Introduction
Oral cancer is the sixth most common cancer
worldwide, and is responsible for 128,000 deaths each year
(1). In south-central Asian
countries, where tobacco use is prevalent, oral cancer is the most
common cancer in men, contributing up to 25% of all new cases of
cancer (2). Delayed patient
presentation combined with lack of effective modalities of
treatment of advanced oral cancers contribute to a 5-year survival
rate of 50% (2) a figure that has
changed little over the last 40 years (3). In the United States, the 5-year
survival rate for patients with localized oral tumors is roughly
80%, however, nearly two-thirds of patients diagnosed with oral
cancer initially present with regional or distant metastases, which
are correlated with survival rates of 57 and 37%, respectively
(3). Because invasion is a key
correlate of patient survival, a more definitive understanding of
the specific mechanisms associated with oral squamous carcinoma
metastasis is critical to improving patient outcomes in both
preventive and therapeutic contexts.
Tumor metastasis is facilitated by a highly
coordinated tandem of increased migratory ability coupled with
increased proteolytic activity towards extracellular matrix
components. The acquisition of such characteristics is one of the
hallmark features of a cellular dedifferentiation program termed
epithelial-to-mesenchymal transition (EMT) (4). An early event in EMT is the
downregulation of E-cadherin, a transmembrane glycoprotein that
plays a critical role in epithelial cell adhesion and in the
maintenance of the polarity of the epithelial layer. Loosening of
cell-cell contacts is part of a coordinated alteration of the
epithelial phenotype that results in the acquisition of mesenchymal
characteristics: loss of polarity, increased motility, fibroblastic
morphology and expression of mesenchymal proteins such as vimentin.
It is this mesenchymal reprogramming that enables cells to
dissociate from the primary tumor nest (5,6) and
eventually invade into the surrounding tissue.
A well-documented phenomena of EMT is a ‘cadherin
switch’, in which E-cadherin loss is accompanied by the de
novo expression of the mesenchymal adhesion protein N-cadherin
(7,8). A growing body of research has
identified a role for N-cadherin in tumor progression that is
causative rather than coincidental. Ectopic expression of
N-cadherin in oral, breast and bladder carcinoma cell lines has
been shown to increase both motility and invasiveness (9–11).
De novo expression of N-cadherin has been found in both
poorly-differentiated tumors and the invasive front of
well-differentiated tumors in several tissue types (12–14).
In oral squamous carcinomas, the presence of N-cadherin has been
strongly correlated with loco-regional invasion and poor patient
prognosis (14,15).
Invasion is facilitated by both increased migration
and by increased activity of matrix metalloproteinases, a family of
zinc-dependent endopeptidases that degrade extracellular matrix
components (16). In several
cohort studies of oral squamous carcinoma, elevated expression of
matrix metalloproteinase-9 (MMP-9) was correlated with regional
lymph node and/or distant metastases (17,18)
and adversely correlated with survival (17). MMP-9 has been identified as a
modulatory target of both E- and N-cadherin-dependent signaling
(11,19–21).
In oral keratinocytes and bronchial cells, MMP-9 expression was
suppressed by E-cadherin-mediated adhesion (20–22),
whereas in breast cells, MMP-9 expression increased in the presence
of N-cadherin (11,19).
Although the role for N-cadherin in conferring
migratory ability to epithelial cells is well established (9,10,23,24),
very few studies have examined the effect of ectopic N-cadherin
expression on matrix metalloproteinase activity. In breast cells,
N-cadherin expression potentiated the MMP-9 expression that
resulted from fibroblast growth factor receptor (FGFR) signaling,
but did not increase basal MMP-9 expression in untreated cells
(19). The means by which
N-cadherin promotes invasion may be tissue-specific, however, as a
similar response to FGF was not seen in N-cadherin expressing
bladder cancer cells (11). Oral
squamous cells are one of many cell types in which the presence of
N-cadherin decreases E-cadherin protein levels (10), thus raising the possibility that
oral tumor progression is facilitated not only by de novo
N-cadherin signaling but also by concomitant decreases in
E-cadherin function.
In the present study, we utilized two oral squamous
carcinoma cell lines to examine the relative roles of N- and
E-cadherin in promoting matrix metalloproteinase expression and
invasive signaling in oral cancer. We also utilized chimeric
constructs consisting of reciprocally substituted E- and N-cadherin
domains to identify features of N-cadherin that are essential for
matrix metalloproteinase expression, migration and invasion in oral
squamous cells. Finally, we have determined the relevance of the
cadherin-associated proteins and transcriptional modulators
β-catenin and p120 in facilitating N-cadherin-dependent invasion.
Our data demonstrate that it is the cytoplasmic portion of
N-cadherin which confers increased MMP-9 expression to oral
squamous carcinoma cells, and suggest a role for the N-cadherin
cytoplasmic binding partner β-catenin in modulating MMP-9
transcription.
Materials and methods
Cell culture
The oral squamous carcinoma cell lines Tu167
(25) and SCC1 cells (26) were maintained at 37°C, 5%
CO2, in minimum essential medium (Sigma) supplemented
with penicillin, streptomycin and 10% fetal bovine serum (PAA).
Murine fibroblast NIH3T3 cells (American Tissue Culture Collection)
were maintained in Dulbecco’s modified Eagle’s medium supplemented
with 10% newborn calf serum, penicillin and streptomycin.
Retroviral transduction
cDNAs encoding full-length human N-cadherin
(27), chimeric EN and NE
cadherins (27) or p120-uncoupled
N-cadherin were subcloned into the retroviral expression vector
LZRS-MS-Pac (28). Construction of
the chimeric EN and NE cadherins has been previously described
(9). The N-cadherin mutant was
generated using a QuickChange site-directed mutagenesis kit. This
cDNA contains three sequential alanine substitutions (E780A, E781A
and D782A) (23,29), which correspond to homologous
mutations in the E-cadherin sequence that abrogate binding of p120
(30). For depletion of endogenous
cadherin transcripts, oligonucleotides directing the formation of
short hairpin RNAs against human N-cadherin (17) or E-cadherin (GGCCTCTACGGTTTCATAA)
were cloned into pSuper. retro.puro retroviral expression vectors
(Oligoengine). Production of amphotropic retrovirus and subsequent
infection of Tu167 and SCC1 cells was performed as previous
described (31).
Immunoblot analysis
Detergent extraction of cell monolayers and SDS-PAGE
was performed as described previously (32). The mouse monoclonal antibodies
directed against N-cadherin (13A9), E-cadherin (4A2), P-cadherin
(6A9), β-catenin (15B8), α-catenin (1G5) have been described
previously (31). The mouse
monoclonal anti-p120 antibody was purchased from BD Biosciences,
and mouse monoclonal anti-β-tubulin from the Developmental Studies
Hybridoma Bank (University of Iowa).
Migration and invasion assays
Tu167 and SCC1 cells were plated in serum free media
in 24-well Matrigel-coated or uncoated Boyden chambers (8 μm pores,
BD Biosciences) at a cell density of 7.5×104
cells/chamber. Media conditioned by NIH3T3 cells (Dulbecco’s
modified Eagle’s medium + 10% newborn calf serum) was used as a
chemoattractant in the lower chamber. Membranes were collected
after 24 (motility) or 48 (invasion) hours, and the upper surface
of each insert membrane was scraped with a cotton swab to remove
cells that had not traversed through to the other side. The
remaining cells were stained with Diff-Quick (Dade) and counted.
