Introduction
Herbal remedies are commonly used in traditional
medicine to treat and prevent human diseases including cancer.
Numerous plant derived flavonoids and phenolic/polyphenolic
compounds with antioxidant and anti-inflammatory activities are
currently used by cancer patients as dietary supplements to
complement chemotherapy. In fact, isolation and identification of
bioactive components from medicinal plants have led to the
synthesis of potent anticancer drugs, including Vinca alkaloids,
taxol, camptothecan, etoposide and retinoids. Triterpenoids are
members of a large family of structurally related compounds known
as cyclosqualenoids that are widely distributed in nature.
Pristimerin (PM) is a quinonemethide triterpenoid present in
various plant species in the Celastraceae and Hippocrateaceae
families (1,2). PM and related compounds have shown
anti-inflammatory, antioxidant and antimalarial activities
(3–5). PM has also shown potent
antiproliferative and apoptosis-inducing activity in glioma,
leukemia, breast, lung and prostate cancer cell lines (6–9).
Induction of apoptosis by PM involves generation of reactive oxygen
species (ROS), activation of caspases, mitochondrial dysfunction,
inhibition of nuclear factor κB (NF-κB), Akt and MAP kinases
(10–13). In addition, PM also inhibits
proteasome activity, tumor cell migration and angiogenesis
(8,14,15).
Carcinoma of the prostate is the most commonly
diagnosed cancer in American males and the second ranked cause of
cancer related mortality (16). An
estimated 233,000 new cases of prostate cancer will be diagnosed
and 29,480 deaths are expected to occur from this disease in the
United States in 2014 (17).
Current therapies (e.g., androgen deprivation, radical
prostatectomy, radiotherapy or brachytherapy) while effective in
treating localized prostate cancer have limited efficacy against
advanced disease and metastatic hormone-refractory disease remains
incurable (18–20). Since the incidence of CaP increases
with advancing age, prostate cancer is expected to be an
increasingly greater problem as life expectancy improves.
In a previous report we have shown that PM induces
apoptosis in CaP cell lines through a ROS-dependent Bcl-2
degradation pathway (21). In the
present study, we demonstrate that induction of apoptosis in CaP
cells by PM is associated with inhibition of cell cycle regulatory
proteins and proteasomal degradation of antiapoptotic survivin, a
member of the inhibitors of apoptosis (IAP) family.
Materials and methods
Reagents
PM was purchased from Sigma Chemicals (St. Louis,
MO). Anti-PARP-1, anti-Bcl-2, anti-Bcl-xL and anti-survivin
antibodies were purchased from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA). The 96 AQueous One Solution Proliferation Assay
System was from Promega (Madison, WI). Annexin V-FITC apoptosis
detection kit was purchased from BD Pharmingen (San Diego, CA).
Stock solution of PM (100 mM) was prepared in DMSO and all test
concentrations were prepared by diluting stock solution in tissue
culture medium.
Cell lines
LNCaP and PC-3 human prostate cancer cell lines were
obtained from the American Type Culture Collection (ATCC,
Rockville, MD). LNCaP were grown in RPMI-1640 supplemented with FBS
and penicillin/streptomycin. PC-3 cells were grown in F-12K
nutrient mixture (Gibco BRL, Rockville, MD) supplemented with 10%
fetal calf serum, 1% penicillin/streptomycin, and 25 mM HEPES
buffer. Both cell lines were cultured at 37°C in a humidified
atmosphere consisting of 5% CO2 and 95% air, and
maintained by subculturing cells twice a week.
Measurement of cell viability (MTS
assay)
Tumor cells (1×104) in 100 μl of tissue
culture medium were seeded into each well of a 96-well plate. After
24-h incubation to allow cells to adhere, cells were treated with
PM at concentrations ranging from 0 to 5 μM. Cultures were
incubated for additional 72 h and cell viability was then
determined by the colorimetric MTS assay using CellTiter 96 AQueous
One Solution Proliferation Assay System from Promega. This assay
measures the bioreduction of the tetrazolium compound MTS by
intracellular dehydrogenases in the presence of electron-coupling
reagent phenazine methosulfate. MTS and phenazine methosulfate were
added to the culture wells, and cultures were incubated for 2 h at
37°C. The absorbance, which is directly proportional to the number
of viable cells in the cultures, was measured at 490 nm using a
microplate reader.
Apoptosis assay
Apoptosis was assessed by the binding of Annexin
V-FITC to phosphotidylserine, which is externalized to the outer
leaflet of the plasma membrane early during induction of apoptosis.
Briefly, untreated cells and cells treated with PM were resuspended
in the binding buffer provided in the Annexin V-FITC apoptosis
detection kit II (BD Biosciences, San Diego, CA) and allowed to
react with 5 μl of Annexin V-FITC reagent and 5 μl of propidium
iodide for 30 min at room temperature in the dark. Stained cells
were analyzed by flow cytometry using Accuri C6 flow cytometer
(Accuri Cytometers Inc., Ann Arbor, MI). The induction of apoptosis
by PM was confirmed by the cleavage of PARP-1 by western blot
analysis.
Western blot analysis
Cell lysates were prepared by detergent lysis [1%
Triton-X 100 (v/v), 10 mM Tris-HCl (pH 7.5), 5 mM EDTA, 150 mM
NaCl, 10% glycerol, 2 mM sodium vanadate, 5 μg/ml leupeptin, 1
μg/ml aprotinin, 1 μg/ml pepstatin A and 10 μg/ml
4-2-aminoethyl-benzenesulfinyl fluoride]. Lysates were clarified by
centrifugation at 14,000 × g for 10 min at 4°C, and protein
concentrations were determined by Bradford assay. Samples (50 μg)
were boiled in an equal volume of sample buffer [20% glycerol, 4%
SDS, 0.2% bromophenol blue, 125 mM Tris-HCl (pH 7.5), and 640 mM
2-mercaptoethanol] and separated on 10% SDS-polyacrilamide gels.
