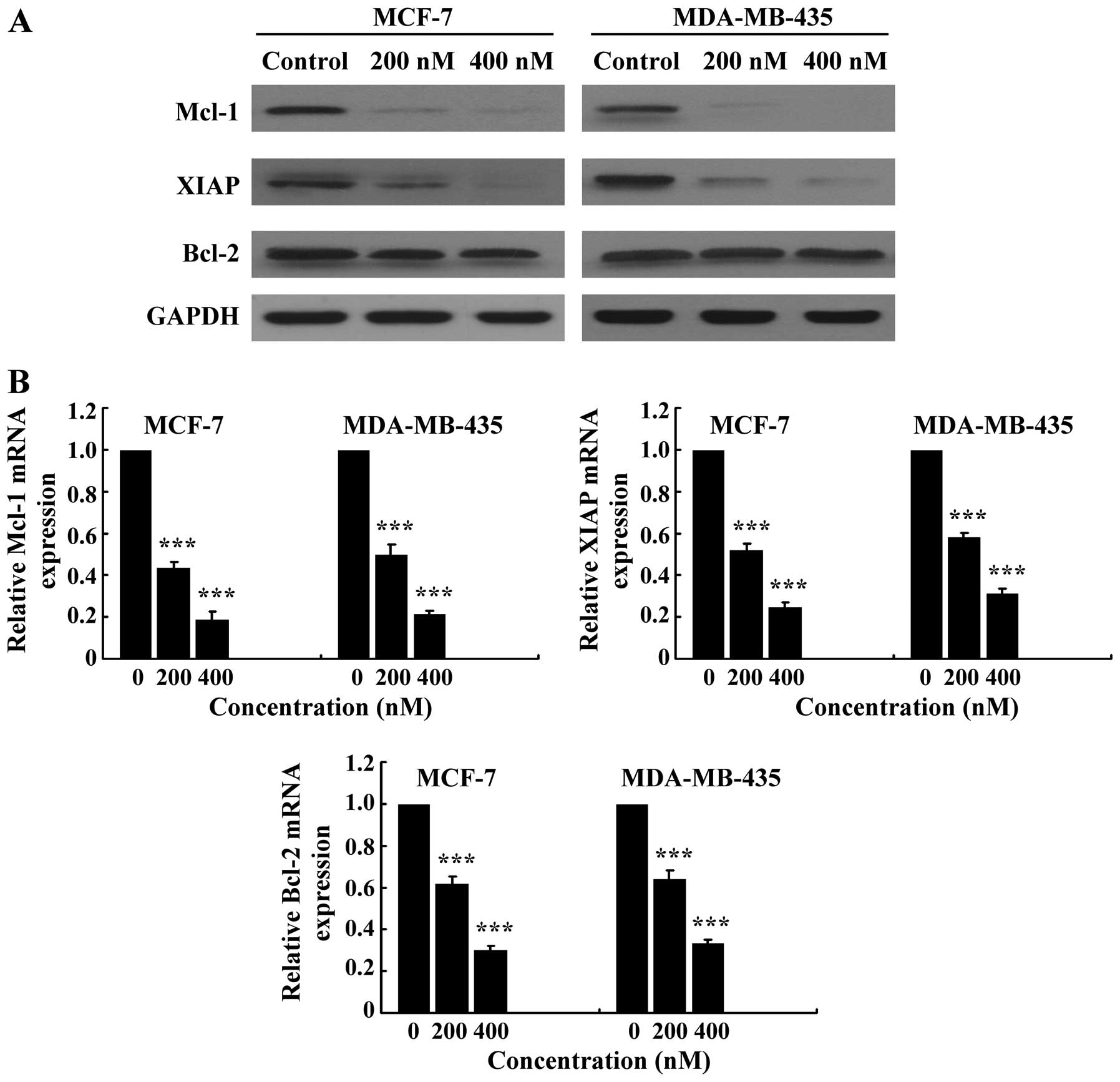
SNS-032 downregulates antiapoptotic
proteins Mcl-1 and XIAP by inhibiting their transcription
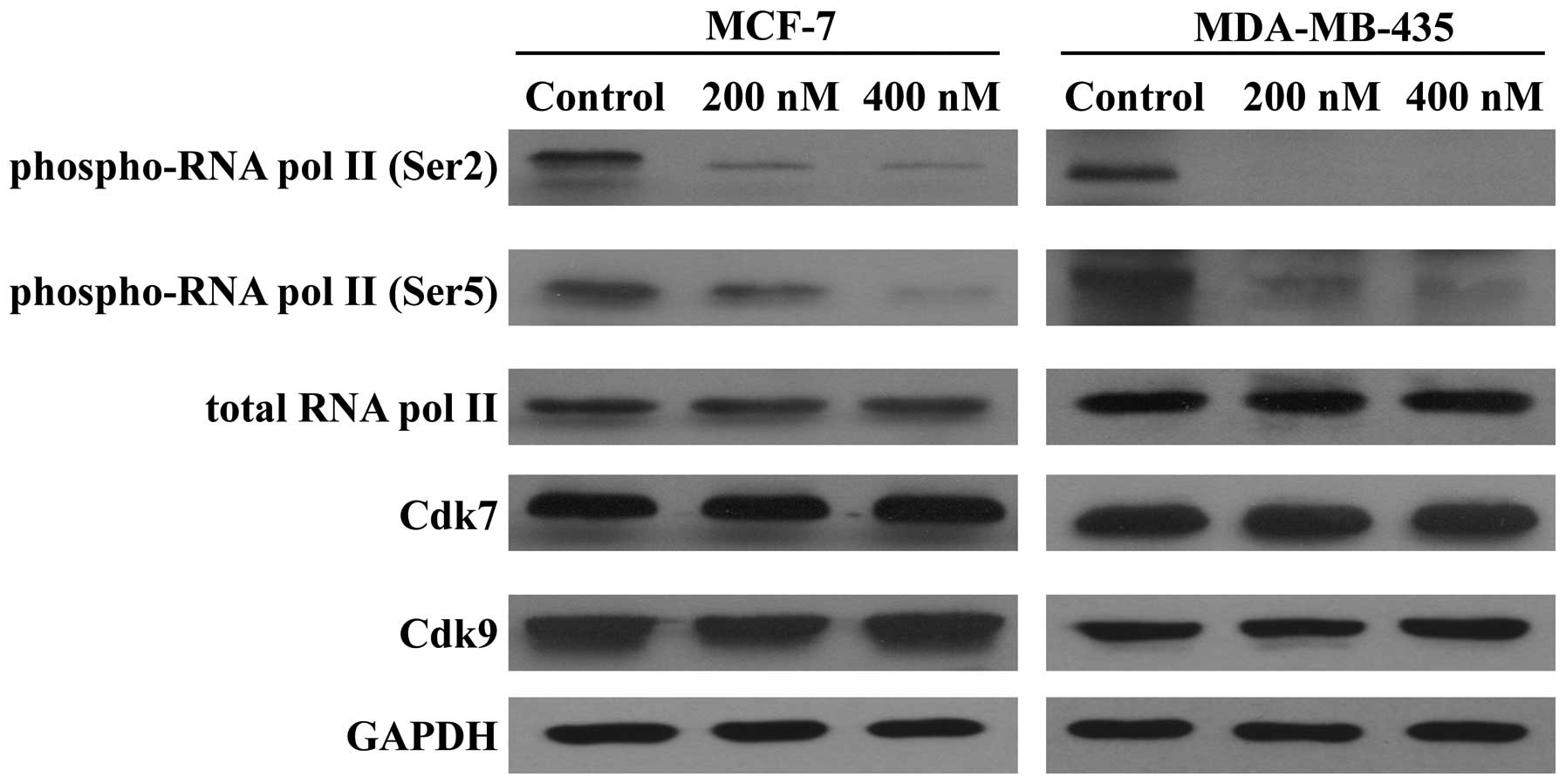
Because SNS-032 is a selective inhibitor of
transcriptional Cdk7 and 9, consequently disabling RNA Pol II and
gene transcription, we further examined its effect on the
expression of antiapoptotic proteins Mcl-1, Bcl-2, and XIAP. As
shown in Fig. 5, exposing MCF-7
and MDA-MB-435 cells to SNS-032 led to a concentration-dependent
decrease in phosphorylated RNA Pol II at Ser2 and Ser5 but not
total protein. The protein levels of Mcl-1 and XIAP were decreased
concentration dependently, while there was no significant change in
the Bcl-2 protein (Fig. 6A),
consistent with a much longer protein half-life (18,20).
