Introduction
Interferons (IFNs) are a family of natural
glycoproteins that consist of IFN-α, -β and -γ. The antiviral
activity of IFNs led to their discovery, but later data revealed
that they also control cell growth and differentiation, inhibit
expression of oncogenes, and activate T lymphocytes, natural killer
cells and macrophages. Therefore, the efficacy of IFN therapy for
various malignancies, including malignant gliomas (1), has been investigated for many
years.
Recently anti-glioma action of IFN-β has been
re-evaluated. IFN-β markedly enhanced sensitivity to temozolomide
(TMZ) via downregulation of MGMT transcription (2,3). The
results of the study suggest that compared to TMZ-based
chemotherapy plus radiotherapy, chemotherapy with IFN-β and TMZ and
concomitant radiotherapy further improve the clinical outcomes of
patients with malignant gliomas. A multicenter phase I clinical
trial established that therapy with IFN-β and TMZ is safe, well
tolerated, and prolongs survival of patients with glioblastoma
(4,5). Taken together, IFN-β increased the
therapeutic efficiency of TMZ in cases of newly diagnosed primary
glioblastoma, particularly in patients with the unmethylated MGMT
promoter (6). A prospective
randomized control trial to compare the clinical outcomes of newly
diagnosed glioblastoma patients treated with TMZ alone or with TMZ
and IFN-β combination therapy is ongoing.
Because glioblastoma is one of the most richly
neovascularized solid tumors in terms of vasoproliferation,
endothelial cell hyperplasia, and endothelial cell cytology
(7), antiangiogenic approach may
be especially suitable for the treatment of malignant gliomas
(8). Recent large clinical study
clearly demonstrated the effectiveness of anti-VEGF antibody
(bevacizumab) for malignant glioma (9,10).
Antiangiogenic activity of IFN-β has been reported previously,
IFN-β inhibits some growth factors (bFGF, interleukin 8) (11,12)
and gelatinase (13) transcription
and/or protein production. In this study, we investigated the
antiangiogenic effect of IFN-β for malignant gliomas in
vitro and in vivo, especially about VEGF production,
angiogenic stimuli and inhibitor balance, and tumor
microcirculation that are not previously proven mechanisms as
antiangiogenic actions of IFN-β.
Materials and methods
Human glioma cell lines and culture
conditions
The human glioma cell line U-87 MG was obtained from
the American Type Culture Collection (Rockville, MD). The human
glioma cell line TK2 was established from glioblastoma at the
Department of Neurosurgery, University of Tsukuba. The human glioma
cell line, Becker, was a generous gift. Cells were maintained in
MEM supplemented with 10% FCS in a humidified atmosphere containing
5% CO2 at 37°C.
Human umbilical cord vein endothelial cells (HUVECs)
harvested from umbilical cords were a generous gift of Dr Okuda
(University of Tsukuba). HUVECs were maintained with collagen
coated flasks (Iwaki Glass, Tokyo, Japan) in E300 medium (Kyokuto,
Tokyo, Japan) which are designed for HUVEC culture containing 2%
fetal calf serum, heparin, aFGF and EGF.
Reagents
Human IFN-β was a gift from Toray Industries, Inc.
(Tokyo, Japan).
Cell proliferation assay (MTT assay)
Cell proliferation assays were performed using the
CellTiter 96™ Aqueous Non-Radioactive Proliferation Assay (Promega
Corp., Madison, WI) as described previously (14). This assay measures the reduction of
a tetrazolium compound
(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium),
by living cells to a formazan product. Briefly, the glioma cells,
1×105/ml in DMEM with 10% FCS, were plated in 96-well
plates (Becton Dickinston, Lincoln Park, NJ) at 5,000 cells. After
24-h incubation, the various doses of IFN-β (10 to 5,000 U/ml) were
added to the wells. The cells were incubated for 48, 96, 144 h. At
the end of incubation period, to the microplate wells were added 20
μl of a freshly prepared combined tetrazolium compound and an
electron coupling reagent (phenazine methosulfate) solution, then
incubated for 2 h at 37°C, and the optical density at 490 nm was
read on an automatic microplate reader (Model 550, Bio-Rad). The
experiment was repeated at least three times in triplicate wells
for each concentration of IFN-β.
Western blot analysis
The tissues and the cell pellets were homogenized
with a ultrasonic homogenizer on ice in 1 ml of extraction buffer
[25 mM Tris, 100 mM NaCl, 20 mM NH4HCO3, pH
7.5, protease inhibitor cocktail; Complete Mini (Roche) one tablet]
per 100 mg wet weight of tissue and the protein lysates were
obtained after centrifugation at 50,000 × g for 30 min at 4°C.
Lysates containing 50 μg of total protein, as estimated by the
method of Bradford using bovine serum albumin as a standard, were
separated on 12% SDS-polyacrylamide gel, electroblotted onto 0.2 μm
nitrocellulose membrane (Bio-Rad) and were immunoassayed with
rabbit polyclonal anti-VEGF antibody (A-20, 100 μg/ml, Santa Cruz
Biotechnology, Santa Cruz, CA) at the dilution of 1:200 and mouse
monoclonal anti-human IP-10 antibody (500 μg/ml, Dako, Glostrup,
Denmark) at the dilution of 1:100. The immunocomplexes formed were
visualized with alkaline phosphatase-conjugated anti-rabbit or
anti-mouse immunoglobulin G (IgG) using ECL western blot analysis
system (Amersham Pharmacia Biotech).
U87 SCID mouse subcutaneous model
After the implantation of 1×105 U87 cells
in the flank of 6 weeks old male SCID mouse (Clea Japan), U87 tumor
tissue fragments were removed and then re-implanted into another
SCID mouse. Harvested tumor fragments, 1 mm3 in size,
were implanted into the flank of 17 SCDI mice. The animals were
divided into 3 groups randomly; 5 control group, 5 IFN-β low dose
group, 7 IFN-β high dose group. IFN-β treatment was started at day
7 after the implantation when the subcutaneous tumor reached 5 mm
in size. IFN-β was locally injected adjacent to the tumor
1×105 U (low dose group) and 5×105 U (high
dose group) once daily for 15 days. Size of the subcutaneous tumor
was measured by caliper. At 15 days after the treatment, the tumor
tissue was removed. A part of the tissue was immediately fixed in
10% phosphate-buffered formalin for 48 h, paraffin-embedded, and
used for routine pathological diagnosis and immunohistochemistry.
The other part of the tissue was immediately frozen with liquid
nitrogen and stored at −70°C. Two mice of the 7 IFN-β high dose
group survived for next 20 days without any treatment and tumor
volume was measured.
Similar set of the U87 subcutaneous tumor
experiments were repeated with IFN-β systemic (intraperitoneal)
injection instead of local injection. Fifteen mice were divided
into 3 groups; 5 control, 5 IFN-β low dose and 5 IFN-β high dose.
This systemic injection experiment was only used to measure the
tumor volume between three groups.
RNA isolation and reverse transcription
polymerase chain reaction (RT-PCR)
Total RNA was extracted from IFN-β treated and
control frozen tumor tissues (4 controls, 4 IFN-β low dose, 4 IFN-β
high dose) and glioma cell lines treated with IFN-β using RNeasy
mini kit (Qiagen GmbH, Germany). Quantitative RT-PCR for IP10 and
VEGF mRNA in glioma cells and glioma tissues has been described
previously (15). We performed
RT-PCR with the GeneAmp™ RNA PCR Kit (Perkin-Elmer Cetus, Norwalk,
CT). Briefly, 1 μg of total RNA was reverse transcribed by MuLV
reverse transcriptase in the presence of random hexamer, followed
by indicated cycles of PCR reaction (95°C for 1 min, 55°C for 1 min
and 72°C for 1 min) in the presence of 2 μM IP10 specific primers
(32 cycles), VEGF specific primers (28 cycles), or the β-actin
specific primers (16 cycles) as a control. The IP10 primers were
designed, the reverse primer (5′-GATTCAGACATCTCTTCTCACCC-3′) is
complementary to positions 295–275, and the forward primer
(5′-TGACTCTAAGTGGCATTCAAGG-3′) corresponds to positions 107–128
(16). The VEGF primers included
the reverse primer (5′-CCTGGTGAGAGATCTGGTTC-3′) spanning bases
861-842 and the forward primer (5′-TCGGGCCTCCGA AACCATGA-3′)
spanning bases 141–160. The β-actin primers included the reverse
primer (5′-GGAGTTGAAGGTAGTTTC GTG-3′) spanning bases 2429-2409 and
the forward primer (5′-CGGGAAATCGTGCGTGACAT-3′) spanning bases
2107–2126. The predicted sizes of the amplified IP10 and β-actin
DNA products were 188 and 214 bp, respectively. The VEGF primers
were chosen because they amplified exons 3 to 8 and allowed for
distinguishing between the different VEGF splicing variants. PCR
products of 516 and 648 bp corresponded with VEGF121 and VEGF165,
respectively. The quantification of these RT-PCR product levels was
performed on a Macintosh computer using the public domain NIH Image
program (developed at the US National Institute of Health).