Proteins resolved on the gels were transferred to nitrocellulose
membranes and probed with antibodies against proteins of interest
followed by HRP-conjugated secondary antibody. Immune complexes
were visualized by chemiluminescence. Protein bands were imaged and
band densities analyzed using the NIH/Scion image analysis
software.
DNA transfection
For expression of HA tagged-survivin, semi-confluent
cultures of PC-3 cells in 60 mm2 cell culture dishes
were transfected with 10 μg of empty or HA-survivin expression
vector (pcDNA3-HA-survivin) (CH3 BioSystems, Amherst, NY) using
Lipofectamine Plus reagent. After incubation for 36 h,
overexpression of survivin in transfected cells was confirmed by
immunoblotting.
Immunoprecipitation
After treatment with PM (5 μM) for 6 h cells were
washed with cold PBS and lysed in NP-40 cell lysis buffer
(Invitrogen, Camarillo, CA) supplemented with 2 mM sodium vanadate,
5 μg/ml leupeptin, 1 μg/ml aprotinin, 1 μg/ml pepstatinin, and 10
μg/ml 4-2-aminoethyl-benzenesulfinyl fluoride for 30 min on ice.
Supernatants were collected after centrifugation at 14,000 × g for
10 min and protein concentration was determined. Each sample (400
μg protein) in 200 μl of antibody binding buffer containing anti-HA
antibody was incubated for 1 h at room temperature followed by
incubation with protein A agarose beads for 1 h. Immune complexes
were washed two times with lysis buffer and analyzed for ubiquitin
by western blot analysis.
Statistical analysis
Data are expressed as mean ± SD. The difference
between control and treatment groups was determined using Dunnett’s
multiple comparison test. Differences with p<0.05 were
considered statistically significant.
Results
Pristimerin inhibits proliferation of CaP
cells
The effect of PM on proliferation of CaP cells
(LNCaP and PC-3 cells) was examined using MTS assay. For this,
cells were treated with PM at concentrations of 0 to 5 μM for 72 h
and the viability of cultures was determined. As shown in Fig. 1, measurable reduction in viability
(~20%) was observed in both cell lines at 0.625 μM PM; however,
significant reduction in viability occurred at 1.25 to 5 μM PM
(47–73%, p<0.05). Thus, PM reduced the proliferation both
androgen-sensitive (LNCaP) and androgen-resistant (PC-3) CaP
cells.
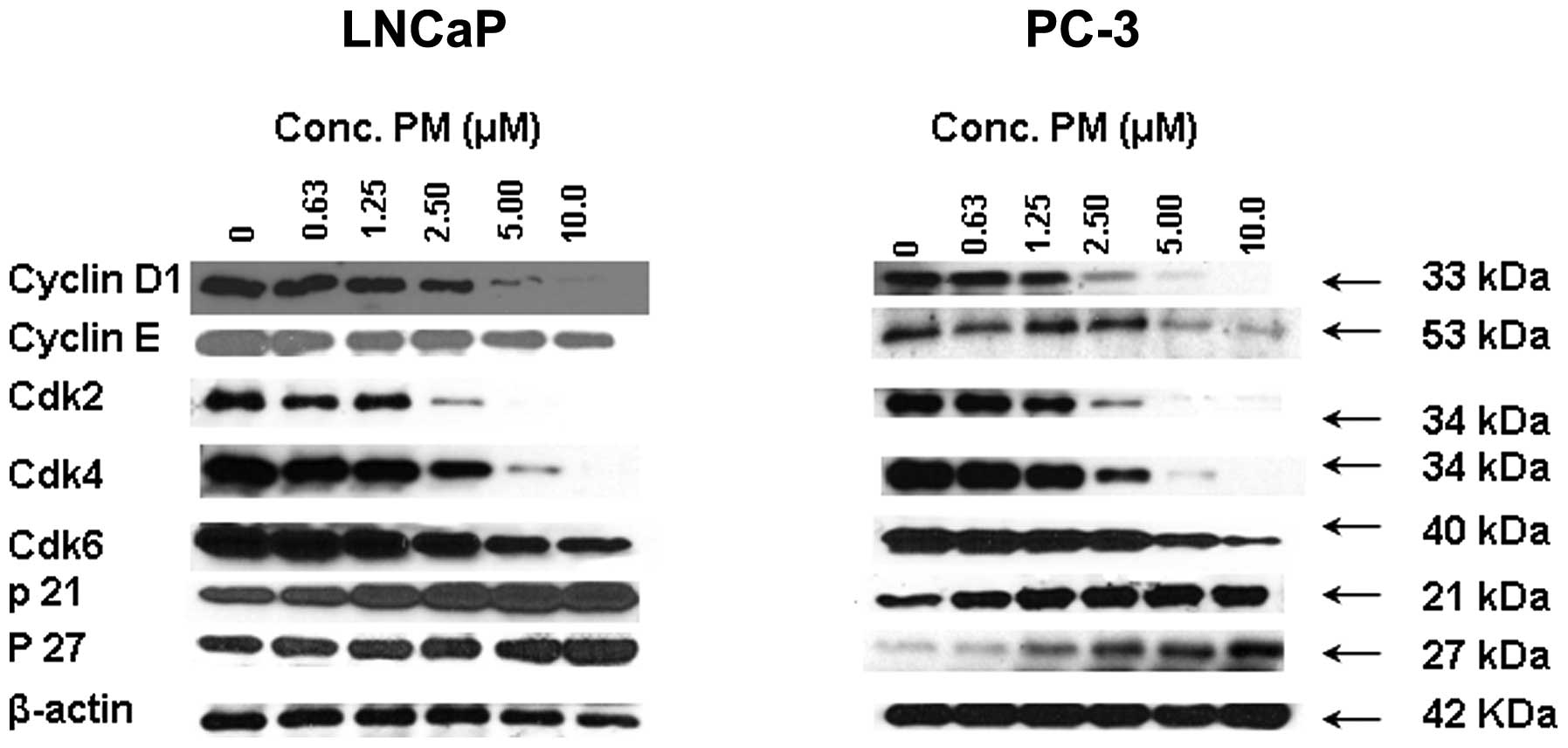
PM inhibits cell cycle regulatory
proteins in CaP cells
Since cell division is regulated by cyclins and
cyclin-dependent kinases (cdks) and cdk inhibitors such as WAF1/21
and KIP1/27, we investigated the effect of PM on these cell cycle
regulators. For this, LNCaP and PC-3 cells were treated with PM
(0–10 μM) for 24 h and levels of cyclin D1, cyclin E, cdk2, cdk4,
cdk6, p21 and p27 were analyzed by western blot analysis. As shown
in Fig. 2, treatment with PM
reduced the level of cyclin D1 and cyclin E in LNCaP cells, whereas
only cyclin E was reduced in PC-3 cells. On the other hand, levels
of cdk2, cdk4 and cdk6 were reduced in both cell lines in a
dose-related manner. Contrary to the inhibition of cyclin D1 cyclin
E and cdks 2, 4 and 6 treatment with PM increased the levels of cdk
inhibitors p21 and p27. Thus, inhibition of cyclin D1 and E and
cdks 2, 4 and 6 suggests arrest of LNCaP and PC-3 cells in G0/G1
cell cycle phase.