RT-PCR revealed that SNS-032 decreased the mRNA levels of Mcl-1 and
XIAP in MCF-7 and MDA-MB-435 cells (Fig. 6B). These results suggest that the
loss of Mcl-1 and XIAP proteins correlates with their
transcriptional inhibition due to the blocking of RNA Pol II
phosphorylation by SNS-032.
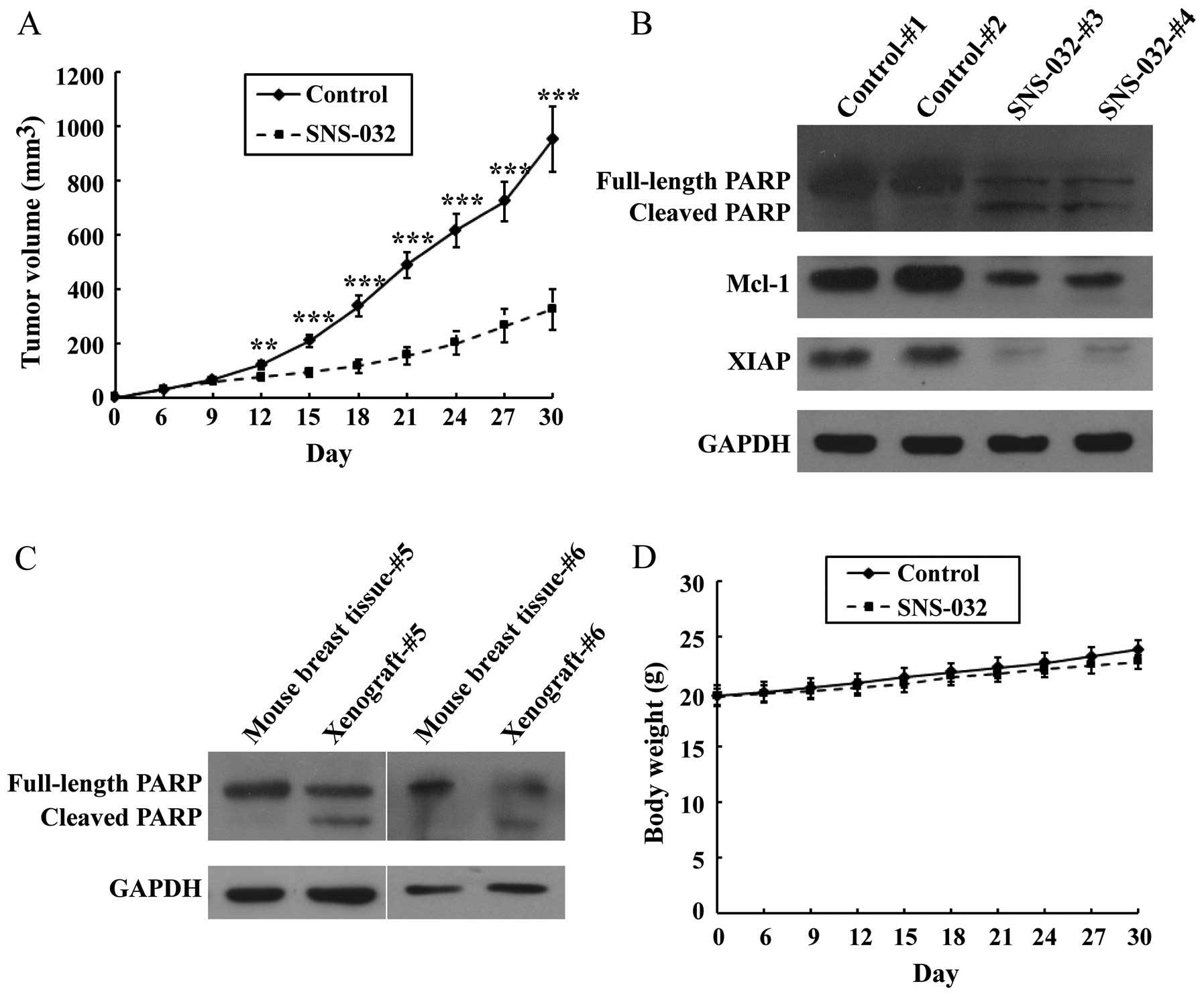
Antitumor effect of SNS-032 on breast
cancer xenografts in vivo
To evaluate the antitumor activity of SNS-032
against breast cancer in vivo, we next tested the
therapeutic effect of SNS-032 on MDA-MB-435 xenografts in a nude
mouse model. When the tumor volumes were assessed on day 6 after
inoculation, all the animals in each group were found to have
developed spinal cord tumors (6/6, or 100%), with a mean volume (±
SD) of ~30 mm3. The growth of the xenograft tumors were
monitored following injection with SNS-032. A marked inhibition of
the growth of the xenografted tumors treated with SNS-032 was
observed. After 30 days of drug administration (eight SNS-032
injections), the volume of the xenografted breast tumor was
significantly inhibited by 65.77% in SNS-032-treated nude mice
(Fig. 7A).
To assess whether this SNS-032-induced tumor
suppression resulted from apoptosis of the grafted cells, western
blot analysis of PARP, Mcl-1 and XIAP was carried out. The results
showed that injection of SNS-032 induced PARP cleavage in the
xenografted breast cancer cells. In contrast, minimal PARP cleavage
was detected in tumors excised from vehicle-treated controls. The
levels of Mcl-1 and XIAP were significantly lower in tumor tissues
from mice treated with SNS-032 than in control treatment (Fig. 7B), so SNS-032 may induce apoptosis
and deplete antiapoptotic proteins Mcl-1 and XIAP in
vivo.