Quantitation was performed by counting cells in 9 random fields of
view at 100× and expressing the average number of cells/field of
view. All experiments were repeated in triplicate. The data are
presented as the average of three independent experiments with the
standard deviation of the average indicated. For presentation of
the Invasion Indices, cell counts for invasion were normalized to
the counts obtained for motility of each respective cell line, and
expressed as a percentage compared to controls.
Substrate gel electrophoresis
For zymographic analysis, confluent cell cultures in
6-well plates were incubated in serum-free MEM for 18 h and then in
newly replaced collection media (serum-free MEM) for an additional
24 h. To normalize to cell number at time of media collection, cell
monolayers were rinsed twice with phosphate-buffered saline and
lysed with RIPA buffer. Volumes of conditioned MEM media
proportional to the total recovered protein from each culture were
analyzed for gelatinase activity according to the protocol of Leber
and Balkwill (33). Gels were
imaged and areas of clearing (representing gelatinase activity)
were quantitated using the Odyssey Infrared Imaging System
(Licor).
Luciferase reporter assays
Cells were transfected with TOPFlash or FOPFlash
firefly luciferase vectors, and a Renilla luciferase control
vector (Promega). Lysates were collected for analysis 24 h after
transfection. Firefly and Renilla luciferase activities were
determined utilizing the Dual Glo Luciferase Assay Kit (Promega).
The values reported were corrected for TCF/LEF-independent
transcriptional activation and transfection efficiency. Data shown
are the average of three independent experiments with standard
deviations indicated.
Real-time PCR
Total RNA was prepared using the High Pure RNA
Isolation Kit (Roche). RNA (2 μg) was reverse-transcribed using the
High Capacity cDNA Synthesis Kit (Invitrogen). Quantitative gene
expression was performed for MMP-9 and glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) using the TaqMan Gene Expression system with
gene-specific primers and probes (for MMP-9, Hs00234579_m1; for
GAPDH, Hs03929097_g1). PCR reactions were performed on a StepOne
Plus Real-Time PCR System (Applied Biosystems) using TaqMan
Universal PCR master mix according to standard manufacturer’s
protocol. The data were then quantitated using the comparative
Ct method for relative gene expression utilizing GADPH
values as an endogenous control.
Results
Effect of altered cadherin expression on
adherens junction components
Oral squamous carcinoma cells are one of many tissue
types that downregulate E-cadherin in response to the
overexpression of mesenchymal cadherins (31,34).
The retention of E-cadherin in invasive, N-cadherin-expressing
cells suggests that the loss of E-cadherin function is
inconsequential to N-cadherin-mediated invasive signaling (10,11).
However, E-cadherin has been shown to be suppressive of invasion in
several tissues, including oral epithelia (20–22).
Such data raise the possibility that decreased E-cadherin function
may independently contribute to increased proteolytic activity. In
order to directly define the specific contributions of E-cadherin
loss and N-cadherin expression to the invasiveness of oral squamous
carcinoma cells, N- or E-cadherin expression was independently
altered in two oral squamous carcinoma lines, Tu167 and UM-SCC1
(SCC1). Retrovirally-encoded N-cadherin cDNA or small hairpin RNAs
(shRNA) were used to alter N- and E-cadherin levels in both
parental cell lines. Endogenous N-cadherin levels in each parental
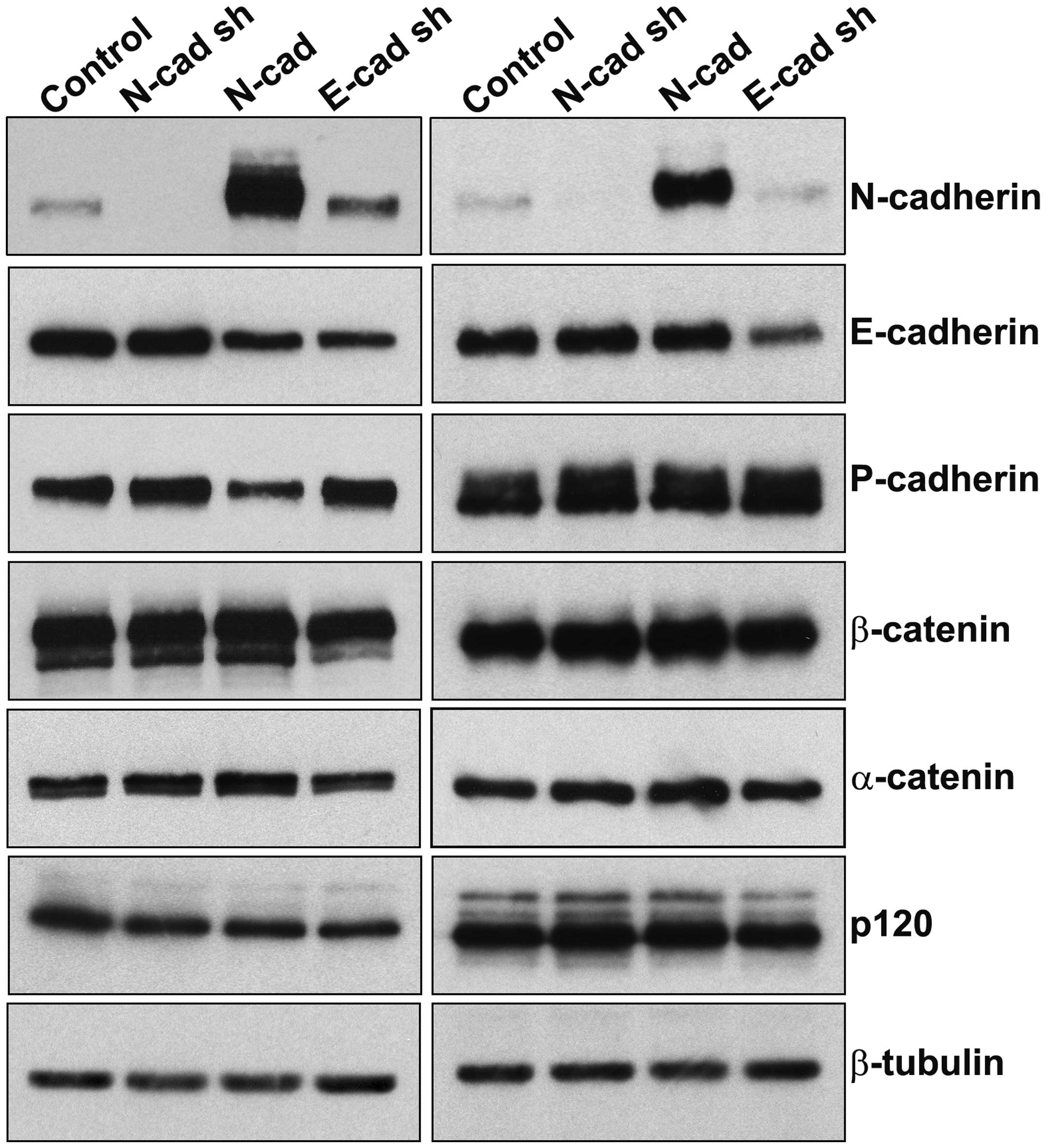
cell line are relatively low en masse (Fig. 1A), but an immunofluorescence
analysis of these cells revealed a marked heterogeneity of
N-cadherin expression (data not shown). To reduce N-cadherin
expression across the entire cell population, an shRNA vector
against N-cadherin was used to deplete N-cadherin from the entire
population.