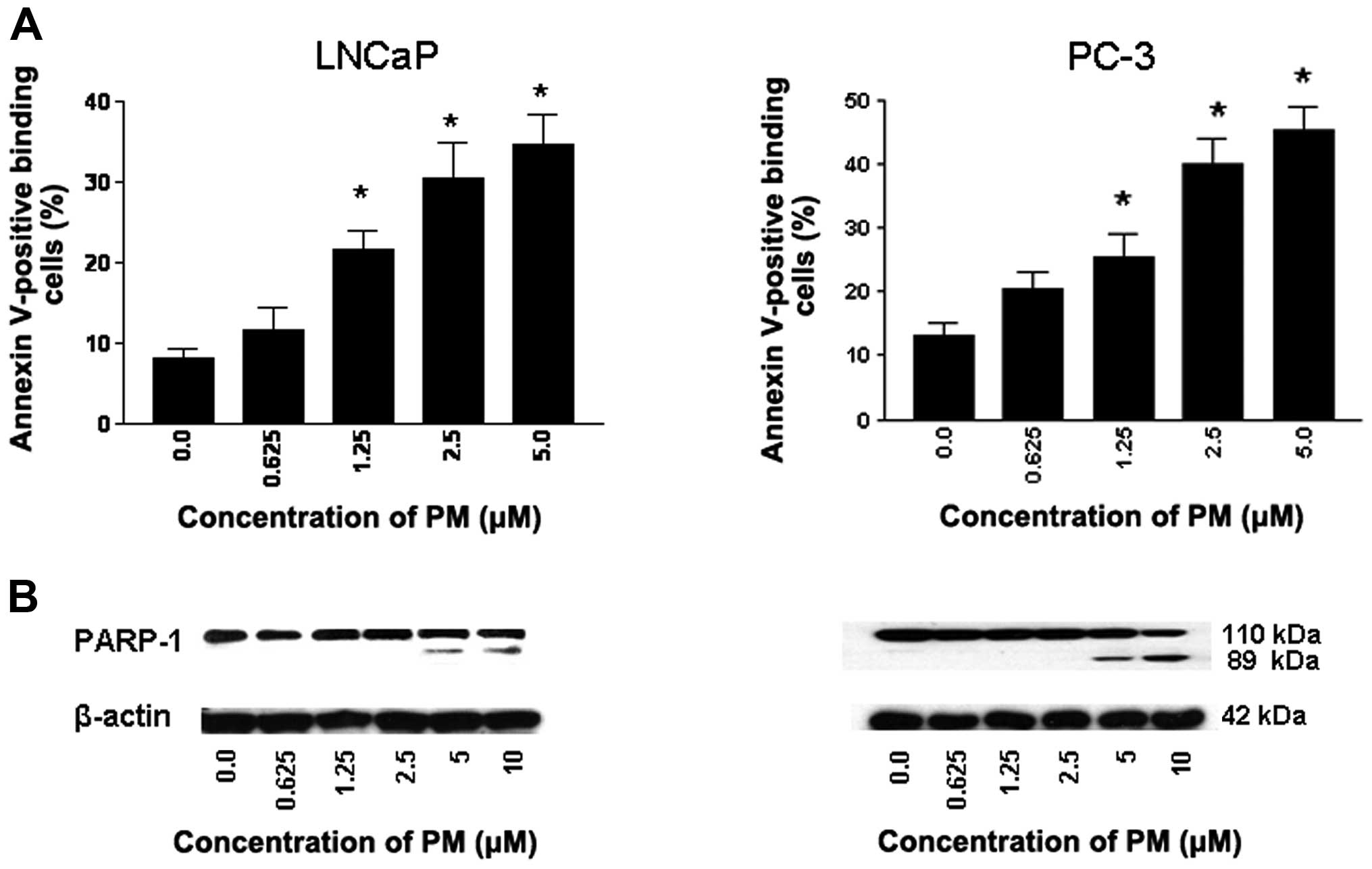
PM induces apoptosis in CaP cells
Whether inhibition of proliferation of CaP cells by
PM leads to induction of apoptosis was investigated next. Thus,
LNCaP and PC-3 cells were treated with PM (0 to 5 μM) for 24 h and
induction of apoptosis was measured from the binding of Annexin
V-FITC by flow cytometry and cleavage of PARP-1 by western blot
analysis. As shown in Fig. 3A,
only a small percentage of untreated LNCaP and PC-3 cells bound
Annexin V-FITC (8 to 12%, respectively). In contrast, the
percentage of Annexin V-FITC binding cells (both cell lines)
increased in a dose-dependent manner after treatment with PM at
0.625 to 5 μM (LNCaP, 11–32%; PC-3, 19–43%).