To assess whether administration of SNS-032 induced
apoptosis in normal breast tissues, western blot analysis of PARP
was carried out using two representative tumor tissues from mice
treated with SNS-032 and their paired adjacent non-tumor breast
tissues. The results showed that SNS-032 did not induce apoptosis
in normal breast tissues (Fig.
7C). Meanwhile, no signs of adverse effects, such as
discomfort, behavioural changes or weight loss (Fig. 7D), were observed in SNS-032-treated
animals. These data strongly suggest that systemically delivered
SNS-032 was able to inhibit the growth of established tumors in
vivo.
Discussion
Despite great advances in screening techniques and
therapy, breast cancer remains a major health problem worldwide,
being the most common cancer and the second leading cause of cancer
death among women (1). Typically,
the treatment of breast cancer involves antihormonal therapy with
the selective estrogen receptor (ER) modulator tamoxifen (21). However, ~30% of ERα (+) tumors do
not respond to tamoxifen or develop resistance in the course of the
treatment (22,23). In addition, the clinical utility of
ER modulators is often limited by side effects and is largely
ineffective against ER-negative breast cancer (24,25).
Therefore, there is an urgent need to explore novel agents that are
relatively safe but can suppress growth of both ER-positive and
ER-negative human breast cancers.
Cdks play a critical role in cancer progression
making them very promising therapeutic targets in human
malignancies. As the development of the pan Cdk inhibitor
flavopiridol, more specific Cdk inhibitors have been developed with
encouraging results (26–29). In this study, we present our
findings on the Cdk inhibitor SNS-032 in breast cancer cells.
