Introduction
Notch is a family of highly conserved cell-surface
receptors that are required in many mammalian developmental
processes (1). The interaction of
Notch receptors and its ligands initiates a cascade of proteolytic
cleavages. TNF-α converting enzyme, a metalloprotease, cleaves the
extracellular subunit to leave membrane-bound form of the
transmembrane subunit of Notch receptors. Then a
presenilin-associated multiprotein complex with γ-secretase
activity recognizes and cleaves a site within the transmembrane
domain to release the intracellular domain of Notch (IC-Notch).
IC-Notch translocates to the nucleus and converts the transcription
factor CBF1/Su(H)/LAG1 (CSL) from a repressor to a transcriptional
activator (2). The downstream
targets of Notch/CSL include Hairy-Enhancer of split (HES) and
Hes-related protein (HERP/HEY) families, which function as
transcriptional repressors suppressing the expression of cell type
specific target genes (3).
To date, four Notch receptors (Notch 1–4) and five
ligands (δ-like 1, 2 and 4; Jagged 1 and 2) have been described in
mammals (2). Notch1 is expressed
in most embryonic and adult tissues. Notch1 protein has an
extracellular domain consisting of multiple EGF-like repeats and
three cysteine-rich Lin12 repeats with heterodimerization (HD)
domain, a transmembrane domain and intracellular domain containing
a CSL associating motif domain, six ankyrin repeats, nuclear
localization signals (NLS), a glutamine-rich domain (OPA) and a
proline-glutamic acid-serine-threonine rich (PEST) sequence
(3). In hematopoietic system,
Notch1 signaling regulates the generation of hematopoietic stem
cells (HSC) from endothelial cells and the self-renewal of HSCs
(4,5). Notch1 signaling is also necessary and
sufficient for T-cell lineage commitment (4,6,7). In
addition, Notch1 signaling participates in later cell fate
decisions promoting CD8+ fate during the CD4/CD8 lineage
decision in T-cells (8). In
adults, excessive Notch1 signaling has been associated with
multiple human cancers (2,9).
The human Notch1 gene was identified from the
t(7;9) (q34;q34.3) chromosomal translocation detected in a subset
of T-cell acute lymphoblastic leukemia (T-ALL) (10). This translocation juxtaposes
Notch1 with the T-cell receptor β gene and leads to the
dis-regulated expression of a truncated form of Notch1, which is
constitutively active. Further studies showed that sustained Notch1
signaling is critical for proliferation and survival of T-ALL cell
lines harboring Notch1 translocations (11). Although the t(7;9) translocation is
present in <1% of human T-ALL, >50% of human T-cell
leukemia/lymphomas have mutations on the extracellular HD domain or
the C-terminal PEST domain of Notch1 (12), with both classes of mutations
resulting in excessive Notch1 signaling. Notch1 is highly expressed
in tumor cells of Hodgkin and anaplastic large cell lymphoma
(ALCL), and directly interacts with the ligand Jagged 1 to induce
proliferation and inhibit apoptosis (13). In addition, the amplification of a
region of chromosome 9q34 which contains c-abl and
Notch1 has been identified as the most frequent genetic
aberration in enteropathy-type T-cell lymphoma (14). Although aberrant Notch1 signaling
is frequently linked to the induction of T-cell
leukemias/lymphomas, the precise roles of Notch1 signaling in
hematopoietic malignancies have not been fully understood.
To study the effect of constitutively active Notch1
signaling in vivo, we established the ZEG-IC-Notch1
transgenic mouse line in which intracellular domain of Notch1
(IC-Notch1) expression can be activated by expression of
Cre-recombinase (15).