The induction of apoptosis was confirmed by the
cleavage of PARP-1 as identified by decrease in 110 kDa native
protein and the emergence of an 89 kDa cleaved PARP-1 fragment in
both cell lines treated with PM (Fig.
3B). Thus, increase in Annexin V-FITC-binding and the cleavage
of PARP-1 demonstrated induction of apoptosis by PM.
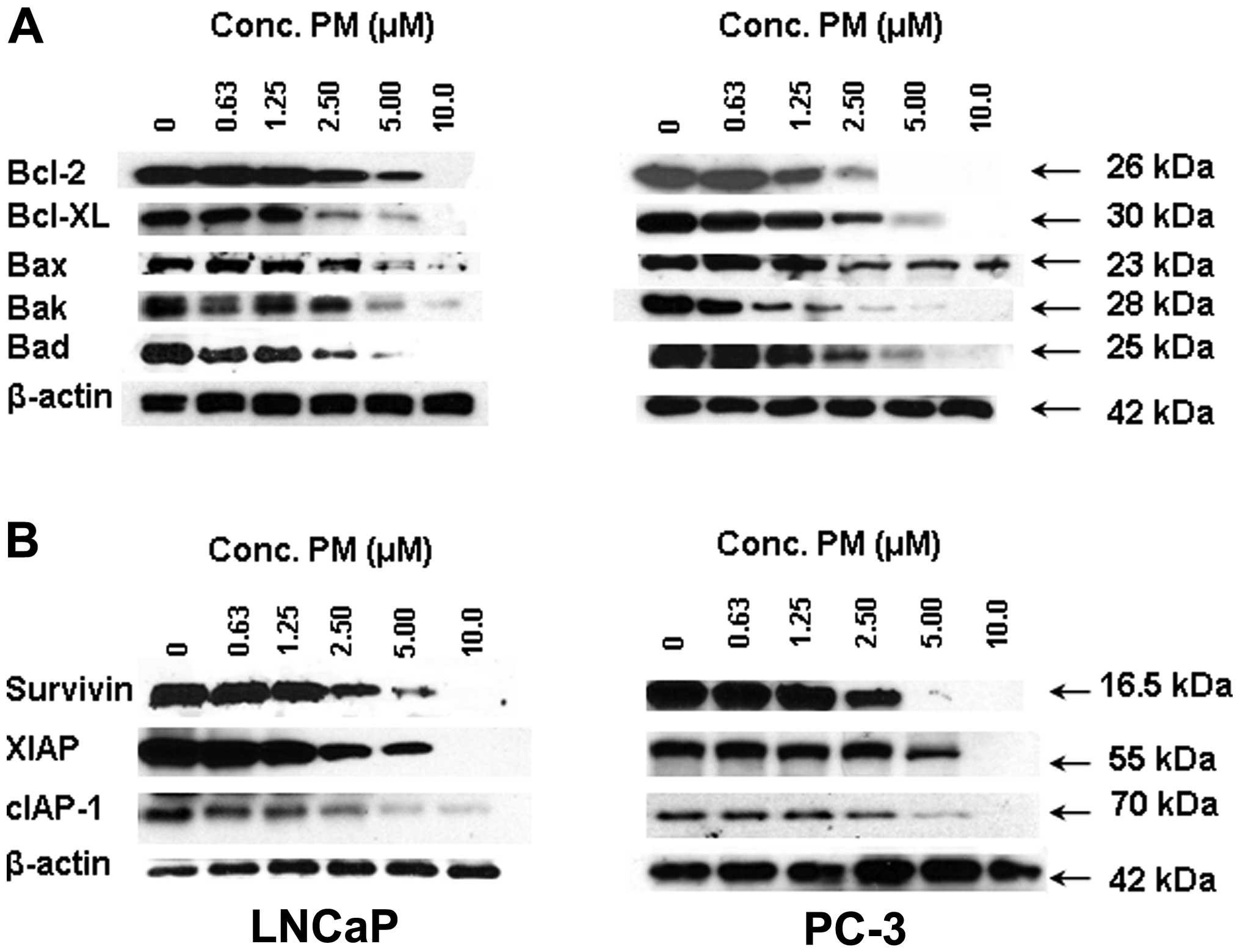
PM inhibits apoptosis-related proteins in
CaP cells
Apotosis is regulated by a number of pro and
anti-apoptotic cellular proteins belonging to the Bcl-2 and IAP
families of proteins. To ascertain the effect of PM on these
apoptosis-regulatory proteins we analyzed the levels of some of the
more prominent members of the Bcl-2 and IAP families by western
blot analysis. Treatment with PM (0–10 μM) decreased the levels of
antiapoptic Bcl-2, Bcl-xL and proapoptotic Bax, Bak and Bad in a
dose-dependent manner in both cell lines (Fig. 4A). A similar inhibitory effect of
PM on the expression of antiapoptotic IAP family members, such as
survivin, XIAP and cIAP-1 was observed (Fig. 4B).