SNS-032 is a highly selective and potent inhibitor
of Cdks 2, 7 and 9 (17). In
addition to its potency and high selectivity, SNS-032 was selected
for development based on its favorable characteristics including
low protein binding in human serum (18) compared with the high degree of
protein binding (92–95%) seen with flavopiridol (30). Despite promising in vitro
activity initially, flavopiridol, the first pan-Cdk inhibitor to
enter clinical trials, demonstrated no significant clinical
activity in phase I/II studies in patients with solid or
hematologic malignancies (31–33).
Subsequent investigations revealed significant binding to human
plasma proteins that altered free drug level, target cell exposure,
and therefore therapeutic activity (30,34).
Subsequently, a pharmacokinetically derived schedule of
flavopiridol administered as a 30-min intravenous bolus followed by
4-h continuous intravenous infusion that sustained half maximal
inhibitory concentration level was active in refractory chronic
lymphocytic leukemia (CLL) (35–37),
which indicates high plasma protein binding may be a key reason for
the previous lack of clinical activity of flavopiridol. In
comparison with flavopiridol, SNS-032 exhibited moderately low
protein binding (63%) in human serum (18). Therefore, SNS-032 has biochemical
and pharmacologic properties that differ from flavopiridol, which
probably predict differing activities in the clinic.
A phase I trial of SNS-032 in advanced solid tumors
including colon cancer, lung cancer, pancreatic cancer and breast
cancer showed that this agent was well tolerated in a total of 21
patients enrolled in this study and oral administration may be
feasible (38). In another phase I
multicenter trial of SNS-032 in patients with advanced B-lymphoid
malignancies, including CLL and multiple myeloma (MM), single-agent
SNS-032 demonstrated mechanism-based target modulation as well as
modest clinical activity in heavily pretreated patients with CLL
and MM (39). In this study, we
present our findings on SNS-032 in breast cancer cells.
As our data show, SNS-032 displayed potent cytocidal
effect on MCF-7 and MDA-MB-435 breast cancer cells, with
IC50 <200 nM. SNS-032, at nanomolar concentrations,
induced significant apoptosis in MCF-7 and MDA-MB-435 cells as
evidenced by activation of caspases, PARP cleavage, Annexin
V-positive binding and TUNEL-positive staining. Of the two breast
cancer cell lines tested, MCF-7 is relatively well differentiated
and estrogen-dependent, whereas MDA-MB-435 is an invasive and
estrogen-independent line. The equally, if not more, effective
inhibition against MDA-MB-435 cells (IC50, 133.6 nM) by
SNS-032 as compared with that against the ER-positive, less
invasive and more differentiated MCF-7 cells (IC50,
184.0 nM) warrants further exploration of the possibility to treat
ER-negative breast cancer.
Cdk inhibitors that function as transcriptional
repressors inhibit RNA Pol II activation by preventing its
phosphorylation, the result is a blockage of gene transcription,
which in turn causes downregulation of short-lived proteins
including some antiapoptotic molecules such as Mcl-1 and XIAP
(18). Mcl-1, an antiapoptotic
member of the Bcl-2 family, is among the most frequently amplified
genes in human cancer and is essential for the survival of
carcinoma cells. Mcl-1 is thought to act by antagonizing
pro-apoptotic proteins such as Bim (40). XIAP is a member of the IAP family
and plays a key role in cell survival. As the most potent human IAP
protein currently identified, XIAP blocks cell death by virtue of
inhibition of distinct caspases (41). In this study, our results with
breast cancer cells treated with SNS-032 showed a
concentration-dependent dephosphorylation of RNA Pol II at serine 5
and 2. In addition, inhibition of transcription substantially
reduced Mcl-1 and XIAP expression, whereas the Bcl-2 protein level
remained stable. The rate of decrease in the protein levels of
Mcl-1 and XIAP was proportional to their half-lives, with Mcl-1
being the most labile. There was no apparent decrease in Bcl-2
protein level, consistent with a much longer protein half-life
(18,20). When Mcl-1 and XIAP are diminished,
the balance between the antiapoptotic and proapoptotic proteins
will be altered, then irreversibly initiating apoptosis. Previous
studies have shown that strategies reducing Mcl-1 or XIAP
expression can sensitize breast cancer cells to other
chemotherapeutic agents such as lapatinib, etoposide and
doxorubicin (42,43). In this regard, our data that
exposure of breast cancer cells to SNS-032 decreased Mcl-1 and XIAP
expression suggest that SNS-032 might be also effective in
enhancing the effects of chemotherapeutics and reducing resistance
to conventional chemotherapy in breast cancer.