As shown in Fig. 1,
in Tu167 cells, overexpression of N-cadherin decreased, but did not
abolish, expression of epithelial E- and P-cadherins. In SCC1
cells, N-cadherin overexpression did not alter levels of E- or
P-cadherin. This decrease in E-cadherin did not affect levels of
N-cadherin expression compared to controls. Levels of β-catenin,
α-catenin, and the isoforms/levels of p120 catenin were unaffected
by the various expression constructs. Most importantly for the
purposes of these studies, shRNA-mediated E-cadherin depletion was
either similar (Tu167) or more extensive (SCC1) than the E-cadherin
depletion seen as a result of N-cadherin overexpression. Thus, the
invasive effects of E-cadherin depletion may be investigated
independently of the consequential E-cadherin depletion caused by
N-cadherin overexpression.
Effect of altered cadherin expression on
colony morphology of oral squamous carcinoma cells
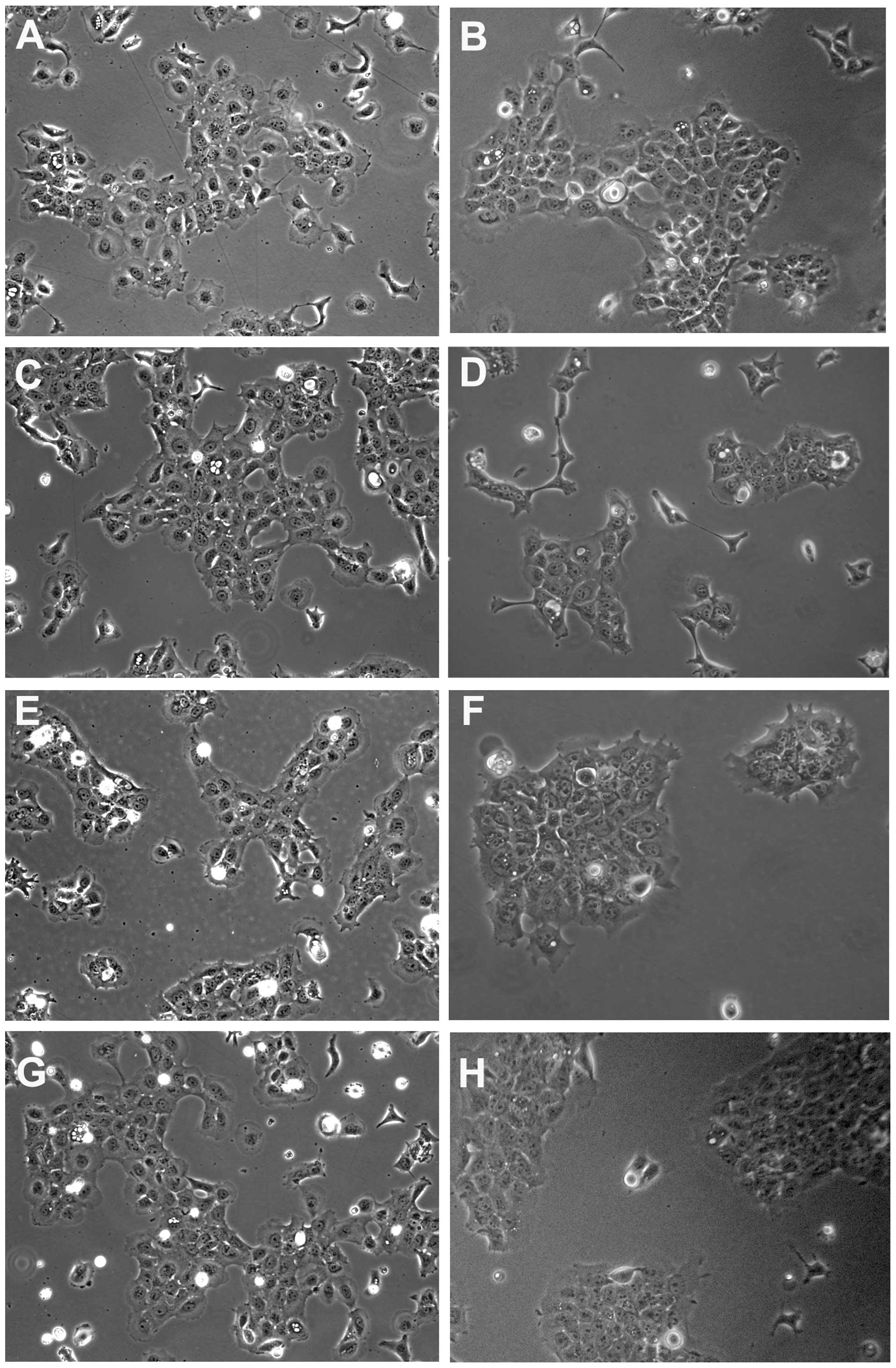
In prior studies utilizing a dexamethasone-inducible
N-cadherin cDNA, SCC1 cells exhibited a scattered phenotype in
response to increased expression of N-cadherin protein (34). Similar studies which utilized a
constitutively-expressed N-cadherin in breast cells showed no
effect on cell or colony morphology (24). In the present study, neither the
constitutive expression of N-cadherin (Fig. 2E and F) nor decreased expression of
E-cadherin (Fig. 2G and H)
resulted in cell scattering. In fact, N-cadherin overexpression
increased colony formation in both cell lines, resulting in fewer
scattered cells within each culture (compare Fig. 2A and B to E and F). E-cadherin
depletion had no effect on cell scattering (Fig. 2G and H).
N-cadherin expression, but not E-cadherin
depletion, increases MMP-9 activity
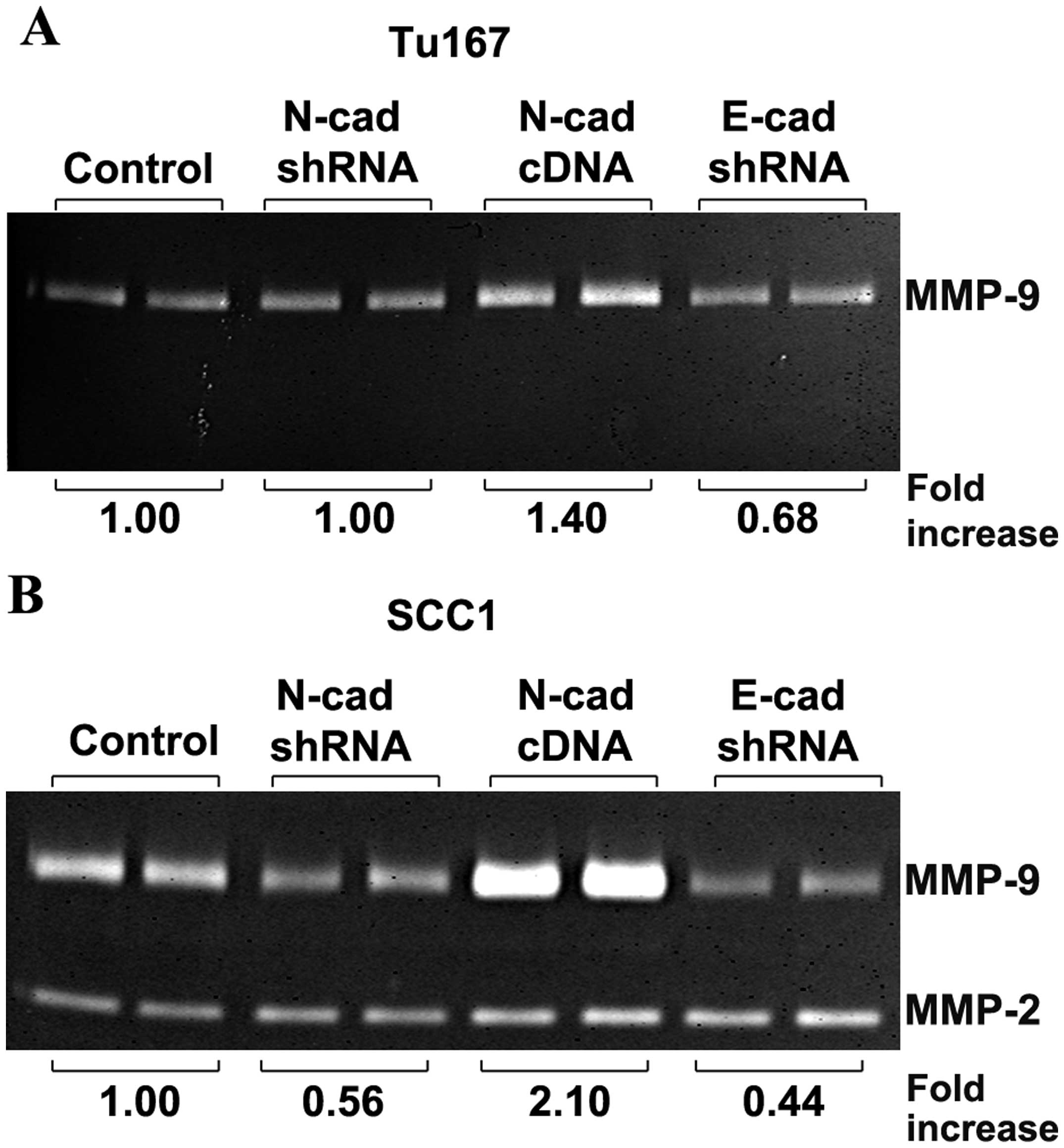
Gelatin zymography of conditioned serum-free media
(Fig. 3) was performed to
quantitate MMP secretion from control cells, cells with depleted