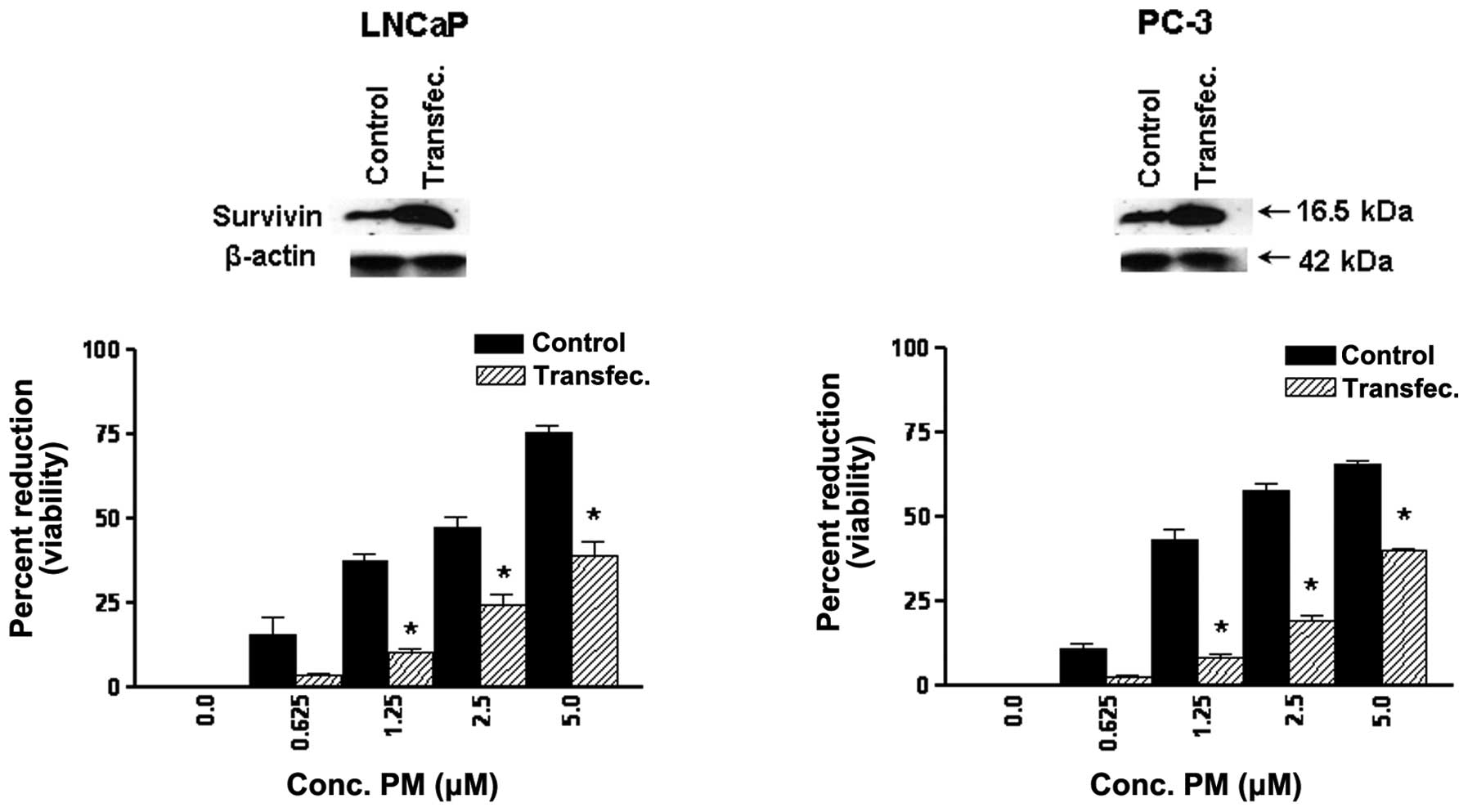
Survivin regulates response to PM in CaP
cells
Survivin is an IAP family member that plays an
important role in cell cycle regulation and inhibition of
apoptosis. To examine the relevance of survivin in antitumor
activity of PM, we measured the response of CaP cells
overexpressing surviving to PM in MTS assay. For this, LNCaP and
CaP cells were transfected with survivin expression vector
(pcDNA3-HA-survivin) and after confirming overabundance of survivin
in transfected cells their response to PM was measured in 72 h MTS
assay. As shown in Fig. 5, there
was significant reduction in the sensitivity of transfected cells
to PM compared to control cells. Transfection with empty plasmid
showed no change in response to PM (not shown). These data
indicated that survivin plays an important role in the response of
CaP cells to PM.