Consistent with our findings in vitro,
SNS-032 significantly suppressed tumor growth in nude mice bearing
MDA-MB-435 tumors, and it was able to induce apoptosis and deplete
antiapoptotic proteins Mcl-1 and XIAP in vivo. It should be
noted that a smaller effect on PARP cleavage was observed in
non-tumor breast tissues. Moreover, no signs of adverse effects,
such as discomfort, behavioural changes or weight loss, were
observed in SNS-032-treated mice. These results suggest that
SNS-032 may be employed as a selective cytotoxic agent for the
elimination of cancer cells.
In conclusion, the results of this study demonstrate
that SNS-032 has significant antitumor activity against human
breast cancer cells both in vitro and in vivo by
inducing apoptosis through activation of both extrinsic and
intrinsic apoptotic pathways. Dephosphorylation of RNA Pol II and
inhibition of Mcl-1 and XIAP RNA synthesis would also contribute to
the apoptotic response induced by SNS-032. Given that SNS-032 has
favorable characteristics including high Cdk inhibitory
selectivity, low protein binding, relatively low toxicity in normal
breast cells and significant antitumor activity in human breast
cancer cells, it is thought that the use of SNS-032 might be a
rational and novel therapeutic strategy for human breast cancer and
warrants further clinical investigation.
Acknowledgements
This study was supported by a grant from the
National Natural Science Foundation of China (no. 81101682), a
Science and Technology Planning Project of Guangzhou Municipal
Health Bureau (no. 201102A213045), a Shenzhen Science and
Technology Planning Project (no. 201303072) and a PhD Start-up Fund
of Guangzhou Women and Children’s Medical Center (no. 201012).
References
|
1
|
Friedenreich CM: Physical activity and
breast cancer: review of the epidemiologic evidence and biologic
mechanisms. Recent Results Cancer Res. 188:125–139. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jafaar ZM, Litchfield LM, Ivanova MM,
Radde BN, Al-Rayyan N and Klinge CM: β-D-glucan inhibits
endocrine-resistant breast cancer cell proliferation and alters
gene expression. Int J Oncol. 44:1365–1375. 2014.
|
|
3
|
Perez EA: Impact, mechanisms, and novel
chemotherapy strategies for overcoming resistance to anthracyclines
and taxanes in metastatic breast cancer. Breast Cancer Res Treat.
114:195–201. 2009. View Article : Google Scholar
|
|
4
|
Gallorini M, Cataldi A and di Giacomo V:
Cyclin-dependent kinase modulators and cancer therapy. BioDrugs.
26:377–391. 2012.PubMed/NCBI
|
|
5
|
Canavese M, Santo L and Raje N: Cyclin
dependent kinases in cancer: potential for therapeutic
intervention. Cancer Biol Ther. 13:451–457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Peterlin BM and Price DH: Controlling the
elongation phase of transcription with P-TEFb. Mol Cell.
23:297–305. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Larochelle S, Amat R, Glover-Cutter K, et
al: Cyclin-dependent kinase control of the initiation-to-elongation
switch of RNA polymerase II. Nat Struct Mol Biol. 19:1108–1115.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cho SJ, Kim YJ, Surh YJ, Kim BM and Lee
SK: Ibulocydine is a novel prodrug Cdk inhibitor that effectively
induces apoptosis in hepatocellular carcinoma cells. J Biol Chem.
286:19662–19671. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang S and Fischer PM: Cyclin-dependent
kinase 9: a key transcriptional regulator and potential drug target
in oncology, virology and cardiology. Trends Pharmacol Sci.