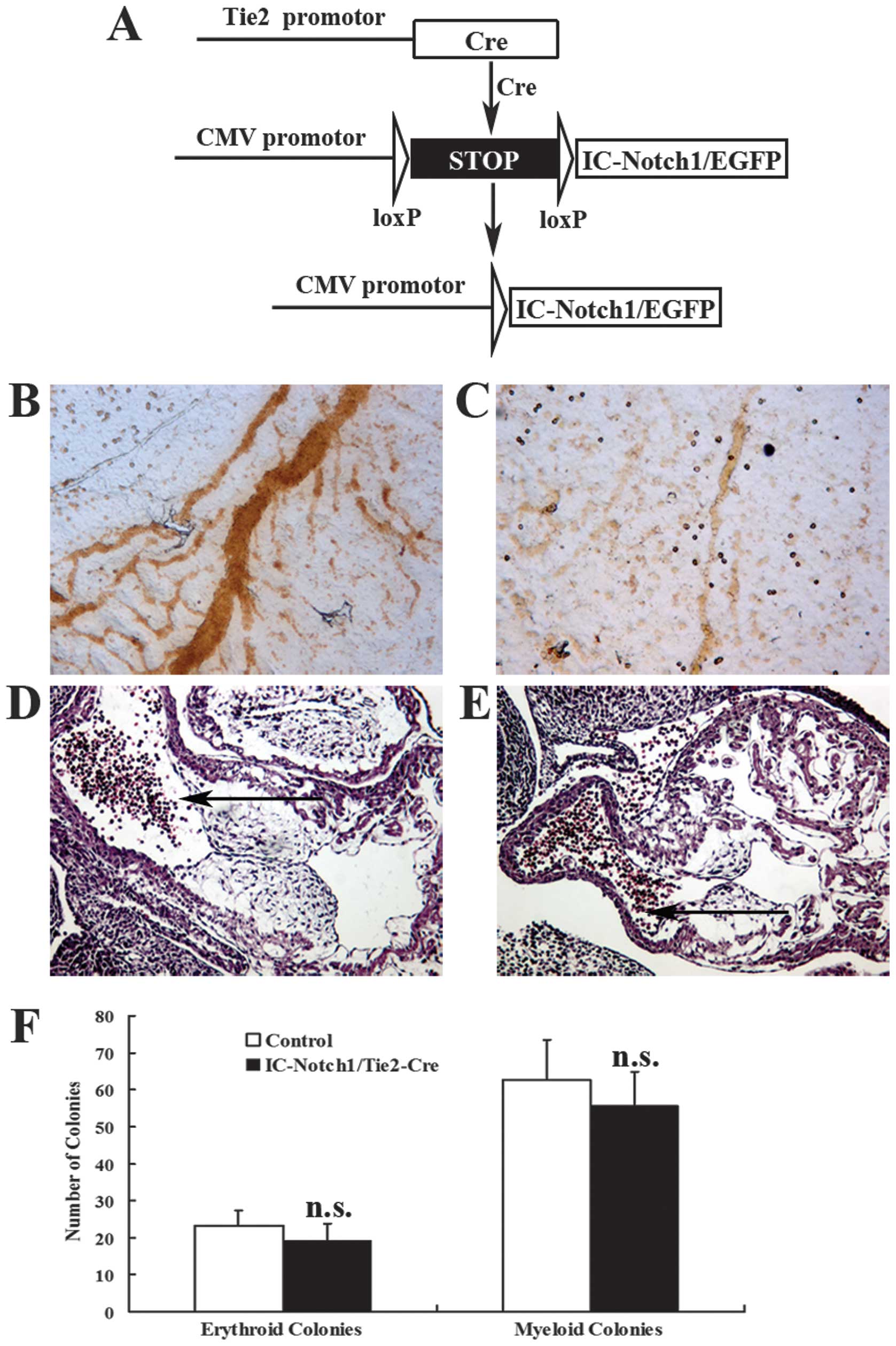
ZEG-IC-Notch1 construct contains a loxP-flanked STOP sequence
consisting of the β-geo fusion gene and three polyadenylation
sequences placed between a CMV promoter and IC-Notch1 coding
sequence with an internal ribosomal entry site (IRES) linked
enhanced green fluorescence protein (EGFP) (Fig. 1A). The efficiency of the
ZEG-IC-Notch1 mice was validated by crossing with pCX-Cre mice, a
global Cre expressing line. Ubiquitous IC-Notch1 expression leads
to embryonic lethality at E9.5 with marked growth arrest and
various developmental defects, including lack of neural tube
closure, disorganized somites, and disrupted vasculature (15).