N-cadherin (N-cad shRNA), overexpressed N-cadherin (N-cad cDNA),
and depleted E-cadherin (E-cad shRNA). In both Tu167 and SCC1 cell
lines, the overexpression of N-cadherin increased MMP-9 activity
1.4-fold and 2-fold, respectively. The depletion of E-cadherin did
not increase MMP-9 activity in either cell line, and in fact
resulted in a moderate suppression of MMP-9 activity (Fig. 3A and B). In SCC1 cells (Fig. 3B), depletion of N-cadherin by shRNA
resulted in a moderate decrease in MMP-9 secretion compared to
control. SCC1 cells also secreted MMP2, which was unaffected by
perturbations in N- or E-cadherin. Tu167 cells did not express
MMP-2 under any conditions analyzed.
N-cadherin expression increases invasion
in a migration-dependent and -independent manner
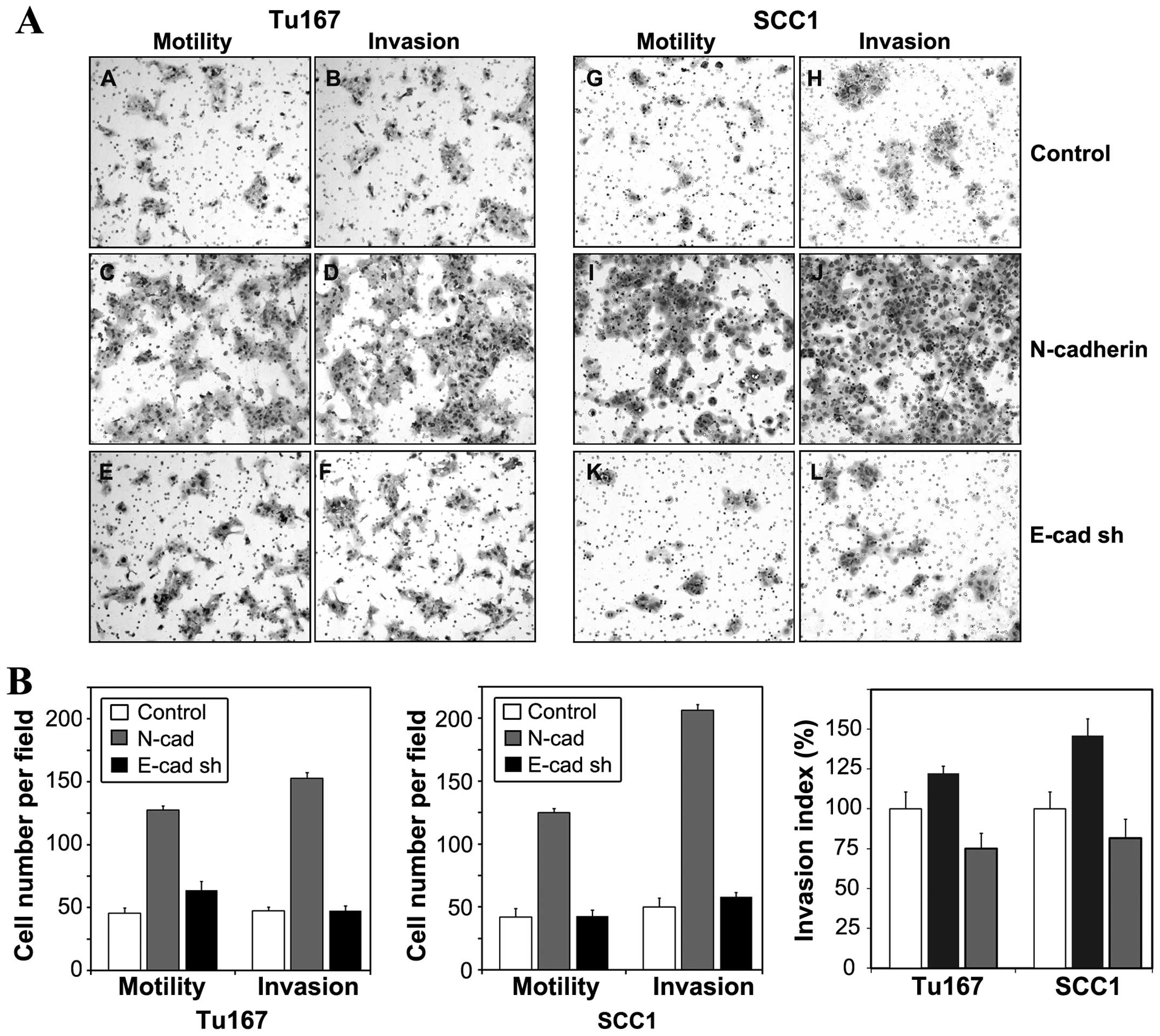
The effect of altered N- and E-cadherin expression
on both cell motility and invasion was examined by Transwell
migration assay (Fig. 4A and B).
Representative images of cells which traversed membrane filters are
shown in Fig. 4A. Quantitation of
cell migration data (Fig. 4B)
revealed increases in motility and invasion in
N-cadherin-expressing cells, but not in E-cadherin depleted cells.
To eliminate the contribution of increased cell motility to the
invasion data, invasion data were normalized to migratory data to
generate an Invasion Index (Fig.
4C), which more accurately depicts the proteolytic contribution
of N-cadherin to cell invasion. The Invasion Indices demonstrated
that overexpression of N-cadherin increased proteolytic activity in
Tu167 and SCC1 cells. The invasive capacity of E-cadherin-depleted
cells was minimal. These findings are consistent with the relative
increases in MMP-9 synthesis demonstrated by zymography (Fig. 3). SCC1 cells displayed a more
robust increase in MMP-9 expression than Tu167 (Fig. 3), and displayed a correspondingly
greater increase in invasive capacity (Fig. 4C).