29:302–313. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lapenna S and Giordano A: Cell cycle
kinases as therapeutic targets for cancer. Nat Rev Drug Discov.
8:547–566. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Diaz-Padilla I, Siu LL and Duran I:
Cyclin-dependent kinase inhibitors as potential targeted anticancer
agents. Invest New Drugs. 27:586–594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu X, Shi S, Lam F, Pepper C, Fischer PM
and Wang S: CDKI-71, a novel CDK9 inhibitor, is preferentially
cytotoxic to cancer cells compared to flavopiridol. Int J Cancer.
130:1216–1226. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shapiro GI: Preclinical and clinical
development of the cyclin-dependent kinase inhibitor flavopiridol.
Clin Cancer Res. 10:S4270–S4275. 2004. View Article : Google Scholar
|
|
14
|
Wang LM and Ren DM: Flavopiridol, the
first cyclin-dependent kinase inhibitor: recent advances in
combination chemotherapy. Mini Rev Med Chem. 10:1058–1070. 2012.
View Article : Google Scholar
|
|
15
|
Gojo I, Zhang B and Fenton RG: The
cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in
multiple myeloma cells through transcriptional repression and
down-regulation of Mcl-1. Clin Cancer Res. 8:3527–3538. 2002.
|
|
16
|
Wittmann S, Bali P, Donapaty S, et al:
Flavopiridol down-regulates antiapoptotic proteins and sensitizes
human breast cancer cells to epothilone B-induced apoptosis. Cancer
Res. 63:93–99. 2003.PubMed/NCBI
|
|
17
|
Conroy A, Stockett DE, Walker D, et al:
SNS-032 is a potent and selective CDK 2, 7 and 9 inhibitor that
drives target modulation in patient samples. Cancer Chemother
Pharmacol. 64:723–732. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen R, Wierda WG, Chubb S, et al:
Mechanism of action of SNS-032, a novel cyclin-dependent kinase
inhibitor, in chronic lymphocytic leukemia. Blood. 113:4637–4645.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xie G, Zhu X, Li Q, et al: SZ-685C, a
marine anthraquinone, is a potent inducer of apoptosis with
anticancer activity by suppression of the Akt/FOXO pathway. Br J
Pharmacol. 159:689–697. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Blagosklonny MV, Alvarez M, Fojo A and
Neckers LM: Bcl-2 protein downregulation is not required for
differentiation of multidrug resistant HL60 leukemia cells. Leuk
Res. 20:101–107. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Du Y, Shi A, Han B, et al: COX-2 silencing
enhances tamoxifen antitumor activity in breast cancer in
vivo and in vitro. Int J Oncol. 44:1385–1393.
2014.PubMed/NCBI
|
|
22
|
Riggins RB, Schrecengost RS, Guerrero MS
and Bouton AH: Pathways to tamoxifen resistance. Cancer Lett.
256:1–24. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang B, Zhang X, Tang B, Zheng P and
Zhang Y: Investigation of elemene-induced reversal of tamoxifen
resistance in MCF-7 cells through oestrogen receptor α (ERα)
re-expression. Breast Cancer Res Treat. 136:399–406.
2012.PubMed/NCBI
|
|
24
|
Núñez M, Medina V, Cricco G, et al:
Glibenclamide inhibits cell growth by inducing G0/G1 arrest in the
human breast cancer cell line MDA-MB-231. BMC Pharmacol Toxicol.
14:62013.PubMed/NCBI
|
|
25
|
Jordan VC and Brodie AM: Development and
evolution of therapies targeted to the estrogen receptor for the
treatment and prevention of breast cancer. Steroids. 72:7–25. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dickson MA and Schwartz GK: Development of
cell-cycle inhibitors for cancer therapy. Curr Oncol. 16:36–43.