Pristimerin downregulates survivin
through ubiquitin-proteasome degradation pathway
To obtain insight into the mechanism by which PM
reduces survivin in CaP cells we investigated the role of
ubiquitin-proteasome degradation pathway in downregulation of
survivin. First, the effect of calpain inhibitor MG101 and
proteasome inhibitors MG132 and lactacystin on PM-mediated
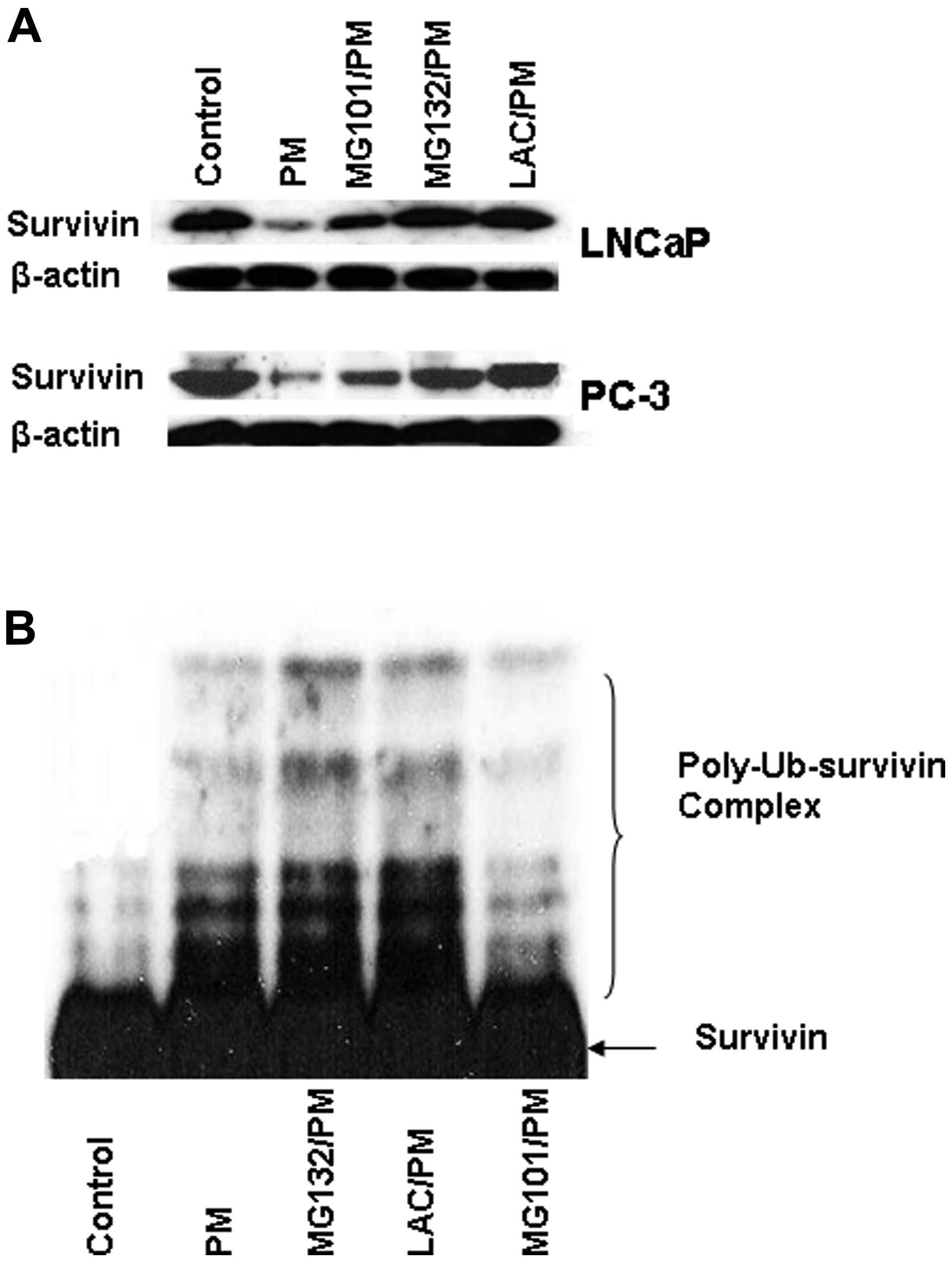
downregulation of survivin was examined. As shown in Fig. 6A, treatment with lysosomal protease
inhibitor MG101 only partially reversed the inhibition of survivin
by PM (~30% reversal). In contrast, pretreatment with proteasome
inhibitors MG132 and lactacystin completely blocked the inhibition
of survivin by PM (Fig. 6A).
The result above indicated the involvement of
proteasome in PM-induced downregulation of survivin. To further
establish the role of proteasome in degradation of survivin, we
investigated the effect of PM on ubiquitination of survivin. For
this, PC-3 cells transfected with survivin expression vector
(pcDNA3-HA-survivin) were treated with PM for 6 h in the presence
or absence of proteasome inhibitors MG132, lactacystin or caplain
inhibitor MG101. Cells were treated with PM for 6 h because
treatment for 6 h was found to induce maximal ubiquitination of
survivin. Cell lysates were subjected to immunoprecipitation with
anti-HA antibody followed by immunoblotting with anti-ubiquitin
antibody to detect ubiquitinated survivin products. As shown in
Fig. 6B, treatment with PM alone
induced ubiquitination of survivin; however, treatment with PM in
the presence of proteasome inhibitors MG132 and LAC resulted in
additional accumulation of the polyubiquitinated survivin products.
On the other hand, treatment with PM in the presence of calpain
inhibitor MG101 did not cause accumulation of the polyubiquitinated
survivin products. Taken together, these data indicated that
survivin downregulation by PM is mediated through an
ubiquitin-proteasomal degradation pathway.
Discussion
There is intense interest in developing novel agents
and treatment strategies to treat hormone-refractory metastatic
prostate cancer. We and others have shown that PM exhibits potent
antitumor activity against a wide range of cancer cell lines,
including prostate cancer cells through multiple mechanisms
(6–14,20).
However, the significance of survivin, a potent inhibitor of
apoptosis and a regulator of cell division in mediating response to
PM in cancer cells has not been investigated. Thus, the present
study was undertaken to examine the role of survivin in apoptotic
cell death of prostate cancer cells by PM. Our results demonstrated
the antiproliferative activity of PM both in androgen-sensitive and
androgen-refractory prostate cancer cells. This result suggested
that the inhibition of tumor cell proliferation by PM may be
attributable to cell cycle inhibition. Indeed, PM has been shown to
arrest cell cycle in G0/G1 phase in pancreatic cancer cells
(21). Cell cycle progression is
controlled by cyclins, cyclin-dependent kinases (cdks) and cdk
inhibitors. In the present study, although a formal cell cycle
analysis was not performed, treatment with PM downregulated levels
of cyclin D1 and E in both CaP cell lines. PM also inhibited the
expression of cdk2, cdk4 and cdk6 in both cell lines. Cyclin D1 and
E in conjunction with cdk2, cdk4 and cdk6 regulate cell cycle
progression through G1 phase. Thus, inhibition of cyclin D1 and E
and cdk2, cdk4 and cdk6 suggests that PM might inhibit
proliferation by arresting prostate cancer cells in G1-phase. Data
also suggest that increase in expression of the cdk inhibitors p21
and p27 by PM may also facilitate G1 arrest by inhibiting the
activity of cyclinE-cdk2 complexes that promotes G1-S phase
progression.