Antibodies and immunohistochemistry
The Dako LSAB Kit for mouse and rabbit primary
antibody (Dako) was used. Tissue sections were deparaffined and
incubated with 10% normal goat serum in PBS for 20 min. The
sections were then incubated with a polyclonal anti-VEGF antibody,
A-20 (Santa Cruz Biotechnology) at a dilution of 1/100 (1 μg/ml
IgG) in PBS overnight at 4°C, and a monoclonal anti-mouse CD31
antibody (BD Pharmingen) at a dilution of 1/200 in PBS for 60 min
at room temperature. Chromatographically purified mouse IgG and
rabbit IgG (Dako) at the same IgG concentration were used as
negative controls. Sections were incubated with biotin-conjugated
goat anti-mouse or anti-rabbit immunogloblin for 10 min, followed
by washing in PBS for 10 min. The sections were then incubated with
peroxidase conjugated streptavidin solution for 5 min, followed by
washing in PBS for 5 min. Sections were then stained with freshly
prepared amino-ethylcarbazole solution for 10 min, followed by
washing for 5 min in tap water. The sections were then
counterstained with hematoxylin and mounted with aqueous mounting
media. The intracellular VEGF immunostaining was assessed
separately for tumor and endothelial cells using a semiquantitative
scale (−, not detected; +, moderate; ++, strong).
Tumor vascular density
Vascular density was scored using the
vasoproliferative component of the MAGS (microscopic angiogenesis
grading system) that has been used to quantify angiogenesis in a
variety of tumors (17). The
number of vessels at 200X field (1.0 mm2) was measured
in microvessel ‘hot spots’ (i.e., microscopic areas containing the
most dense collections of microvessels, as initially identified
under low power magnification) with the use of an Olympus
microscope, AHBT3 (Olympus, Tokyo, Japan) on CD31 stained tissue
sections. Vascular density was defined by averaging the number of
vessels in the three most vascularised areas.
Histochemical detection of apoptotic
cells and determination of apoptotic index
Apoptotic cells were visualized using the ApopTag
in situ detection kit (Oncor, Gaithersburg, MD) as described
previously (15). The staining
procedures were modified based on the manufacturer’s instructions.
Briefly, after deparaffinization and rehydration, the tissues were
digested with proteinase K (20 μg/ml in PBS; Wako, Osaka, Japan)
for 20 min at room temperature and washed. Slides were then put
into 3% H2O2 for 5 min and washed with PBS.
After adding the equilibration buffer for 10 min. TdT enzyme was
pipetted onto the sections, which were then incubated at 37°C for 1
h. The reaction was stopped by putting sections in stop/wash
buffer. After washing, anti-digoxigenin-peroxidase was added to the
slides. Slides were washed, stained with diaminobenzidine (DAKO)
substrate, and counterstained with hematoxylin. A specimen known to
be positive for apoptotic cells was used as positive control for
subsequent staining. Substitution of TdT with distilled water was
used as a negative control. The apoptotic index was expressed as
the ratio of positively staining tumor cells to all tumor cells,
given as a percentage for each case. At least five representative
areas without necrosis in a section were selected by light
microscopy using 40- to 200-fold magnification. A minimum of 3,000
cells was counted under a 400-fold magnification. Positively
staining tumor cells with the morphological characteristics of
apoptosis were identified using standard criteria, including
chromatin condensation, nucleolar disintegration, and formation of
crescentic caps of condensed chromatin at the nuclear
periphery.
Glioma conditioned medium induction of
HUVEC migration
Glioma cells (1×105) were plated into a
6-well plate. After incubation for 24 h in MEM with 10% FCS, the
medium was changed to MCDB107 with 0.5% FCS containing various
concentrations of IFN-β. After 48 h incubation, the conditioned
medium was harvested and the concentration of VEGF in glioma
conditioned medium was measured using Quantikine™ Human VEGF
Immunoassay (R&D Systems, Minneapolis, MN). Endothelial cell
migration was evaluated by 24-well modified Boyden chamber (Coster,
Cambridge, MA) as described previously. The chamber contains
Nucleopore polycarbonate membranes (8-μm pore size) that had been
soaked overnight in 0.1% gelatin in 0.1% acetic acid. A total of
100 μl of HUVECs, 2×106 cells/ml in MCDB107 with 0.5%
FBS, was plated in upper well and 600 μl of collected conditioned
medium was added to lower wells. The assembly was incubated for 6
h. The membrane was removed, fixed in methanol, stained with
hematoxylin and the cells in upper surface were gently wiped with
cotton swab. The insert was mounted on glass slide. The number of
migrated cells was counted from at random five fields using ×25
magnification. Data were expressed as cells per field. One field
corresponded to 0.09 mm2 (width, 309 μm × height, 291
μm) of the membrane area. The experiment was repeated two times in
quadruplicate for each concentration.
SCID mouse U87 implant cranial window
model and quantitation of intravital tumor microcirculation
U87 tumor tissue fragment (1 mm3) was
implanted on the surface of the SCID mouse cranial window (n=3).
IFN-β was injected intraperitoneally for 7 days, and then the
cranial window was evaluated for tumor microcirculation. Three
series of experimental studies to visualize blood flow dynamics of
the tumor microcirculation and to quantify their microhemodynamic
parameters were performed (18).
First, by labeling plasma component, the tumor
microvasculature was visualized and mapped to obtain information on
vascular architecture and dimensions of microvessels. To enhance
the contrast of microvessel images against a dark background, a
solution of FITC-labeled dextran (FITC-Dx, MW 150,000; Sigma, St.
Louis, MO) was intravenously injected (20 mg/ml, 2 ml/kg). This
permitted bright fluorescence images of the vascular lumen, and
enabled mapping of the vascular architecture and accurate
measurements of luminal diameter. The diameter of microvessels was
measured carefully with a vernier caliper on the standstill frame
of the video-recorded images by playback of a high quality
video-cassette recorder (Model BR-S605B). Their average values were
calculated from five measurements in each vessel.
Secondary, to visualize the flow behavior of
erythrocytes in vivo and to measure their velocities, a part
of erythrocytes was labeled fluorescently and injected
intravenously. The arterial blood (0.2 ml) of a donor mouse was
collected from a tail vein into a 1.5 ml test tube containing
heparin (100 units) for anticoagulation. The erythrocytes were
separated from the plasma by centrifugation and were washed twice
with pysiological saline solution. These erythrocytes were then
incubated at room temperature with a phosphate-buffered saline
(PBS) solution, adjusted to pH 7.8, containing 1 mg/ml fluorescein
isothiocyanate (FITC). After 60 min of incubation, the labeled
cells were washed twice with a saline solution containing 1% bovine
serum albumin to remove uncombined fluorescent dyes. The final
volume percent of the labeled cells was adjusted to 50% by adding
an isotonic saline solution. These suspensions were injected
intravenously through a tail vein. The erythrocyte velocity was
calculated by a frame-by-frame analysis and averaged for at least
10 measurements.
Thirdly, to analyze the leukocyte behavior and the
leukocyte-endothelium interaction, leukocytes were also labeled
fluorscently. A working solution of rhodamine 6G (Sigma) was
prepared by dissolving 10 mg of the dye in 40 ml of physiological
saline. This solution was diluted with saline until a final
concentration of 50 μg/ml. The optimal concetration of the dye for
imaging leukocytes was determined from several preliminary
experiments. Each solution was freshly prepared on the day of the
experiment and filtered through a 0.22 μm filter before each
experiment. Leukocytes were found to be visualized by injecting a
small bolus of 2 ml/kg of the solution intravenously. Arolling
leukocyte was defined as one that marginates along the vessel wall
and is clearly dissociated from the bulk of the blood flow. An
adhering leukocyte was defined as one that stays stationary during
at least 15 sec of the 30-sec observation period (19).