In this study, we extend our previous findings by
generating and characterizing two additional transgenic mouse
lines: i) an ZEG-IC-Notch1/Tie2-Cre double-transgenic line in which
IC-Notch1 expression is restricted to the endothelial and
hematopoietic compartments; and ii) an inducible
ZEG-IC-Notch1/Tie2-tTA/tet-O-Cre triple-transgenic mouse in which
Cre-mediated excision is controlled by a tetracycline-inducible
system and which facilitates activation of IC-Notch1 expression
post-natally. These mouse lines provide significant insights into
the role of tissue-specific and post-natal IC-Notch1 expression in
malignant transformation.
Materials and methods
Transgenic mice
The ZEG-IC-Notch1 transgenic mice were previously
generated in our laboratory (15).
The Tie2-tTA mice express the tetracycline transactivator (tTA)
(tet repressor fused to a VP16 activator) in endothelial and
hematopoietic cells under the regulation of 2.1b Tie2
promoter (16) (provided by Dr
Urban Deutsch, Theodor-Kocher-Institute). The tet-O-Cre mice carry
the Cre coding sequence downstream of a minimal CMV promoter and
tetracycline operator (17)
(provided by Dr Andras Nagy, Samuel Lunenfeld Research Institute).
Tie2-Cre mice have multiple copies of a transgene comprised of the
Tie2 promoter, Cre cDNA, and MT-1 polyA followed by Tie2
intron 1 enhancer (18) (provided
by Dr Masashi Yanagisawa, University of Texas Southwestern Medical
Center). All mouse strains were maintained on mixed backgrounds and
genotyped as previously described in literature. Experiments
complied with ethical standards of the Sunnybrook Health Sciences
Center Research Institute Animal Care Committee.
Yolk sac hematopoietic colony assay
Intact yolk sacs were taken from the whole litter of
E8.5–9 (18–20 somites) of ZEG-IC-Notch1/Tie2-Cre mouse embryos and
washed with Iscove’s modified Dulbecco’s medium (IMDM). To obtain
single cell suspension, individual yolk sacs were incubated with
0.2 ml 0.05% Trypsin in 37°C for 3 min and suspended in 0.5 ml of
IMDM. The cell suspension were then plated in 1.5 ml of 1.0%
methylcellulose in IMDM supplemented with 15% (vol/vol) FCS, 100 μM
2-mercaptoethanol, 2 mM L-glutamine, 1% BSA, 10 μg/ml pancreatic
insulin, 200 μg/ml human transferring, 3 U/ml erythropoietin, 10
ng/ml IL-3, 10 ng/ml IL-6, and 50 ng/ml stem cell factor (MethoCult
GFM3434; StemCell Technologies) (19). Colonies were incubated in a
humidified CO2 atmosphere at 37°C and scored by
microscopy at day 10. The erythroid colonies were identified
according to positive benzidine staining (20).
Tetracycline inducible Cre system
We previously developed a tetracycline inducible Cre
system that combines the tetracycline-off and Cre/loxP
systems to gain both temporal and spatial control of transgene
expression (21). In this study,
ZEG-IC-Notch1 transgenic mice were bred with Tie2-tTA mice and
double transgenic offspring were then crossed with tet-O-Cre mice.
The breeding pairs were maintained with 0.1 mg/ml doxycycline
(tetracycline analogue; Sigma) in drinking water with 5% sucrose
(22). To maintain the efficiency
of doxycycline, the drinking water was protected from light and
replaced every 24 h. Doxycycline was withdrawn on the day the pups
were born, and genotyped to identify triple transgenic offspring
(ZEG-IC-Notch1/Tie2-tTA/tet-O-Cre).