The cytoplasmic region of N-cadherin is
required for induction of MMP-9 expression
An oral squamous carcinoma cell model has previously
been utilized to identify the fourth extracellular repeat domain
(EC4) of N-cadherin as the region responsible for conferring
increased motility (9). In breast
cells, this same region of N-cadherin mediated increased MMP-9
synthesis in an FGF2-dependent manner (19). Because N-cadherin overexpression
increased both MMP-9 and invasion in oral squamous cells (Figs. 3 and 4), studies utilizing chimeric cadherin
constructs were undertaken to better define the region(s) of
N-cadherin that may mediate invasive signaling. Oral squamous cells
were independently transduced with retroviral vectors that coded
for chimeric cadherin molecules. The EN chimera consisted of the
extracellular and transmembrane domains of E-cadherin, and the
cytoplasmic portion of N-cadherin. The NE chimera consisted of the
extracellular and transmembrane regions of N-cadherin, and the
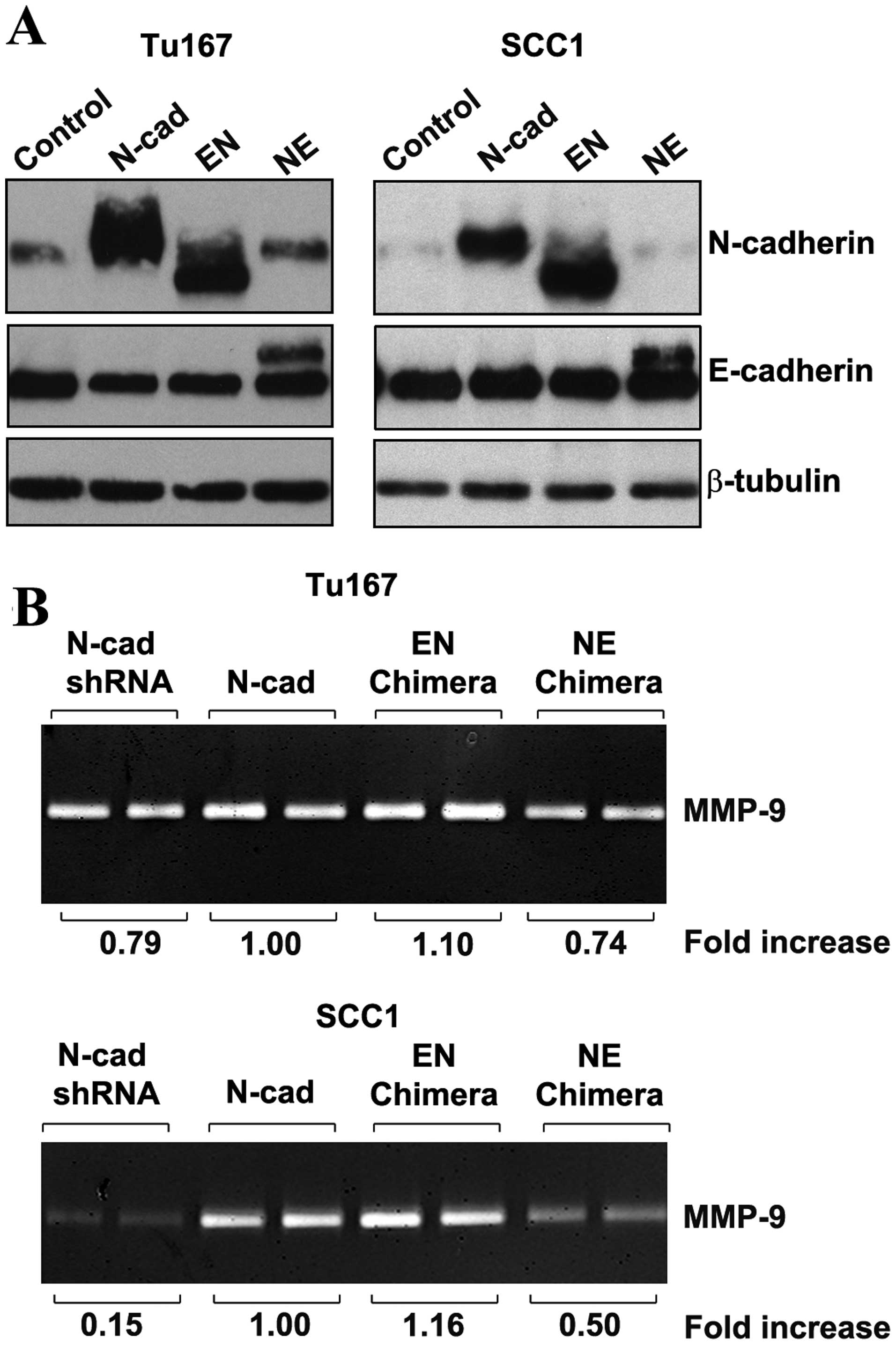
cytoplasmic portion of E-cadherin. Western blot analysis (Fig. 5A) was performed to confirm
expression of chimeric proteins in transduced cells. Antibodies
directed against cytoplasmic epitopes of E- and N-cadherin were
utilized to verify overexpression of chimeric molecules (Fig. 5A).
The EN chimera (EN), which lacked the N-cadherin
extracellular domain, retained the ability to induce MMP-9 in both
Tu167 and SCC1 cells (Fig. 5B).
When the cytoplasmic domain of N-cadherin was lacking (i.e., the NE
chimera), MMP-9 levels were reduced to control levels in Tu167
cells and markedly decreased in SCC1 cells (Fig. 5B). These results suggest that
invasive signaling is conferred by the cytoplasmic domain of
N-cadherin, and that, in oral squamous cells, the N-cadherin EC4
domain is dispensable for increased MMP-9 synthesis.
P120 is necessary for N-cadherin-induced
motility, but not invasion
The cytoplasmic region of N-cadherin interacts with
two proteins that are known to play key roles in tumor progression:
p120 catenin and β-catenin (35,36).
P120 has previously been shown to be integral to the ability of
ectopically expressed mesenchymal cadherins to increase motility
and, by consequence, invasiveness of epithelial cells (23). This phenomenon is dependent upon
proper binding of p120 to the cadherin juxtamembrane domain
(23,29,30).