2009.PubMed/NCBI
|
|
27
|
Rizzolio F, Tuccinardi T, Caligiuri I,
Lucchetti C and Giordano A: CDK inhibitors: from the bench to
clinical trials. Curr Drug Targets. 11:279–290. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McInnes C: Progress in the evaluation of
CDK inhibitors as anti-tumor agents. Drug Discov Today. 13:875–881.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: a changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rudek MA, Bauer KS Jr, Lush RM III, et al:
Clinical pharmacology of flavopiridol following a 72-hour
continuous infusion. Ann Pharmacother. 37:1369–1374. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Byrd JC, Peterson BL, Gabrilove J, et al:
Treatment of relapsed chronic lymphocytic leukemia by 72-hour
continuous infusion or 1-hour bolus infusion of flavopiridol:
results from Cancer and Leukemia Group B study 19805. Clin Cancer
Res. 11:4176–4181. 2005. View Article : Google Scholar
|
|
32
|
Flinn IW, Byrd JC, Bartlett N, et al:
Flavopiridol administered as a 24-hour continuous infusion in
chronic lymphocytic leukemia lacks clinical activity. Leuk Res.
29:1253–1257. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aklilu M, Kindler HL, Donehower RC, Mani S
and Vokes EE: Phase II study of flavopiridol in patients with
advanced colorectal cancer. Ann Oncol. 14:1270–1273. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Christian BA, Grever MR, Byrd JC and Lin
TS: Flavopiridol in the treatment of chronic lymphocytic leukemia.
Curr Opin Oncol. 19:573–578. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Byrd JC, Lin TS, Dalton JT, et al:
Flavopiridol administered using a pharmacologically derived
schedule is associated with marked clinical efficacy in refractory,
genetically high-risk chronic lymphocytic leukemia. Blood.
109:399–404. 2007. View Article : Google Scholar
|
|
36
|
Phelps MA, Lin TS, Johnson AJ, et al:
Clinical response and pharmacokinetics from a phase 1 study of an
active dosing schedule of flavopiridol in relapsed chronic
lymphocytic leukemia. Blood. 113:2637–2645. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lin TS, Ruppert AS, Johnson AJ, et al:
Phase II study of flavopiridol in relapsed chronic lymphocytic
leukemia demonstrating high response rates in genetically high-risk
disease. J Clin Oncol. 27:6012–6018. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Heath EI, Bible K, Martell RE, Adelman DC
and Lorusso PM: A phase 1 study of SNS-032 (formerly BMS-387032), a
potent inhibitor of cyclin-dependent kinases 2, 7 and 9
administered as a single oral dose and weekly infusion in patients
with metastatic refractory solid tumors. Invest New Drugs.
26:59–65. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tong WG, Chen R, Plunkett W, et al: Phase
I and pharmacologic study of SNS-032, a potent and selective Cdk2,
7, and 9 inhibitor, in patients with advanced chronic lymphocytic
leukemia and multiple myeloma. J Clin Oncol. 28:3015–3022. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Naumann K, Schmich K, Jaeger C, Kratz F
and Merfort I: Noxa/Mcl-1 balance influences the effect of the
proteasome inhibitor MG-132 in combination with anticancer agents
in pancreatic cancer cell lines. Anticancer Drugs. 23:614–626.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li QQ, Lee RX, Liang H, Wang G, Li JM,
Zhong Y and Reed E: β-Elemene enhances susceptibility to cisplatin
in resistant ovarian carcinoma cells via downregulation of ERCC-1
and XIAP and inactivation of JNK. Int J Oncol. 43:721–728.
2013.
|
|
42
|
Martin AP, Mitchell C, Rahmani M, Nephew
KP, Grant S and Dent P: Inhibition of MCL-1 enhances lapatinib
toxicity and overcomes lapatinib resistance via BAK-dependent
autophagy. Cancer Biol Ther. 8:2084–2096. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lima RT, Martins LM, Guimarães JE, Sambade
C and Vasconcelos MH: Specific downregulation of bcl-2 and xIAP by
RNAi enhances the effects of chemotherapeutic agents in MCF-7 human
breast cancer cells. Cancer Gene Ther. 11:309–316. 2004. View Article : Google Scholar : PubMed/NCBI
|