Immunohistochemistry
Tissue preparation and immunohistochemistry were
performed as previous described (23). Briefly, tissue samples were fixed
with 4% paraformaldehyde before embeded in Tissue-Tek OCT (Sakura
Finetechnical) over dry ice. The frozen blocks were cryosectioned
at 7 μm, placed onto L-polylysine-coated slides (Thermo Fisher
Scientific), dried, and stored at −80°C. All tissue sections were
treated with 3% H2O2 in methanol for 30 min
to block endogenous peroxidase activity and incubated in a
humidified chamber in blocking buffer of 5% normal goat serum
(Vector Laboratories) for 1 h. Slides were then incubated at 4°C
overnight with the rat anti-CD90 primary antibody (1:500; BD
Pharmingen). The next day, slides were exposed for 30 min to the
biotinylated secondary antibody (1:200; Vector Laboratories).
Antibody binding was visualized via the streptavidin-horseradish
peroxidase (HRP) together with diaminobenzidine (DAB) detection
systems (Vector Laboratories). The slides were then washed,
counterstained with hematoxylin, dehydrated, and mounted for
examination. All slides were photographed using a Leica DFC300
camera with the Leica FireCam 120 program.
Flow cytometry
Single cell suspensions were prepared by
mechanically dissociating tissues in FACS buffer (PBS with
Ca2+/Mg2+, 0.1% NaN3 and 5% FCS)
and filtering through a 40 μm cell filter. Spleen samples were
treated with freshly prepared red blood cell lysis buffer (9:1 mix
of 0.83% NH4Cl and 1 M Tris-HCl pH 7.65) and
re-filtered. Cells were resuspended in FACS buffer. Fluorescence of
EGFP was detected on the FL1 channel of a FACSCalibur
(Becton-Dickinson Biosciences). Results were then analyzed using
FlowJo (TreeStar, Inc.).
Western blot analysis
The lymphoma and lymph node tissues were lysated in
ice cold RIPA buffer (20 mM Tris pH 7.5, 150 mM NaCl, 50 mM NaF, 1%
NP40, 0.1% DOC, 0.1% SDS, 1 mM EDTA and supplemented with 1 mM PMSF
and 1 μg/ml leupeptin). The protein concentration was determined
using the BCA assay (Bio-Rad). Then equal amounts of protein were
separated by a 10% SDS-PAGE and transferred onto PVDF membrane.
Membranes were blocked with 2.5% BSA, and incubated with the
primary antibodies at 4°C overnight in PBS-T. Primary antibodies
included rabbit anti-p53 antibody, rabbit anti-ARF antibody (both
from Santa Cruz Biotechnology), and mouse anti-β-actin antibody
(Sigma). Immunoreactivity was visualized with HRP-linked secondary
antibodies and chemiluminescence (Millipore). β-actin levels were
used as controls for protein loading.
Semi-quantitative PCR analysis
Total RNA isolation from embryo hearts was performed
using TRIzol reagent (Invitrogen Life Technologies) according to
the manufacturer’s protocol. An aliquot of 2 μg total RNA from each
sample was used for synthesis of cDNA using a High-Capacity cDNA
Reverse Transcription kits (Applied Biosystems). The first-strand
cDNA was amplified in a final volume of 20 μl with 1 unit of Taq
DNA polymerase (Invitrogen Life Technologies and 10 pmol of each
primer. Oligonucleotide primer sequences are listed in Table I. The thermal-cycle program was:
95°C for 5 min (1 cycle), 94°C for 1 min, 58°C for 1 min 72°C for 1
min (30 cycles), 72°C for 5 min (1 cycle). The PCR products were
visualized by ethidium bromide staining following a 1.5% agarose
gel electrophoresis.
 | Table IPCR primer sequences. |
Table I
PCR primer sequences.
| Gene | Sequence
(5′→3′) | Size (bp) | Tm (°C) |
|---|
| m-p53 | F:
GGATTTGTATCCCGAGTATCTG | 183 | 57.21 |
| R:
GTCTCAGTGTGATGATGGTA | | 58.17 |
| m-ARF | F:
GCGCACGATCCT | 66 | 59.36 |
| R:
TTGAGCAGAGAGCTGCTACGT | | 61.98 |
| β-actin | F:
GGCACACACTCTACAATG | 352 | 59.19 |
| R:
GTGTGTGAGCTGTAGCC | | 60.96 |
Statistical analysis
Data are expressed as mean ± standard error (SE).