Statistical analyses
Vascular density, MIB-1 positivity, apoptosis index,
tumor volumes, densitometric value of VEGF, IP10 and β-actin, and
the parameter of tumor microcirculation (diameter, velocity, number
of leukocyte) were expressed as mean ± SD. Statistically
significant differences between the groups were determined using a
one-way analysis of variance and the Tukey’s test. All p-values are
two-sided; values are considered statistically significant for
p<0.05.
Results
Antiangiogenic activity of IFN-β in
vitro
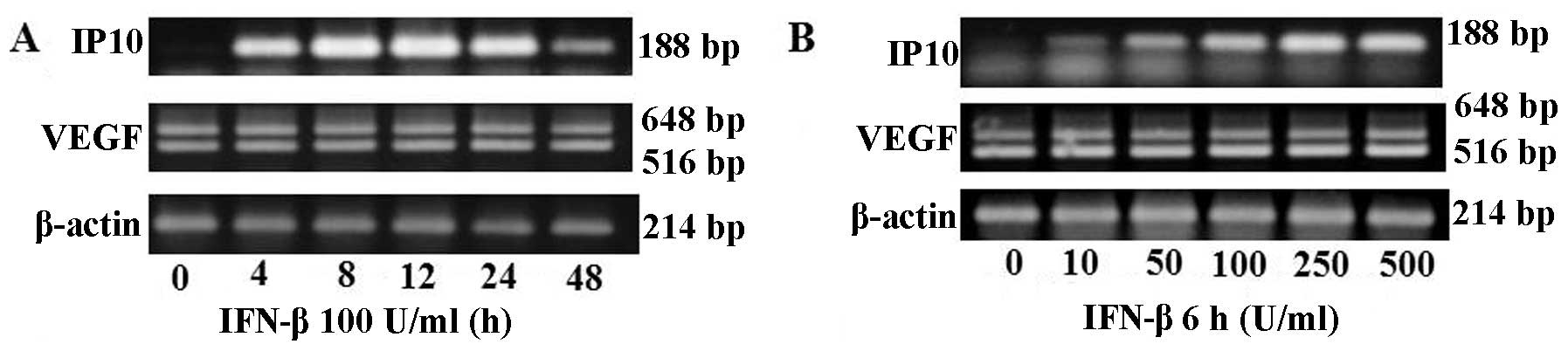
RT-PCR analysis demonstrated IFN-β upregulated IP10
mRNA expression time- (4–48 h) and dose- (10–500 U/ml) dependently,
but not VEGF mRNA expression (Fig.
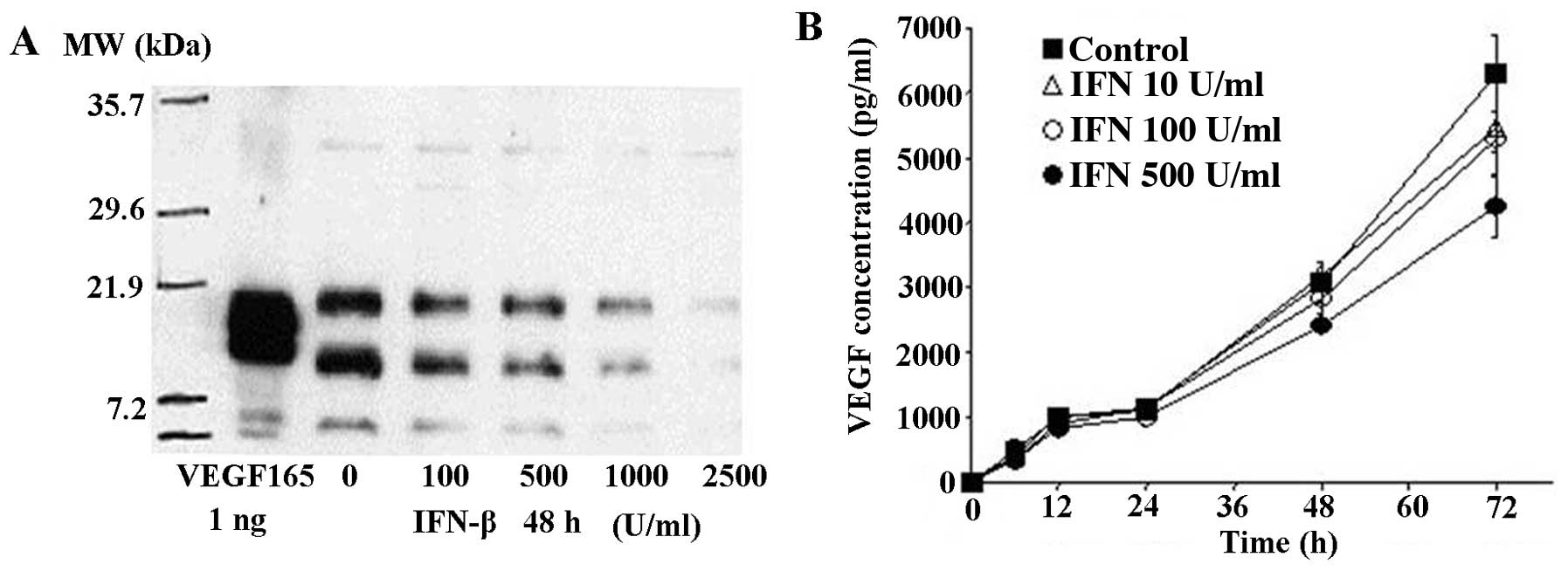
1). However, VEGF protein concentration and secretion in the
conditioned medium was decreased time- and dose-dependently by
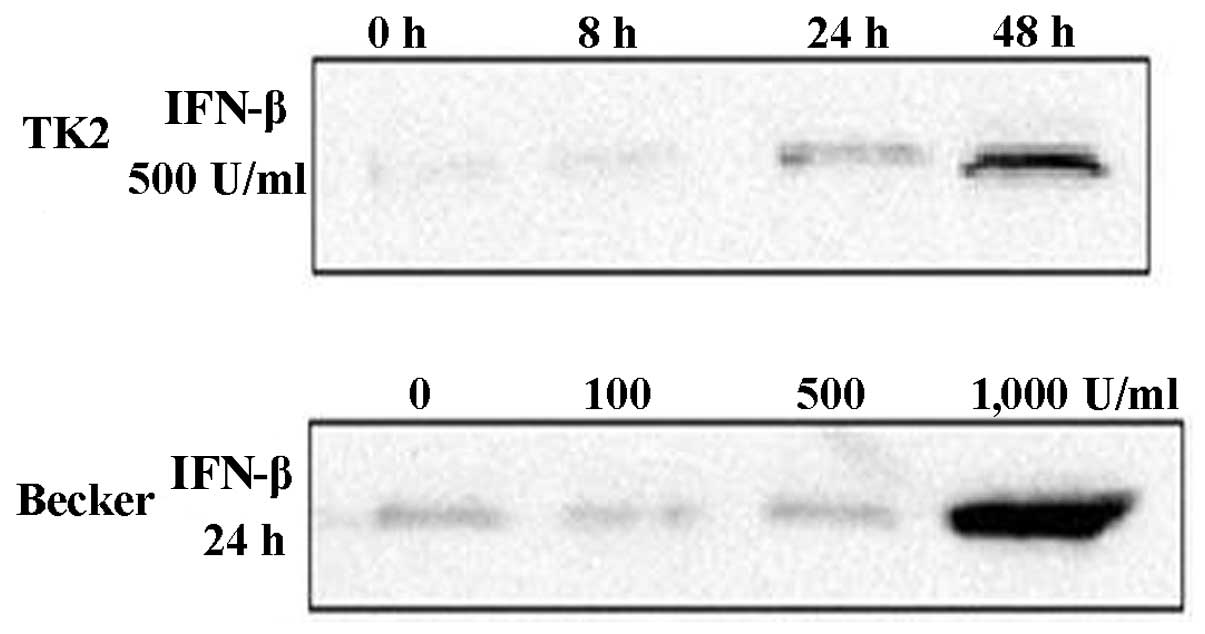
IFN-β treatment (Fig. 2). IP10
protein in cell extracts was increased time- and dose-dependently
with IFN-β treatment (Fig. 3).