To determine the relevance of such interactions with respect to
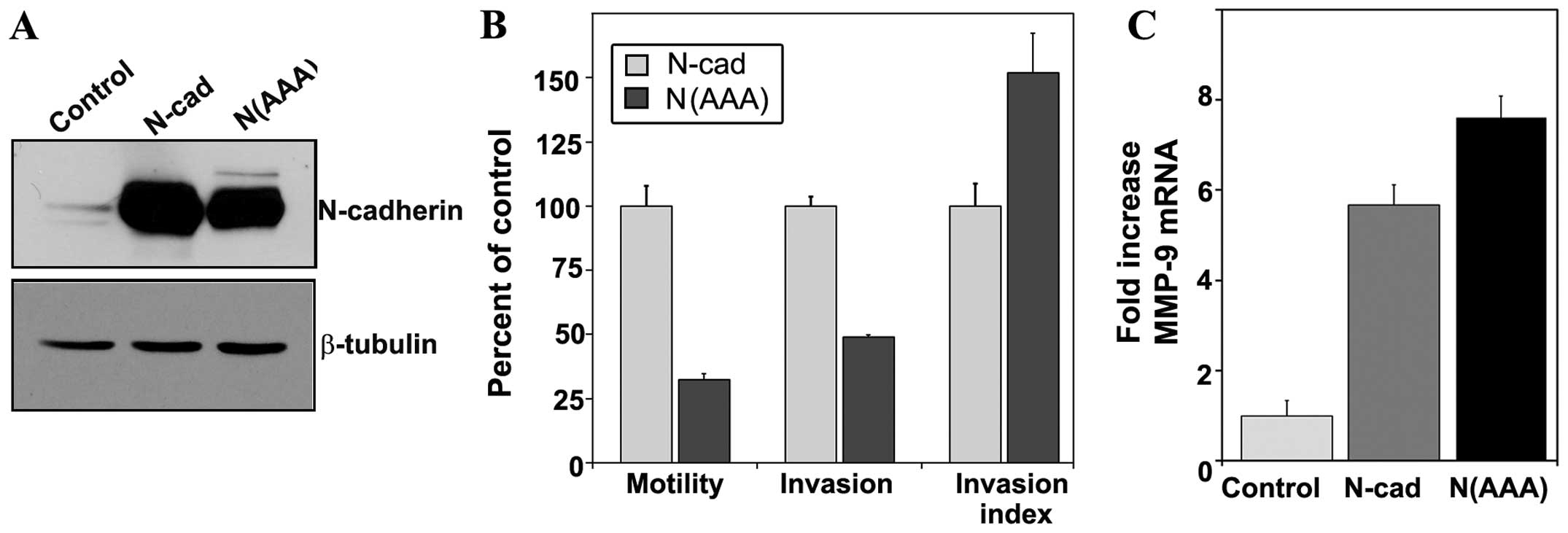
MMP-9 expression, SCC1 cells were transduced with a mutated version
of N-cadherin that contained three proximal alanine substitution
mutations in the p120 binding domains N(AAA). For both E- and
N-cadherin (29,30) homologous mutations have been shown
to abrogate binding of each cadherin to p120, and functionally
interfere with p120-dependent activities (23). Western blot analysis was used to
confirm expression of the mutated N-cadherin protein (Fig. 6A).
Consistent with previous studies (23,29)
cells expressing p120-uncoupled N-cadherin exhibited a four-fold
decrease in motility compared to cells with wild-type N-cadherin,
which corresponded to a decrease in invasion (Fig. 6B). However, the Invasion Index
value for cells expressing the N(AAA) mutant demonstrated a 50%
increase in invasive capacity compared to control cells (Fig. 6B). An analysis of MMP-9 transcript
levels in these cells also revealed no impairment in MMP-9
expression as a result of the N(AAA) mutation (Fig. 6C). These data suggest that although
p120 may be critical for N-cadherin mediated motility in oral
squamous carcinoma cells, it does not play a role in the
proteolytic aspect of invasion.
N-cadherin overexpression increases
transcriptional activation of β-catenin target genes
One of the most well characterized interactions of
the cytoplasmic portion of N-cadherin is with that of the protein
β-catenin. β-catenin has been shown to function as a modulator of
both cytoskeletal attachment and TCF/LEF-dependent transcription
(37,38). β-catenin has also been shown to
increased transcription of several matrix-metalloproteinase genes,
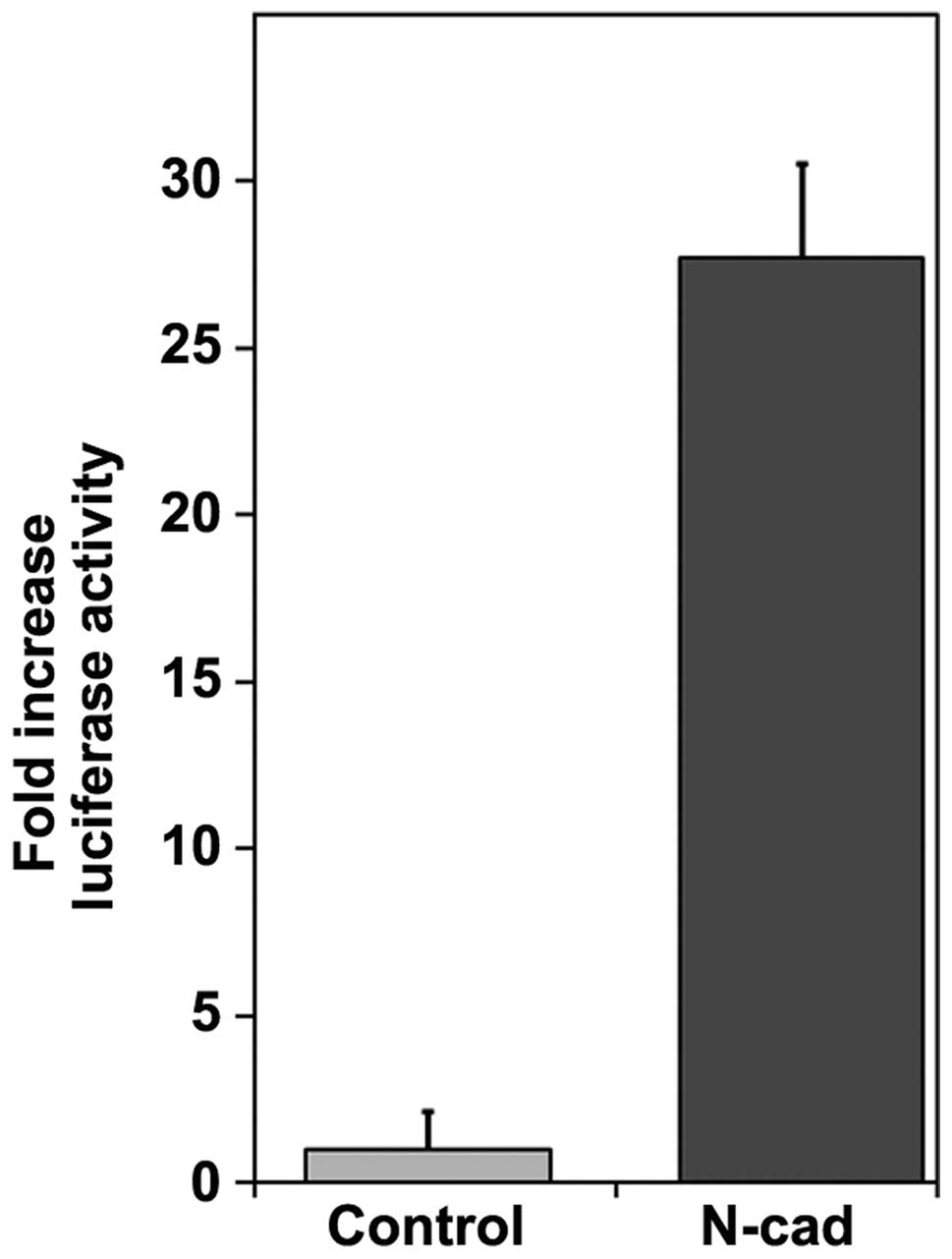
including MMP-9 (39,40). Overexpression of wild-type
N-cadherin increased β-catenin-dependent transcriptional activation
of a luciferase reporter by 27-fold compared to control cells
(Fig. 7).
Discussion
The present study demonstrated that in oral squamous
cells, the overexpression of N-cadherin, but not loss of
E-cadherin, is sufficient to increase levels of MMP-9 transcript
and protein levels (Figs. 3 and
6C). N-cadherin expression also
increased invasion in a manner that was not entirely the result of
increased migratory capacity. These data are in contrast to studies
by Suyama et al, who demonstrated robust
N-cadherin-dependent increases in MMP-9 only upon administration of
fibroblast growth factor (19). In
our hands, FGF was not necessary for increased levels of MMP-9 in
oral squamous cells, nor did treatment of these cells with FGF
produce further increases in MMP-9 gelatinase activity (unpublished
data). Additional studies performed in breast cells have
demonstrated that N-cadherin expression stimulates both invasion
and motility to relatively the same degree compared to control
cells (10), further supporting
the notion that N-cadherin expression in breast cells drives
motility but may play less of a role in modulating proteolytic
enzyme expression than it does in oral squamous carcinoma. The
mechanism by which N-cadherin increases MMP-9 expression and
invasion in oral squamous carcinoma cells appears to be a function
of N-cadherin that is independent of E-cadherin downregulation. The
shRNA-mediated decreases in E-cadherin were not able to substitute
for N-cadherin overexpression with regard to increasing invasive
characteristics (Figs. 3 and
4).