In most instances, inhibition of cell proliferation
by anticancer agents forces tumor cells to undergo apoptosis. PM
increased Annexin V binding and cleaved PARP-1 in both cell lines
indicating induction of apoptosis. This result corroborates our
previous findings demonstrating induction of apoptosis by PM in
epithelially-derived ovarian and pancreatic cancer cells via the
inhibition of antiapoptotic (prosurvival) signaling molecules such
as Akt, NF-κB and mTOR (14,20).
In these tumor systems, induction of apoptosis involved cleavage of
caspases-8, -9 and -3, loss of mitochondrial membrane potential and
generation of free radicals supporting results of studies reported
by others.
The intrinsic (mitochondrial) pathway of apoptosis
is regulated by members of the Bcl-2 family of proteins that
includes both pro- and anti-apoptotic molecules (22). In addition, members of the IAP
family are potent inhibitors of apoptosis (23). Bcl-2 and Bcl-xL are two major
antiapoptotic Bcl-2 family members that reside in the mitochondrial
membrane and inhibit apoptosis by preventing the activation of
inner mitochondrial permeability transition pore and release of
proapotogenic mitochondrial contents including cytochrome c
(24). PM inhibited Bcl-2 and
Bcl-xL in both cell lines in a dose-related manner (Fig. 4). Interestingly, proapoptotic Bax,
Bak and Bad were also inhibited by PM. Normally, proapoptotic Bax,
Bak and Bad counteract antiapoptotic Bcl-2 and Bcl-xL and if the
ratio of the antiapoptotic and proapoptotic members is tilted in
favor of proapoptotic proteins, apoptosis ensues. Since PM reduced
both anti- and pro-apoptotic Bcl-2 family members the exact role of
Bcl-2 family of proteins in induction of apoptosis by PM in
prostate cancer cells remains unresolved.
cIAP-1, XIAP and survivin are members of the
inhibitor of apoptosis family of proteins (IAP) that block
apoptosis by blocking activation or neutralizing the activity of
caspases 3, 7 and 9 (23,25). cIAP-1 interferes with the
activation of caspases, whereas XIAP binds to and inhibits caspase
3, 7 and 9. Survivin also inhibits caspase activation. Treatment
with PM reduced the expression of these IAP members in prostate
cancer cells, thereby contributing to the induction of apoptosis by
PM.
Besides inhibiting apoptosis, survivin also
regulates cell division and cytokinesis (26,27).
Survivin is only expressed in the G2-M phase and during mitosis it
localizes to the mitotic spindle by interaction with tubulin.
Because of the prominent role survivin plays in the inhibition of
apoptosis and regulation of cell division, we investigated the
significance of survivin in mediating response to PM and the
mechanism by which PM down-regulates its expression in CaP cells.
The former was addressed by evaluating the response of tumor cells
expressing abundance of survivin. Overexpression of survivin
increased the resistance of tumor cells to PM (Fig. 5), implicating survivin in mediating
the response to PM.
Levels of many short-lived proteins associated with
apoptosis and cell cycle including survivin are regulated by
ubiquitin-proteosome degradation pathway (28,29).
Whether PM-mediated reduction in levels of survivin occurred
through protesomal degradation was examined using pharmacological
inhibitors of proteasomes. As shown in Fig. 6, proteasome inhibitors MG132 and
lactacystin completely blocked the inhibition of survivin by PM
whereas calpain inhibitor MG101 only partially reversed the
inhibitory effect of PM, indicating that degradation of survivin by
PM is primarily by proteasomes.
The degradation of proteins by 26S proteasome
requires ubiquitination of target proteins through addition of
multiple ubiquitin moieties at lysine residues. To confirm the
involvement of proteasomes in PM-induced degradation of surviving,
we analyzed ubiquitin-survivin complexes in tumor cells treated PM
in the presence of proteasome inhibitors MG132 and lactacystin or
calpain inhibitor MG101. Treatment with PM in the presence of MG132
or LAC resulted in accumulation of polyubiquitinated survivin
products compared to treatment with PM alone. On the other hand,
treatment with PM in the presence of MG101 did not cause
accumulation of polyubiquitinated survivin. Taken together, these
data demonstrated that downregulation of survivin by PM is mediated
through the ubiquitin-proteasome degradation pathway. Thus,
understanding the role and mechanism by which PM downregulates
survivin may facilitate development of PM for the
prevention/treatment of prostate cancer.
Acknowledgements
This study was supported by NIH grant 1R01 CA130948
from the National Cancer Institute.
References
|
1
|
Buffa Filho W, Corsino J, Bolzani da SV,
Furlan M, Pereira AM and Franca SC: Quantitative determination for
cytotoxic Friedo-nor-oleanane derivatives from five morphological
types of Maytenus ilicifolia(Celastraceae) by reverse-phase
high-performance liquid chromatography. Phytochem Anal. 13:75–78.
2002.
|
|
2
|
Chang FR, Hayashi K, Chen IH, et al:
Antitumor agents. 228. five new agarofurans, Reissantins A-E, and
cytotoxic principles from Reissantia buchananii. J Nat Prod.
66:1416–1420. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sassa H, Kogure K, Takaishi Y and Terada
H: Structural basis of potent antiperoxidation activity of the
triterpene celastrol in mitochondria: effect of negative membrane
surface charge on lipid peroxidation. Free Radic Biol Med.