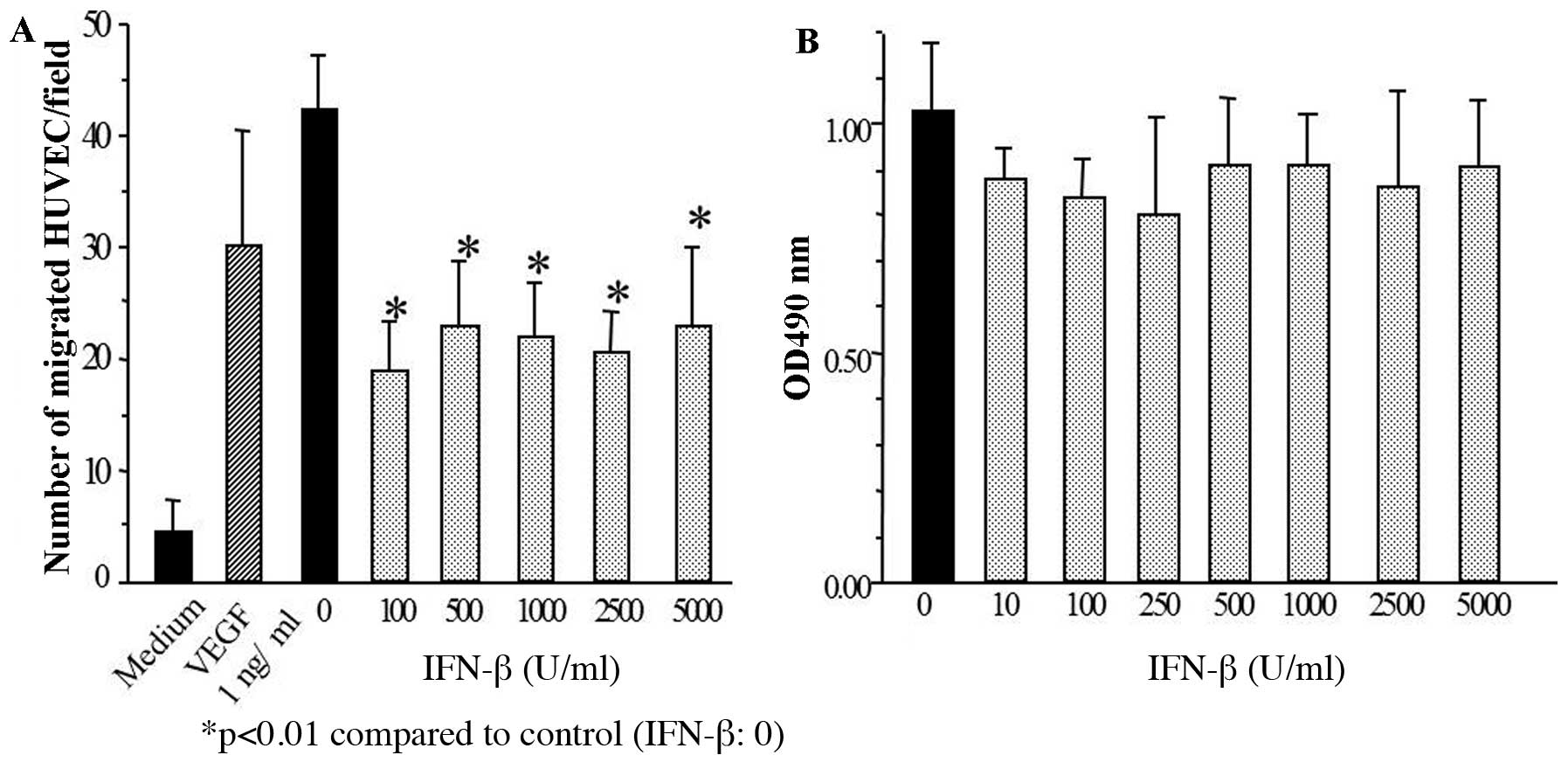
Increased VEGF and decreased IP10 protein expression of glioma
cells treated with IFN-β at 100 U/ml for 48 h resulted in the
inhibition of HUVECs migration (Fig.
4A). By contrast, glioma cell proliferation was not affected by
IFN-β treatment for 48 h at the dose ranging from 10 to 5,000 U/ml
(Fig. 4B).
IFN-β inhibition of glioma growth in
subcutaneous tumor
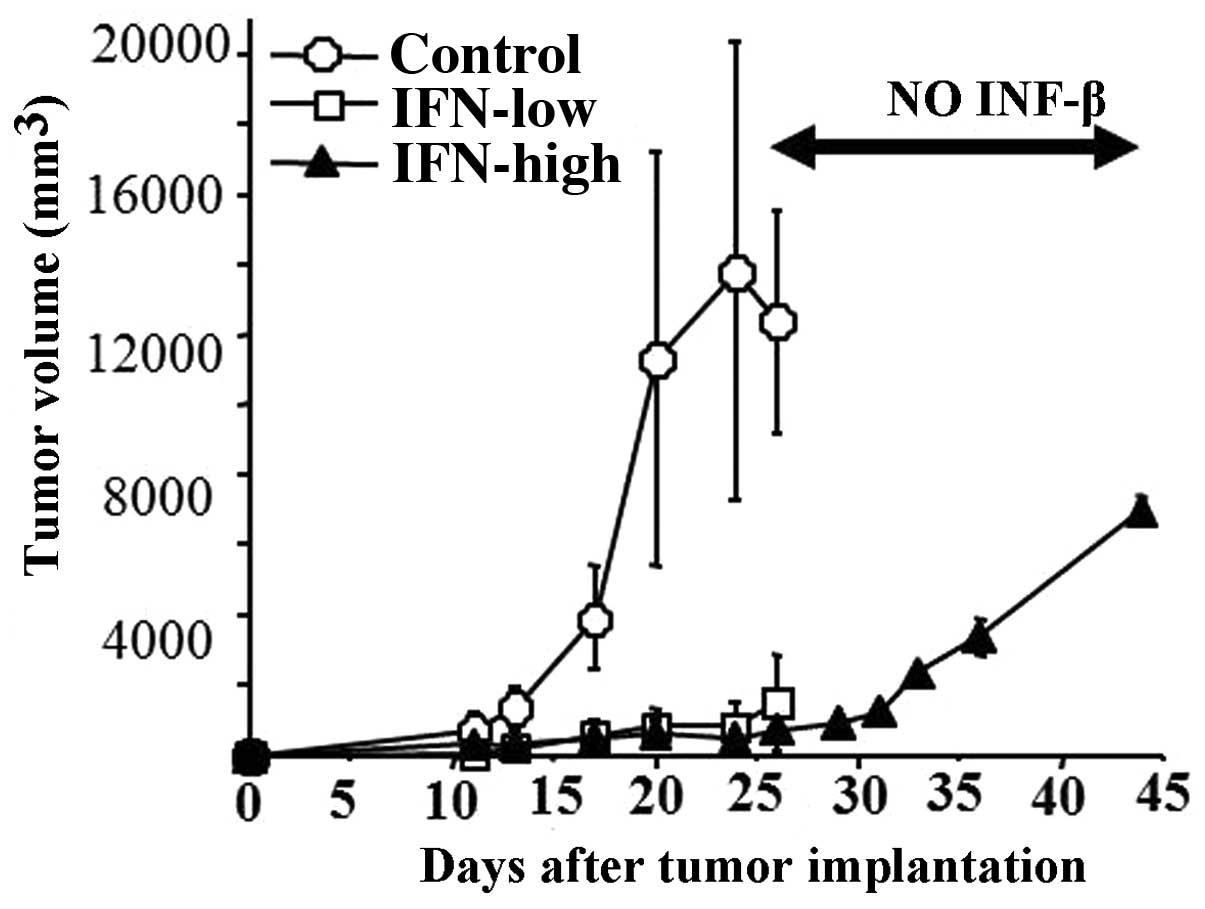
IFN-β local injection as well as systemic injection
for 15 days significantly inhibited U87 subcutaneous growth
(Fig. 5). VEGF protein expression
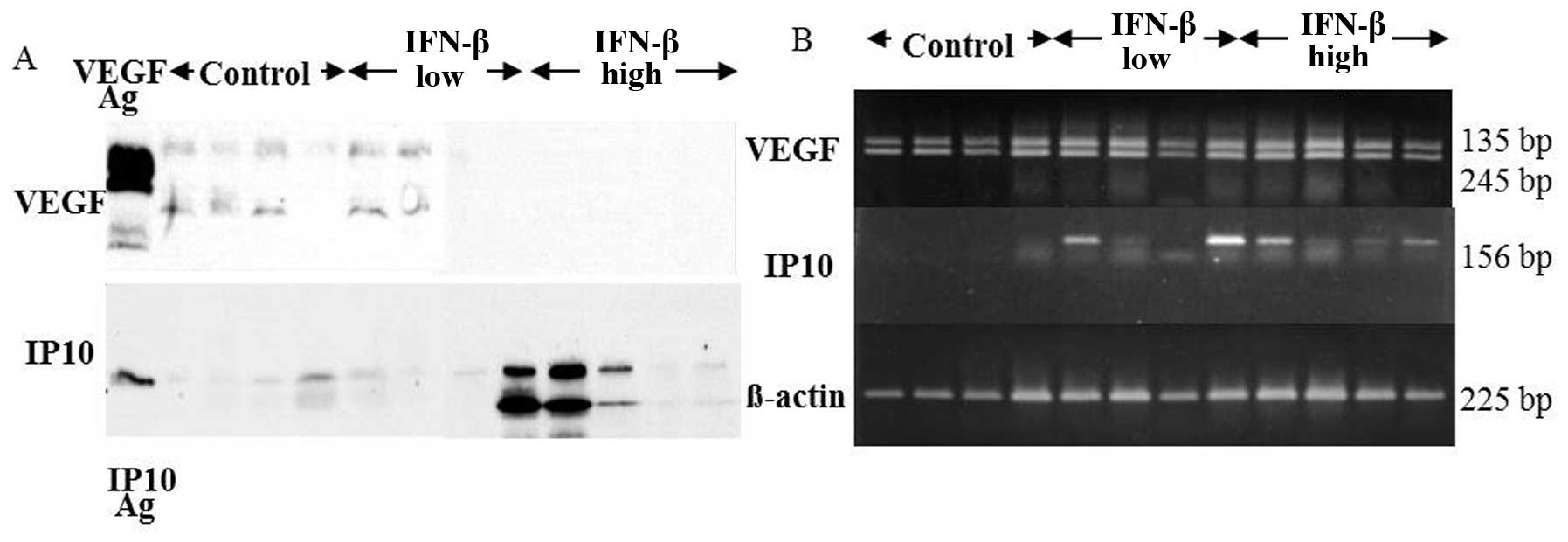
of U87 tumor tissues was decreased dramatically in the IFN-β high
dose treatment, while IP10 protein expression increased (Fig. 6A). RT-PCR analysis demonstrated
that IP10 mRNA expression of the tumor tissues was not detected in
any of control group. Upregulation of IP10 mRNA expression of the
tumor tissues was observed in 2 of 4 IFN-β low dose group and all 4
in IFN-β high dose group. VEGF mRNA expression of the tumor tissues
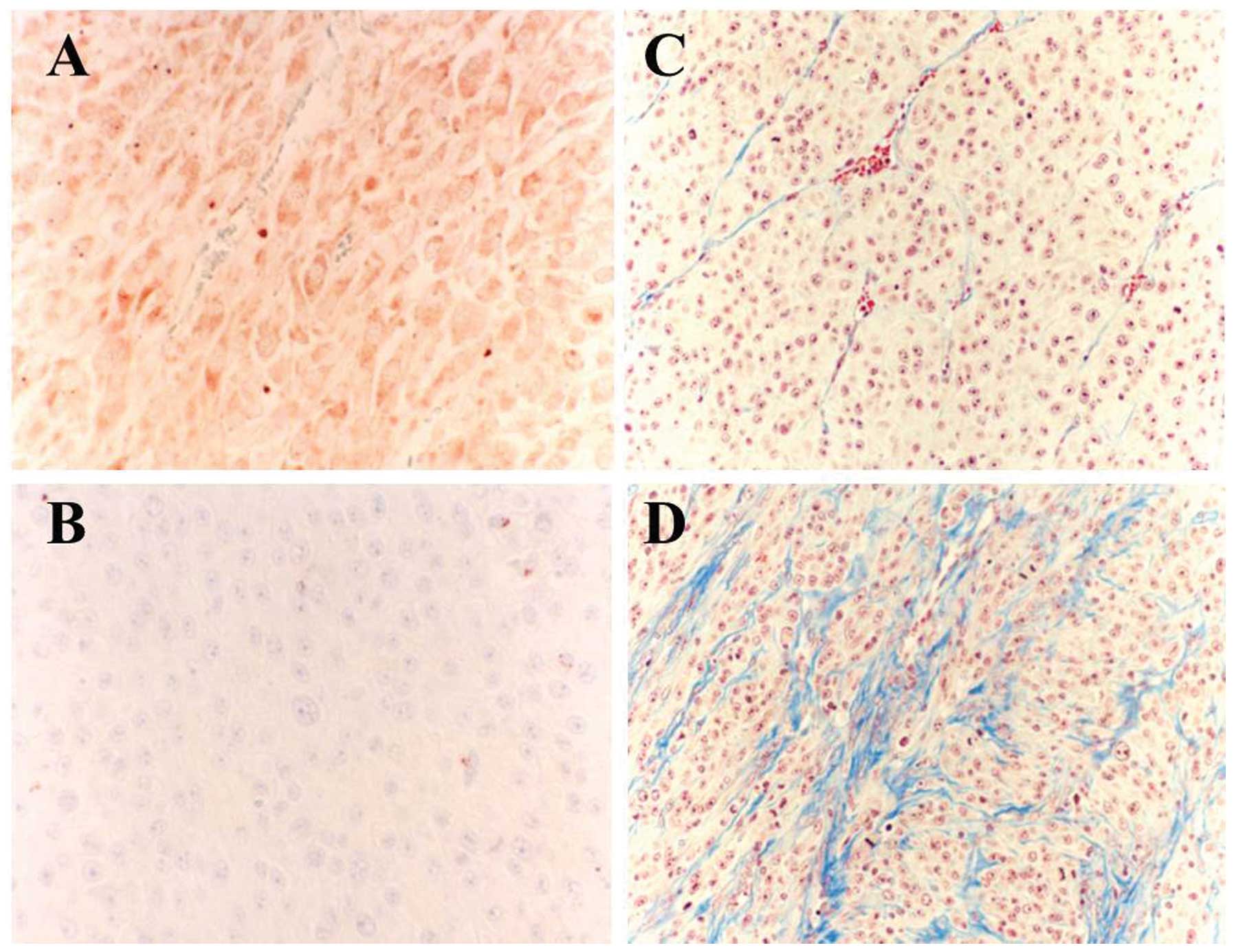
was not affected by RT-PCR analysis (Fig. 6B). Immunohistochemistry clearly
demonstrated high VEGF expression in the control tumor tissues and
decreased VEGF expression in the IFN treated tumor tissues
(Fig. 7). VEGF expression was
strong in 5 of 5 control group and in none of IFN-β high and low
dose group. CD31 positive vessel densities were significantly
decreased both in the IFN-β low and high treated groups compared to
control group (Table I). In IFN-β
high treated group, MIB-1 positivities were significantly low and
apoptosis indices were significantly high compared to the control
animals (Table I). The growth
inhibitory effect of IFN-β was reversible because the tumor did
re-start to grow after the discontinuation of the IFN-β treatment
at similar degree to control tumors (Fig. 5A). In addition, IFN-β treatment
increased collagen fiber deposition in the tumor tissues, which was
demonstrated by Masson’s Trichrome stain (Fig. 7C and D). The increase was not
observed in the group of systemic IFN-β treatment.
 | Table IU87 tumor pathology 25 days after
interferon-β treatment. |
Table I
U87 tumor pathology 25 days after
interferon-β treatment.