Because the extracellular domains of N-cadherin did
not appear to play a role in MMP-9 induction (Fig. 5), we turned our investigation to
cytoplasmic N-cadherin binding partners that have previously been
shown to mediate aggressive signaling. P120 catenin interacts with
the N-cadherin juxtamembrane region and facilitates lateral
clustering of surface cadherins to provide strength to the adherens
junction (41). P120 has also been
shown to act as a critical modulator of N-cadherin-dependent
invasion and migration through its activation of Rho family GTPases
(23). Consistent with other
studies demonstrating the critical role of p120 in
mesenchymal-cadherin-induced motility, migration conferred by the
N(AAA) N-cadherin mutant, which is known to be deficient in binding
to p120, was markedly reduced compared to controls. However, cells
expressing the N(AAA) mutant exhibited an invasive capacity and
MMP-9 expression level that was even greater than that seen with
expression of wild-type N-cadherin (Figs. 6B and 7). These data suggest that the
p120-binding domain of N-cadherin was not a critical element in the
induction of MMP-9.
A second binding partner of the cytoplasmic portion
of N-cadherin is β-catenin, a growth-associated protein that is
dually resident within the adherens junction and in the nucleus,
where it functions as a coactivator of the Tcf/Lef family of
transcription factors (38). In
the present study, N-cadherin overexpression increased
β-catenin-dependent transcriptional activity 27-fold (Fig. 7D) in the absence of increased
β-catenin synthesis (Fig. 1A).
MMP-9 transcripts were also increased in cells expressing either
wild-type N-cadherin or the mutant N(AAA) cadherin (Fig. 6C), which is known to retain
functional binding of β-catenin (30). Previous studies suggest a high
likelihood that the increased MMP-9 activity in N-cadherin
overexpressing cells is due to increased β-catenin transcriptional
activity. The promoter region of MMP-9 contains Tcf/Lef consensus
sequences (39,40), and β-catenin transcriptional
activity has been positively correlated with increased MMP-9
transcript levels in several cell types, including oral squamous
cells (39,42).
The N-cadherin-dependent increase in β-catenin
transcriptional activation is in discordance with previously
proposed hypotheses that the sequestration of β-catenin within
adherens junctions interfered with the ability of β-catenin to
function as a transcriptional activator (43,44).
Such a mechanism is unlikely in oral squamous carcinoma cells,
however, as depletion of E-cadherin did not increase invasive
capacity (Fig. 4B) or synthesis of
MMP-9 (Fig. 3), and overexpression
of N-cadherin potentiated both MMP-9 synthesis (Fig. 3) and β-catenin-dependent
transcriptional activity (Fig.
7).
How then might N-cadherin overexpression positively
regulate the nuclear activities of β-catenin? Recent research
suggests that the initial association of β-catenin with cadherins
may be vital to the ability of β-catenin to function as a
transcriptional activator. In MDCK cells, the association of
β-catenin with E-cadherin and the internalization of
membrane-resident E-cadherin complexes were necessary to maintain
β-catenin-mediated transcriptional activation of reporter genes
(45). This mechanism may be
applicable to overexpression of N-cadherin as well, as the
depletion of N-cadherin in N-cadherin-expressing HEK293 cells and
in murine embryos significantly decreased β-catenin-mediated
transcriptional activity (45).
Howard et al (45) have
proposed a model whereby β-catenin is rendered transcriptionally
competent as the result of its adhesion to cadherin molecules at
the adherens junction. The subsequent internalization of
membrane-resident cadherins allows dissociation of this ‘primed’
form of β-catenin, enabling transcriptional activation of Tcf/Lef
target sequences. It is worth noting that distinct pools of
adhesive versus transcriptionally active β-catenin have been
structurally characterized (44).
Recent studies in neural cells suggest that N-cadherin may play a
pivotal role in altering the balance of these pools by promoting
the phosphorylation of β-catenin by the AKT protein kinase
(46).
The present study demonstrates distinct roles of
N-cadherin in promoting motility and invasion-related proteolysis.
Although p120 is critical for increased motility, p120 does not
appear to function in the induction of matrix metalloproteinases.
Our data suggest that increased transcriptional activation of
matrix metalloproteinases, and increased invasive capacity, was
instead mediated by increased β-catenin transcriptional activity.
Such regulation of β-catenin may have implications beyond increased
matrix metalloproteinase activity, as β-catenin-dependent
transcription is integral to a number of signaling pathways
essential to tumor progression (36,37).
Further studies will be necessary to elucidate the mechanisms by
which N-cadherin-dependent increases in β-catenin function may
modulate tumor invasion in oral squamous carcinoma cells.
Acknowledgements
The authors wish to thank Dr Keith Johnson and Dr
Margaret Wheelock, for advice in developing the initial stages of
this work. This study was partially supported by the National
Institute of Health grant 1F32DE17516, and by intramural funding
from Midwestern University.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Warnakulasuriya S: Global epidemiology of
oral and oropharyngeal cancer. Oral oncol. 45:309–316. 2009.
View Article : Google Scholar
|
|
3
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2013. CA Cancer J Clin. 63:11–30. 2013. View Article : Google Scholar
|
|
4
|
Savagner P: Leaving the neighborhood:
molecular mechanisms involved during epithelial-mesenchymal
transition. Bioessays. 23:912–923. 2001. View Article : Google Scholar
|
|
5
|
Takeichi M: Cadherins in cancer:
implications for invasion and metastasis. Curr Opin Cell Biol.
5:806–811. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Birchmeier W and Behrens J: Cadherin
expression in carcinomas: role in the formation of cell junctions
and the prevention of invasiveness. Biochim Biophys Acta.
1198:111994.PubMed/NCBI
|
|
7
|
Hazan RB, Qiao R, Keren R, Badano I and
Suyama K: Cadherin switch in tumor progression. Ann NY Acad Sci.
1014:155–163. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wheelock MJ, Shintani Y, Maeda M, Fukumoto
Y and Johnson KR: Cadherin switching. J Cell Sci. 121:727–735.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim JB, Islam S, Kim YJ, Prudoff R, Sass
K, Wheelock M and Johnson K: N-Cadherin extracellular repeat 4
mediates epithelial to mesenchymal transition and increased
motility. J Cell Biol. 151:1193–1206. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nieman MT, Prudoff RS, Johnson KR and
Wheelock MJ: N-cadherin promotes motility in human breast cancer
cells regardless of their E-cadherin expression. J Cell Biol.
147:631–644. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rieger-Christ KM, Lee P, Zagha R, et al:
Novel expression of N-cadherin elicits in vitro bladder cell
invasion via the Akt signaling pathway. Oncogene. 23:4745–4753.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nagi C, Guttman M, Jaffer S, et al:
N-cadherin expression in breast cancer: correlation with an
aggressive histologic variant-invasive micropapillary carcinoma.
Breast Cancer Res Treat. 94:225–235. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nguyen PT, Kudo Y, Yoshida M, Kamata N,
Ogawa I and Takata T: N-cadherin expression is involved in
malignant behavior of head and neck cancer in relation to
epithelial-mesenchymal transition. Histol Histopathol. 26:147–156.