17:201–207. 1994. View Article : Google Scholar
|
|
4
|
Dirsch VM, Kiemer AK, Wagner H and Vollmar
AM: The triterpenoid quinonemethide pristimerin inhibits induction
of inducible nitric oxide synthase in murine macrophages. Eur J
Pharmacol. 336:211–217. 1997. View Article : Google Scholar
|
|
5
|
Figueiredo JN, Raz B and Sequin U: Novel
quinone methides from Salacia kraussii with in vitro
antimalarial activity. J Nat Prod. 61:718–723. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Costa PM, Ferreira PM, da Bolzani VS, et
al: Antiproliferative activity of pristimerin isolated from
Maytenus ilicifolia ( Celastraceae) in human HL-60 cells.
Toxicol In Vitro. 22:854–863. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang H, Landis-Piwowar KR, Lu D, et al:
Pristimerin induces apoptosis by targeting the proteasome in
prostate cancer cells. J Cell Biochem. 103:234–244. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yan YY, Bai JP, Xie Y, Yu JZ and Ma CG:
The triterpenoid pristimerin induces U87 glioma cell apoptosis
through reactive oxygen species-mediated mitochondrial dysfunction.
Oncol Lett. 5:242–248. 2013.PubMed/NCBI
|
|
9
|
Lee JS, Yoon IS, Lee MS, et al: Anticancer
activity of pristimerin in epidermal growth factor receptor
2-positive SKBR3 human breast cancer cells. Biol Pharm Bull.
36:316–325. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu CC, Chan ML, Chen WY, Tsai CY, Chang FR
and Wu YC: Pristimerin induces caspase-dependent apoptosis in
MDA-MB-231 cells via direct effects on mitochondria. Mol Cancer
Ther. 4:1277–1285. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lu Z, Jin Y, Chen C, Li J, Cao Q and Pan
J: Pristimerin induces apoptosis in imatinib-resistant chronic
myelogenous leukemia cells harboring T315I mutation by blocking
NF-kappaB signaling and depleting Bcr-Abl. Mol Cancer. 9:1122010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Byun JY, Kim MJ, Eum DY, et al: Reactive
oxygen species-dependent activation of Bax and poly(ADP-ribose)
polymerase-1 is required for mitochondrial cell death induced by
triterpenoid pristimerin in human cervical cancer cells. Mol
Pharmacol. 76:734–744. 2009. View Article : Google Scholar
|
|
13
|
Deeb D, Gao X, Liu YB, Pindolia K and
Gautam SC: Pristimerin, a quinonemethide triterpenoid, induces
apoptosis in pancreatic cancer cells through the inhibition of
pro-survival Akt/NF-κB/mTOR signaling proteins and anti-apoptotic
Bcl-2. Int J Oncol. 44:1707–1715. 2014.PubMed/NCBI
|
|
14
|
Mu XM, Shi W, Sun LX, et al: Pristimerin
inhibits breast cancer cell migration by up-regulating regulator of
G protein signaling 4 expression. Asian Pac J Cancer Prev.
13:1097–1104. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mu X, Shi W, Sun L, Li H, Jiang Z and
Zhang L: Pristimerin, a triterpenoid, inhibits tumor angiogenesis
by targeting VEGFR2 activation. Molecules. 17:6854–6868. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cooperberg MR, Park S and Carroll PR:
Prostate cancer 2004: insights from the national disease
registries. Oncology (Williston Park). 18:1239–1258.
2004.PubMed/NCBI
|
|
17
|
Prostate Cancer. National Cancer
Institute, U.S. National Institutes of Health Cancer.gov.
http:www.cancer.gov/cancer-topics/types/prostate.
Accessed Feb 7, 2014
|
|
18
|
Garnick MB: Hormonal therapy in the
management of prostate cancer: From Higgins to the present.
Urology. 49:5–15. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hanks GE: Long-term control of prostate
cancer with radiation. Urol Clin North Amer. 23:605–616. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu Y, Gao X, Deeb D, Ali S and Gautam SC:
Pristimerin induces apoptosis in prostate cancer cells by
down-regulating Bcl-2 through ROS-dependent ubiquitin-proteasomal
degradation pathway. J Carcinogen Mutagen. S6:005doi:
104172/2157–2518. S6–005. 2013.PubMed/NCBI
|
|
21
|
Wang Y, Zhou Y, Zhou H, et al: Pristimerin
causes G1 arrest, induces apoptosis, and enhances the
chemosensitivity to gemcitabine in pancreatic cancer cells. PLOS
One. 7:e438262012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chao DT and Korsmeyer SJ: BCL-2 family:
regulators of cell death. Annu Rev Immunol. 16:395–419. 1998.
View Article : Google Scholar
|
|
23
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar
|
|
24
|
Deveraux QL and Reed JC: IAP family
proteins - suppressors of apoptosis. Genes Dev. 13:239–252. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tamm I, Wang Y, Sausville E, Scudiero DA,
Vigna N, Oltersdorf T and Reed JC: IAP-family protein survivin
inhibits caspase activity and apoptosis induced by Fas (CD95), Bax,
caspases, and anticancer drugs. Cancer Res. 58:5315–5320.
1998.PubMed/NCBI
|
|
26
|
Zhao J, Tenev T, Martins LM, Downward J
and Lemoine NR: The ubiquitin-proteasome pathway regulates survivin
degradation in a cell cycle-dependent manner. J Cell Sci.
113:4363–4371. 2000.PubMed/NCBI
|
|
27
|
Li F1, Ambrosini G, Chu EY, Plescia J,
Tognin S, Marchisio PC and Altieri DC: Control of apoptosis and
mitotic spindle checkpoint by surviving. Nature. 396:580–584. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Koepp DM, Harper JW and Elledge SJ: How
the cyclin became a cyclin: regulated proteolysis in the cell
cycle. Cell. 97:431–434. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aberle H, Bauer A, Stappert J, Kispert A
and Kemler R: beta-catenin is a target for the ubiquitin-proteasome
pathway. EMBO J. 16:3797–3804. 1997. View Article : Google Scholar : PubMed/NCBI
|