| Tumor volume
(mm3) | MIB-1 (%) | Apoptosis index
(%) | Vessel density
(no./mm2) |
|---|
| Control |
12,371.4±3,195.3 | 38.2±3.9 | 0.20±0.001 | 89.0±11.4 |
| IFN-β low |
1,523.8±1,354.4b | 31.4±6.4 | 0.56±0.104 | 54.0±11.1b |
| IFN-β high | 769.0±339.0b | 21.8±2.7a | 1.00±0.024b | 78.3±8.8b |
IFN-β inhibition of glioma
microcirculation
With the cranial window model, 7 days after the U87
fragment implantation, tortuous vessels grew around the fragment
where red blood cell velocity and the degree of leukocyte adhesion
and rolling significantly decreased compared to those of the normal
cortical vein at the diameter of 20–80 μm. IFN-β systemic treatment
for 7 days dramatically reversed the decreased leukocyte adhesion
and rolling in the U87 glioma graft, while tortuous vessel
morphology and red blood cell velocity were not unchanged (Table II).
| Table IIU87 tumor microcirculation with
interferon-beta treatment. |
Table II
U87 tumor microcirculation with
interferon-beta treatment.
| Vessel diameter
(μm) | Red blood cell
velocity (μm/sec) | Vessel diameter
(μm) | Leukocyte adhesion,
rolling (no./600 sec) |
|---|
| No tumor | 23.9±10.5 | 475.6±175.3 | 45.6±34.7 | 112.1±80.5 |
| U87 tumor | 19.8±8.9 | 248.0±91.2 | 35.0±21.0 | 27.9±14.7 |
| U87 tumor, IFN | 20.0±0.2 | 277.2±8.7 | 66.0±15.2 | 306.8±131.5a |
Discussion
Our study clearly demonstrated that IFN-β inhibited
glioma angiogenesis in three different aspects, in vitro,
subcutaneous tumor, and intracerebral tumor microcirculation. The
local and systemic administration of IFN-β to SCID mouse bearing
human glioblastoma cells decreased expression of VEGF, induced
expression of IP10, reduced vascular density and inhibited
reversible tumor growth. Also the systemic administration of IFN-β
significantly inhibited glioma growth and reversed the
microcirculation of the glioma tissues to that of the non-tumor
brain tissue, suggesting the anti-proliferative effect is not due
to only high local concentrations of the protein. Among these
manifestations, simultaneous action with upregulation of VFGF and
downregulation of IP10 is unique. Because angiogenesis is
influenced by the balance between stimulatory and inhibitory
molecules released by the tumor and its microenvironment, any
decrease in a stimulatory molecule or an increase in an inhibitory
molecule should reduce the level of neovascularization within the
tumor.
Antiangiogenic action of IFN-β
Clinical data concluded that the systemic chronic
administration of IFN-α or IFN-β accelerate the regression of
richly vascularized tumors, e.g., life-threatening hemangiomas of
infancy (20),
hemangio-endotheliomas (21),
hemangiopericytoma (22) and
Kaposi’s sarcomas (23). The
mechanisms responsible for this remarkable clinical outcome
remained unclear. IFN-β can inhibit angiogenesis by several
mechanisms.
In vitro, IFN-β inhibits endothelial cell
proliferation (11), endothelial
cell migration (24),
downregulation of transcription and production of bFGF protein
(11), interleukin 8 (25) and collagenase type IV (13), all of which are involved in the
angiogenic response. In addition, our data demonstrate IFN-β
inhibits production of VEGF protein, although the effects is
marginal, and induces production of IP10, endogenous angiogenesis
inhibitor, resulting in inhibition of HUVEC migration induced by
glioma conditioned medium.
However, the antiangiogenic mechanism of IFN-β for
malignant gliomas has not been investigated comprehensively.
Boethius et al (26)
reported that systemic administration of IFN-α to patients with
glioblastoma multiforme induced marked changes in the tumor
vasculature, which supports the notion that IFN-α may have an
effect on tumor vessels. We demonstrated that IFN-β inhibits glioma
cell induced endothelial cell migration, VEGF secretion in the
glioma cell conditioned medium, and VEGF expression and vessel
densities in the glioma tissues. IFN-β is superior to IFN-α in
terms of anti-angiogenic effects (27). The above strongly suggests that the
in vivo antitumor effect of IFN-β in malignant gliomas may
be mediated, at least in part, via the angiogenesis inhibition
rather than the antiproliferative activity on tumor cells. Hong
et al (28) demonstrated
the level of VEGF and bFGF expression of U87 cells was not
influenced by IFN-β treatment at concentrations from 10–500 IU/ml
for 24 and 72 h. They measured the expression of cell extract and
we measured secreted protein of VEGF.
IFN-β inhibits VEGF secretion
We reported that IFN-β treatment inhibited glioma
cell VEGF secretion in vitro and glioma angiogenesis with
downregulation of VEGF in glioma tissue. The investigations
concerning to relationship of VEGF inhibition and IFN-β are very
limited. VEGF promotes phosphorylation-dependent ubiquitination and
degradation of IFN receptor and ensuing attenuation of IFN-α/β
signaling; these processes appear to be required for efficient
angiogenesis (29). In another
report, the antitumor effect of IFN-β was offset by the
tumor-progressive character of endothelial progenitor cells (EPCs)
and the tumor growth, and the vascular density of tumor tissues
increased by the co-implanted EPCs were decreased upon IFN-β
treatment. In addition, overall expression levels of VEGF in tumor
tissues that were decreased upon IFN-β treatment (30).
Recent clinical studies indicate that anti-VEGF
agents are important for the treatment of angiogenesis-dependent
diseases. The approaches used today are mainly based on the
development and administration of functional recombinant protein
antagonists that either neutralize the extracellular VEGF function
or block VEGF signaling in target cells. The disadvantages of
current therapeutic strategies are many, including difficulties in
manufacturing active recombinant protein, high-dose requirements,
high costs for manufactures and consumers, and the probable need
for lifetime treatment of the patient. A novel anti-VEGF strategy
by blocking its secretion in tumor cells is reported by retaining a
VEGF binding protein in the cell (31), by inhibiting VEGF promoter activity
on neuroendocrine tumor cell lines (32), by a PI3 kinase inhibitor in
melanoma (33) and by IFN-α in
melanoma cell line (34). PI3
kinase activation may occur via loss of phosphatase and tensin
homolog (PTEN) that is closely related to IFN-β sensitivity in
glioma (35). Taken together,
IFN-β treatment appears to be one of novel strategies of
anti-angiogenesis on glioma by preventing VEGF secretion.
IP10 antiangiogenic action
IFN-β upregulates IP10 from a number of cells,
including keratinocytes, fibroblasts, endothelial cells,
mononuclear phagocytes and cancer cells. IFN-β may shift the
biological balance of ELR+ (IL-8) and ELR-CXC (IP10) chemokines,
leading to reduced net angiogenic activity (36). IP10 is a potent inhibitor of not
only ELR+ CXC chemokine (IL-8) but also the unrelated angiogenic
factors bFGF and VEGF (36).
The production of IP10 from adenocarcinoma and
squamous carcinoma tumors was inversely correlated with their
growth. SCID mouse bearing tumors were given intratumor injection
of recombinant human IP10. IP10 treatment resulted in a >40%
reduction in tumor size and mass, respectively. The mechanism of
growth inhibition by intratumor administration of IP10 was found to
be correlated with a reduction in primary tumor-derived angiogenic
activity and neovasculature (37).
IP10 protein expression was inversely correlated with vascular
density and clinical behavior in endometrial cancer (38). We demonstrated IFN-β treatment
shifted the balance of VEGF/IP-10 into angiostatic state in glioma
cells and tissue. Interestingly, IP-10 has been identified as a
major biological marker mediating cancer severity and may be
utilized as a prognostic indicator for various cancers (39). IP-10 shows potential as a
biological response marker of IFN-β in glioma.
IFN-β affects tumor microcirculation in
gliomas
Yuan et al (40) and Foltz et al (19) reported the intravital microscopic
analysis of malignant gliomas transplanted into a cranial window
preparation. Although cranial window model to visualize tumor
microcirculation is not a new method, this method is superior to
the present MRI based evaluation of tumor vasculature, such as
perfusion MRI (41) and vessel
architecture imaging (42).
Intravital analysis with the cranial window model can provide us
information of the tumor microcirculation, such as leukocyte
adhesion to vessel wall in addition to vessel architecture.