2011.PubMed/NCBI
|
|
14
|
Di Domenico M, Pierantoni G, Feola A, et
al: Prognostic significance of N-cadherin expression in oral
squamous cell carcinoma. Anticancer Res. 31:4211–4218.
2011.PubMed/NCBI
|
|
15
|
Zhao D, Tang XF, Yang K, Liu JY and Ma XR:
Over-expression of integrin-linked kinase correlates with aberrant
expression of Snail, E-cadherin and N-cadherin in oral squamous
cell carcinoma: implications in tumor progression and metastasis.
Clin Exp Metastasis. 29:957–969. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Folgueras AR, Pendás AM, Sánchez LM and
López-Otín C: Matrix metalloproteinases in cancer: from new
functions to improved inhibition strategies. Int J Dev Biol.
48:411–424. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Katayama A, Bandoh N, Kishibe K, Takahara
M, Ogino T, Nonaka S and Harabuchi Y: Expressions of matrix
metalloproteinases in early-stage oral squamous cell carcinoma as
predictive indicators for tumor metastases and prognosis. Clin
Cancer Res. 10:634–640. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Patel BP, Shah PM, Rawal UM, Rawal UM,
Desai AA, Shah SV, Rawal RM and Patel PS: Activation of MMP-2 and
MMP-9 in patients with oral squamous cell carcinoma. J Surg Oncol.
90:81–88. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Suyama K, Shapiro I, Guttman M and Hazan
RB: A signaling pathway leading to metastasis is controlled by
N-cadherin and the FGF receptor. Cancer Cell. 2:301–314. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Munshi HG, Ghosh S, Mukhopadhyay S, et al:
Proteinase suppression by E-cadherin-mediated cell-cell attachment
in premalignant oral keratinocytes. J Biol Chem. 277:38159–38167.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nawrocki-Raby B, Gilles C, Polette M, et
al: E-Cadherin mediates MMP down-regulation in highly invasive
bronchial tumor cells. Am J Pathol. 163:653–661. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wong AST and Gumbiner BM:
Adhesion-independent mechanism for suppression of tumor cell
invasion by E-cadherin. J Cell Biol. 161:1191–1203. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yanagisawa M and Anastasiadis PZ: p120
catenin is essential for mesenchymal cadherin-mediated regulation
of cell motility and invasiveness. J Cell Biol. 174:1087–1096.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hazan RB, Phillips GR, Qiao RF, Norton L
and Aaronson SA: Exogenous expression of N-cadherin in breast
cancer cells induces cell migration, invasion, and metastasis. J
Cell Biol. 148:779–790. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Myers JN, Holsinger FC, Jasser SA, Bekele
BN and Fidler IJ: An orthotopic nude mouse model of oral tongue
squamous cell carcinoma. Clin Cancer Res. 8:293–298.
2002.PubMed/NCBI
|
|
26
|
Brenner JC, Graham MP, Kumar B, et al:
Genotyping of 73 UM-SCC head and neck squamous cell carcinoma cell
lines. Head Neck. 32:417–426. 2010.PubMed/NCBI
|
|
27
|
Tamura I, Sakaki T, Chaqour B, Howard PS,
Ikeo T and Macarak EJ: Correlation of P-cadherin and β-catenin
expression and phosphorylation with carcinogenesis in rat tongue
cancer induced with 4-nitroquinoline 1-oxide. Oral Oncol.
39:506–514. 2003.
|
|
28
|
Kim YJ, Johnson KR and Wheelock MJ:
N-cadherin-mediated cell motility requires cis dimers. Cell Commun
Adhes. 12:23–39. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen H, Paradies NE, Fedor-Chaiken M and
Brackenbury R: E-cadherin mediates adhesion and suppresses cell
motility via distinct mechanisms. J Cell Sci. 110:345–356.
1997.PubMed/NCBI
|
|
30
|
Thoreson MA, Anastasiadis PZ, Daniel JM,
et al: Selective uncoupling of p120ctn from E-cadherin disrupts
strong adhesion. J Cell Biol. 148:189–202. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Maeda M, Johnson E, Mandal S, et al:
Expression of inappropriate cadherins by epithelial tumor cells
promotes endocytosis and degradation of E-cadherin via competition
for p120ctn. Oncogene. 25:4595–4604. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Maeda M, Johnson KR and Wheelock MJ:
Cadherin switching: essential for behavioral but not morphological
changes during an epithelium-to-mesenchyme transition. J Cell Sci.
118:873–887. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Leber TM and Balkwill FR: Zymography: a
single-step staining method for quantitation of proteolytic
activity on substrate gels. Anal Biochem. 249:24–28. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Islam S, Carey TE, Wolf GT, Wheelock MJ
and Johnson KR: Expression of N-cadherin by human squamous
carcinoma cells induces a scattered fibroblastic phenotype with
disrupted cell-cell adhesion. J Cell Biol. 135:1643–1654. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Anastasiadis PZ and Reynolds AB: The p120
catenin family: complex roles in adhesion, signaling and cancer. J
Cell Sci. 113:1319–1334. 2000.PubMed/NCBI
|
|
36
|
Clevers H: Wnt/β-catenin signaling in
development and disease. Cell. 127:469–480. 2006.
|
|
37
|
Behrens J: Control of β-catenin signaling
in tumor development. Ann NY Acad Sci. 910:21–35. 2000.
|
|
38
|
Korinek V, Barker N, Morin PJ, et al:
Constitutive transcriptional activation by a β-catenin-Tcf complex
in APC−/− colon carcinoma. Science. 275:1784–1787. 1997.
|
|
39
|
Wu B, Crampton SP and Hughes CC: Wnt
signaling induces matrix metalloproteinase expression and regulates
T cell transmigration. Immunity. 26:227–239. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Crampton SP, Wu B, Park EJ, et al:
Integration of the β-catenin-dependent Wnt pathway with integrin
signaling through the adaptor molecule Grb2. PLoS One.
4:e78412009.
|
|
41
|
Niessen CM and Gumbiner BM: The
juxtamembrane region of the cadherin cytoplasmic tail supports
lateral clustering, adhesive strengthening, and interaction with
p120ctn. J Cell Biol. 141:779–789. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li S, Jiao J, Lu Z and Zhang M: An
essential role for N-cadherin and β-catenin for progression in
tongue squamous cell carcinoma and their effect on invasion and
metastasis of Tca8113 tongue cancer cells. Oncol Rep. 21:1223–1233.
2009.
|
|
43
|
Orsulic S, Huber O, Aberle H, Arnold S and
Kemler R: E-cadherin binding prevents beta-catenin nuclear
localization and beta-catenin/LEF-1-mediated transactivation. J
Cell Sci. 112:1237–1245. 1999.PubMed/NCBI
|
|
44
|
Gottardi CJ and Gumbiner BM: Distinct
molecular forms of β-catenin are targeted to adhesive or
transcriptional complexes. J Cell Biol. 167:339–349. 2004.
|
|
45
|
Howard S, Deroo T, Fujita Y and Itasaki N:
A positive role of cadherin in Wnt/β-catenin signalling during
epithelial-mesenchymal transition. PLoS One. 6:e238992011.
|
|
46
|
Zhang J, Shemezis JR, McQuinn ER, Wang J,
Sverdlov M and Chenn A: AKT activation by N-cadherin regulates
beta-catenin signaling and neuronal differentiation during cortical
development. Neural Dev. 8:72013. View Article : Google Scholar : PubMed/NCBI
|