Malignant glioma caused decreased number of leukocyte adhesion to
endothelial cells, which is a key factor in the tumor
microcirculation. We demonstrated that IFN-β influenced the glioma
microcirculation with reversal of the inhibition of leukocyte
adhesion. IFN-β inhibits activated leukocyte migration through
human brain microvascular endothelial cell monolayer (43). Implantation of IFN-β producing
cells upregulated the adhesion molecules, ICAM-1 and VCAM-1
(27). TNF-α by co-treatment with
IFN-β increased soluble VCAM-1 in human cerebral endothelial cells
(44). VEGF antagonist, such as
soluble Flt1 (soluble form of VEGF receptor) increases ICAM1, VCAM1
and leukocyte adhesion in endothelial cells (45). Our data suggest IFN-β increased
ICAM1 and VCAM1 directly or indirectly through inhibition of VEGF
production with glioma cells, resulting reversal of leukocyte
adhesion.
Taken together, the reversal effect of IFN-β on the
tumor microcirculation that is considered as a concept for vascular
normalization (46), could be one
of the mechanisms by which IFN-β treatment exerts antiangiogenic
effects in malignant glioma.
IFN-β affects matrix reaction
Matrix modifications were characterized on
histological sections stained with Masson’s Trichrome stain.
Collagen deposition or structure of IFN-β treated animals appeared
to be thicker than in the non-treated ones. The collagen deposition
was prominently observed with local injection of IFN-β, but not
with systemic treatment. Combined with the similar increase of
collagen fiber in the stromal tissue surrounding the IFN-producing
tumor cells (27), high
concentration of IFN-β in the tissues is needed for this thicker
collagen deposition. Thicker collagen deposition may increase the
interstitial pressure in the tumor and decrease diffusion of
angiogenic molecules, e.g., VEGF, to the endothelial cells,
resulting in inhibition of angiogenesis (47). However IFNs are species specific.
Our studies on the effect of the IFNs concerning the host
microenvironment (immune, endothelial cells and extracellular
matrix) have some limitations.
In conclusion, one of the important roles of IFN-β
for malignant glioma growth inhibition was anti-angiogenesis by
directly inhibiting angiogenesis through downregulation of VEGF and
upregulation of IP-10 and indirectly changing the tumor
microcirculation and regulating the interstitial pressure. At
present, the clinical effectiveness of IFN-β for human malignant
gliomas is limited. Several published studies have shown that
anti-angiogenic agents act synergistically with chemotherapy or
radiation therapy. Also the growth inhibitory effect was reversible
and non-toxic even with high dose of IFN-β in our study, suggesting
combination therapy with IFN-β and chemotherapy or radiation
therapy for long-term usage will overcome many of the limitations
of individual treatment.
Acknowledgements
We gratefully acknowledge Norio Ohshima, Department
of Biomedical Engineering, University of Tsukuba, Koji Tsuboi,
Proton Medical Research Center, University of Tsukuba, and Youji
Mitsui, Biophysiology, Tokushima Bunri University for the excellent
discussion, Toray Industries, Inc. and Daiichi Pharmaceutical Co.
for supply of human interferon-β, and Yoshiko Tsukada, Makiko
Miyagawa, and Momoyo Ito for the excellent technical assistance.
This study was supported in part by a Grant-in-Aid for Scientific
Research from the Ministry of Education, Culture, Sports, Science
and Technology of Japan (S.T.), Japan Brain Foundation (S.T.) and
Japanese Foundation for Multidisciplinary Treatment of Cancer
(S.T.).
References
|
1
|
Fine HA, Wen PY, Robertson M, O’Neill A,
Kowal J, Loeffler JS and Black PML: A phase I trial of a new
recombinant human β-interferon (BG9015) for the treatment of
patients with recurrent gliomas. Clin Cancer Res. 3:381–387.
1997.
|
|
2
|
Natsume A, Ishii D, Wakabayashi T, Tsuno
T, Hatano H, Mizuno M and Yoshida J: IFN-beta down-regulates the
expression of DNA repair gene MGMT and sensitizes resistant glioma
cells to temozolomide. Cancer Res. 65:7573–7579. 2005.PubMed/NCBI
|
|
3
|
Yoshino A, Ogino A, Yachi K, Ohta T,
Fukushima T, Watanabe T, Katayama Y, Okamoto Y, Naruse N and Sano
E: Effect of IFN-beta on human glioma cell lines with temozolomide
resistance. Int J Oncol. 35:139–148. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wakabayashi T, Kayama T, Nishikawa R,
Takahashi H, Hashimoto N, Takahashi J, Aoki T, Sugiyama K, Ogura M,
Natsume A and Yoshida J: A multicenter phase I trial of combination
therapy with interferon-beta and temozolomide for high-grade
gliomas (INTEGRA study): the final report. J Neurooncol.
104:573–577. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Colman H, Berkey BA, Maor MH, Groves MD,
Schultz CJ, Vermeulen S, Nelson DF, Mehta MP and Yung WK; Radiation
Therapy Oncology Group. Phase II Radiation Therapy Oncology Group
trial of conventional radiation therapy followed by treatment with
recombinant interferon-beta for supratentorial glioblastoma:
results of RTOG 9710. Int J Radiat Oncol Biol Phys. 66:818–824.
2006. View Article : Google Scholar
|
|
6
|
Motomura K, Natsume A, Kishida Y, Higashi
H, Kondo Y, Nakasu Y, Abe T, Namb H, Wakai K and Wakabayashi T:
Benefits of interferon-beta and temozolomide combination therapy
for newly diagnosed primary glioblastoma with the unmethylated MGMT
promoter: a multicenter study. Cancer. 117:1721–1730. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brem S, Cotran R and Folkman J: Tumor
angiogenesis: a quantitative method for histologic grading. J Natl
Cancer Inst. 48:347–356. 1972.PubMed/NCBI
|
|
8
|
Norden AD, Drappatz J and Wen PY: Novel
anti-angiogenic therapies for malignant gliomas. Lancet Neurol.
7:1152–1160. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nagane M, Nishikawa R, Narita Y, Kobayashi
H, Takano S, Shinoura N, Aoki T, Sugiyama K, Kuratsu J, Muragaki Y,
Sawamura Y and Matsutani M: Phase II study of single-agent
bevacizumab in Japanese patients with recurrent malignant glioma.
Jpn J Clin Oncol. 42:887–895. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chinot OL, Wick W, Mason W, Henriksson R,
Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea
D, Brandes AA, Hilton M, Abrey L and Cloughesy T: Bevacizumab plus
radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl
J Med. 370:709–722. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Singh RK, Gutman M, Bucana CD, Sanchez R,
Llansa N and Fidler IJ: Interferons α and β down-regulate the
expression of basic fibroblast growth factor in human carcinomas.
Proc Natl Acad Sci USA. 92:4562–4566. 1995.
|
|
12
|
Dinney CPN, Bieleberg DR, Perrotte P,
Reich R, Eve BY, Bucana CD and Fidler IJ: Inhibition of basic
fibroblast growth factor expression, angiogenesis, and growth of
human bladder carcinoma in mice by systemic interferon-α
administration. Cancer Res. 58:808–814. 1998.PubMed/NCBI
|
|
13
|
Gohji K, Fidler IJ, Tsan R, Radinsky R,
von Eschenbach AC, Tsuruo T and Nakajima M: Human recombinant
interferon-beta and -gamma decrease gelatinase production and
invasion by human KG-2 renal-carcinoma cells. Int J Cancer.
58:380–384. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Takano S, Gately S, Neville ME, Herblin
WF, Gross JL, Engelhard H, Perricone M, Eidsvoog K and Brem S:
Suramin, an anticancer and angiosuppressive agent, inhibits
endothelial cell binding of basic fibroblast growth factor,
migration, proliferation, and induction of urokinase-type
plasminogen activator. Cancer Res. 54:2654–2660. 1994.
|
|
15
|
Takano S, Tsuboi K, Matsumura A, Tomono Y,
Mistui Y and Nose T: Expression of the angiogenic factor thymidine
phosphorylase in human astrocytic tumors. J Cancer Res Clin Oncol.
126:145–152. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Boorsma DM, de Haan P, Willemze R and
Stoof TJ: Human growth factor (huGRO), interleukin-8 (IL-8) and
interferon-gamma-inducible protein (gamma-IP-10) gene expression in
cultured normal human keratinocytes. Arch Dermatol Res.
286:471–475. 1994. View Article : Google Scholar
|
|
17
|
Takano S, Yoshii Y, Kondo S, Maruno T,
Shirai S and Nose T: Concentration of vascular endothelial growth
factor in the serum and tumor tissue of brain tumor patients.
Cancer Res. 56:2185–2190. 1996.PubMed/NCBI
|
|
18
|
Suzuki T, Yanagi K, Ookawa K, Hatakeyama K
and Ohshima N: Flow visualization of microcirculation in solid
tumor tissues: intravital microscopic observation of blood
circulation by use of a confocal laser microscope. Front Med Biol
Eng. 7:253–263. 1996.
|
|
19
|
Foltz RM, McLendon RE, Friedman HS, Dodge
RK, Bigner DD and Dewhirst MW: A pial window model for the
intracranial study of human glioma microvascular function.
Neurosurgery. 36:976–984. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ezekowitz RAB, Mulliken JB and Folkman J:
Interferon alfa 2a therapy for life-threatening hemangiomas in
infancy. N Engl J Med. 324:1456–1463. 1992. View Article : Google Scholar
|
|
21
|
Orchard PJ, Smith CM, Woods WG, Day DL,
Dehner LP and Shapiro R: Treatment of haemangio endotheliomas with
alpha interferon. Lancet. 2:565–567. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kirn DH and Kramer A: Long-term function
from disease progression with interferon alfa therapy in two
patients with malignant hemangiopericytoma. J Natl Cancer Inst.
88:764–765. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Real FX, Qettgen HF and Kroun SE: Kaposi’s
sarcoma and the acquired immunodeficiency syndrome: treatment with
high and low doses of recombinant leukocyte α interferon. J Clin
Oncol. 4:544–551. 1986.
|
|
24
|
Stout AJ, Gresser I and Thompson WD:
Inhibition of wound healing in mice by local interferon-β
injection. Int J Exp Pathol. 74:79–85. 1993.
|
|
25
|
Oliveira IC, Sciavolino PJ, Lee TH and
Vilcek J: Downregulation of interleukin 8 gene expression in human
fibroblasts: unique mechanism of transcriptional inhibition by
interferon. Proc Natl Acad Sci USA. 89:9049–9053. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boethius J, Blomgren H, Collins VP, Greitz
T and Strander H: The effect of human interferon-α administration
to patients with glioblastoma multiforme. Acta Neurochir.
68:239–251. 1983.
|
|
27
|
Rozera C, Carlei D, Lollini PL, De
Giovanni C, Musiani P, Di Carlo E, Belardelli F and Ferrantini M:
Interferon (IFN)-β gene transfer into TS/A adenocarcinoma cells and
comparison with IFN-α. Am J Pathol. 154:1211–1222. 1999.
|
|
28
|
Hong YK, Chung DS, Joe YA, Yang YJ, Kim
KM, Park YS, Yung WKA and Kang JK: Efficient inhibition of in vivo
human malignant glioma growth and angiogenesis by interferon-β
treatment at early stage of tumor development. Clin Cancer Res.
6:3354–3360. 2000.PubMed/NCBI
|
|
29
|
Zheng H, Qian J, Carbone CJ, Leu NA, Baker
DP and Fuchs SY: Vascular endothelial growth factor-induced
elimination of the type 1 interferon receptor is required for
efficient angiogenesis. Blood. 118:4003–4006. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xiao HB, Zhou WY, Chen XF, Mei J, Lv ZW,
Ding FB, Li GQ, Zhong H and Bao CR: Interferon-β efficiently
inhibited endothelial progenitor cell-induced tumor angiogenesis.
Gene Ther. 19:1030–1034. 2012.
|
|
31
|
Björndahl M, Cao R, Eriksson A and Cao Y:
Blockage of VEGF-induced angiogenesis by preventing VEGF secretion.
Circ Res. 94:1443–1450. 2004.PubMed/NCBI
|
|
32
|
Von Marschall A, Scholz A, Cramer T,
Schafer G, Schirner M, Oberg K, Wiedenmann B, Hocker M and Rosewicz
S: Effects of interferon alpha on vascular endothelial growth
factor gene transcription and tumor angiogenesis. J Natl Cancer
Inst. 95:421–437. 2003.PubMed/NCBI
|
|
33
|
Bedogni B, O’Neill MS, Welford SM, Bouley
DM, Giaccia AJ, Denko NC and Powell MB: Topical treatment with
inhibitors of the phosphatidylinositol 3′-kinase/Akt and
Raf/mitogen-activated protein kinase kinase/extracellular
signal-regulated kinase pathways reduces melanoma development in
severe combined immunodeficient mice. Cancer Res. 64:2552–2560.
2004.
|
|
34
|
Raig ET, Jones NB, Varker KA, Benniger K,
Go MR, Biber JL, Lesinski GB and Carson WE III: VEGF secretion is
inhibited by interferon-alpha in several melanoma cell lines. J
Interferon Cytokine Res. 28:553–561. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yoshino A, Tashiro S, Ogino A, Yachi K,
Ohta T, Fukushima T, Watanabe T, Katayama Y, Okamoto Y, Sano E and
Tsumoto K: Gene expression profiles predicting the response to
IFN-β and a combination of temozolomide and IFN-β in malignant
gliomas. Int J Oncol. 39:529–542. 2011.
|
|
36
|
Keane MP, Arenberg DA, Moore BB, Addison
CL and Strieter RM: CXC chemokines and angiogenesis/angiostasis.
Proc Assoc Am Physicians. 110:288–296. 1998.PubMed/NCBI
|
|
37
|
Arenberg DA, Kunkel SL, Polverini PJ,
Morris SB, Burdick MD, Glass MC, Taub DT, Iamettoni MD, Whyte RI
and Strieter RM: Interferon-γ-inducible protein 10 (IP-10) is an
angiostatic factor that inhibits human non-small cell lung cancer
(NSCLC) tumorigenesis and spontaneous metastases. J Exp Med.
184:981–982. 1996.
|
|
38
|
Sato E, Fujimoto J and Tamaya T:
Expression of interferon-gamma-inducible protein 10 related to
angiogenesis in uterine endometrial cancers. Oncology. 73:246–251.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu M, Guo S and Stiles JK: The emerging
role of CXCL10 in cancer (Review). Oncol Lett. 2:583–589.
2011.PubMed/NCBI
|
|
40
|
Yuan F, Salehi HA, Boucher Y, Vasthrae US,
Tuma RF and Jain RK: Vascular permeability and microcirculation of
gliomas and mammary carcinomas transplanted in rat and mouse
cranial windows. Cancer Res. 54:4564–4568. 1994.PubMed/NCBI
|
|
41
|
Fatterpekar GM, Galheigo D, Narayana A,
Johnson G and Knopp E: Treatment-related change versus tumor
recurrence in high-grade gliomas: a diagnostic conundrum - use of
dynamic susceptibility contrast-enhanced (DSC) perfusion MRI. AJR
Am J Roentgenol. 198:19–26. 2012. View Article : Google Scholar
|
|
42
|
Emblem KE, Mouridsen K, Bjornerud A,
Farrar CT, Jennings D, Borra RJ, Wen PY, Ivy P, Batchelor TT, Rosen
BR, Jain RK and Sorensen AG: Vessel architectural imaging
identifies cancer patient responders to anti-angiogenic therapy.
Nat Med. 19:1178–1183. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lou J, Gasche Y, Zheng L, Giroud C, Morel
P, Clements J, Ythier A and Grau GE: Interferon-β inhibits
activated leukocyte migration through human brain microvascular
endothelial cell monolayer. Lab Invest. 79:1015–1025. 1999.
|
|
44
|
Kallmann B, Hummel V, Lindenlaub T,
Ruprecht K, Toyka KV and Rieckmann P: Cytokine-induced modulation
of cellular adhesion to human cerebral endothelial cells is
mediated by soluble vascular cell adhesion molecule-1. Brain.
123:687–697. 2000. View Article : Google Scholar
|
|
45
|
Cindrova-Davies T, Sanders DA, Burton GJ
and Charnock-Jones DS: Soluble FLT1 sensitizes endothelial cells to
inflammatory cytokines by antagonizing VEGF receptor-mediated
signalling. Cardiovasc Res. 89:671–679. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Goel S, Wong AH and Jain RK: Vascular
normalization as a therapeutic strategy for malignant and
nonmalignant disease. Cold Spring Harb Perspect Med. 2:a0064862012.
View Article : Google Scholar
|
|
47
|
Boucher E, Unemori B, Seed B and Jain RK:
Relaxin increases the transport of large molecules in high collagen
content tumors. Proc Am Assoc Cancer Res. 41:abs 642000.
|