The significance of differences was estimated by one-way ANOVA. All
statistical analyses were performed with SPSS software (SPSS,
Inc.).
Results
ZEG-IC-Notch1/Tie2-Cre embryos was lethal
at mid-gestation without significant alterations in hematopoietic
system
The Tie2 promoter directs expression
specifically in endothelial cells and hematopoietic cells (18). The ZEG-IC-Notch1 mice were crossed
with Tie2-Cre mice to activate IC-Notch1 expression in these two
cell types (Fig. 1A). No
ZEG-IC-Notch1/Tie2-Cre positive offspring were obtained, and we
found that the double transgenic embryos died before E10.5 with
defects in vascular development and widespread hemorrhaging. These
observations were consistent with our previous studies showing the
lethality of ZEG-IC-Notch1/Tie2-Cre embryos with lack of angiogenic
remodeling and branching in the yolk sacs and embryonic bodies
(24,25). In these IC-Notch1 expressing
embryos, the circulation system of hematopoietic cells have been
developed before the death of the embryos. On the yolk sac,
hematopoietic cells are still present in the disrupted vessels
(Fig. 1C). Histological analysis
revealed the presence of hematopoietic cells within the lumen of
dorsal aorta and cardinal vein (Fig.
1E). We observed a fraction of hematopoietic cells expressing
second reporter EGFP in the double transgenic embryos at E9.5,
suggesting Cre excision under Tie2 promoter is effective in
hematopoietic cells at this stage (18).
Notch signaling involves in erythroid vs. myeloid
lineage decision by inhibiting erythroid differentiation (26,27),
thus we measured the extent of primitive hematopoiesis by in
vitro colony assay of yolk-sac cells from E9.5 embryos
(Table II). The average number of
erythroid, myeloid and total colonies derived from
ZEG-IC-Notch1/Tie2-Cre yolk sacs were comparable to the control
(Fig. 1F; p=0.17, p=0.31, p=0.38,
respectively). The morphology of the colonies did not show obvious
abnormalities. These data suggested that primitive hematopoiesis
was not significantly affected in the ZEG-IC-Notch1/Tie2-Cre
embryos at mid-gestation and the embryonic lethal phenotype
resulted from disruption of the vasculature.
| Table IINumber of hematopoietic colonies
generated from yolk sacs in methylcellulose colony forming
assays. |
Table II
Number of hematopoietic colonies
generated from yolk sacs in methylcellulose colony forming
assays.
| Mouse Genotype | Erythroid
Colonies | Myeloid
Colonies | Total Colonies |
|---|
| Control | 23.4±4 | 62.6±10.8 | 86.5±12.3 |
|
ZEG-IC-Notch1/Tie2-Cre | 19.2±4.5a | 55.8±9.1b | 74.4±10.4c |
IC-Notch1 expression was activated in
hematopoietic cells postnatally by using tetracycline regulated Cre
system
To activate IC-Notch1 in adults, ZEG-IC-Notch1 were
bred with Tie2-tTA/Tet-O-Cre mice and maintained on tetracycline.
The Tie2 promoter drives tTA expression in endothelial cells
and hematopoietic cells but the tTA protein is bound and disabled
by tetracycline, which prevents the expression of Cre recombinase.
After the pups were born, tetracycline was withdrawn to release the
tTA protein, which activates the tet-operator and induces Cre
expression. Cre recombinase excises the STOP sequence of the
ZEG-IC-Notch1 transgene, leading to the expression of IC-Notch1 and
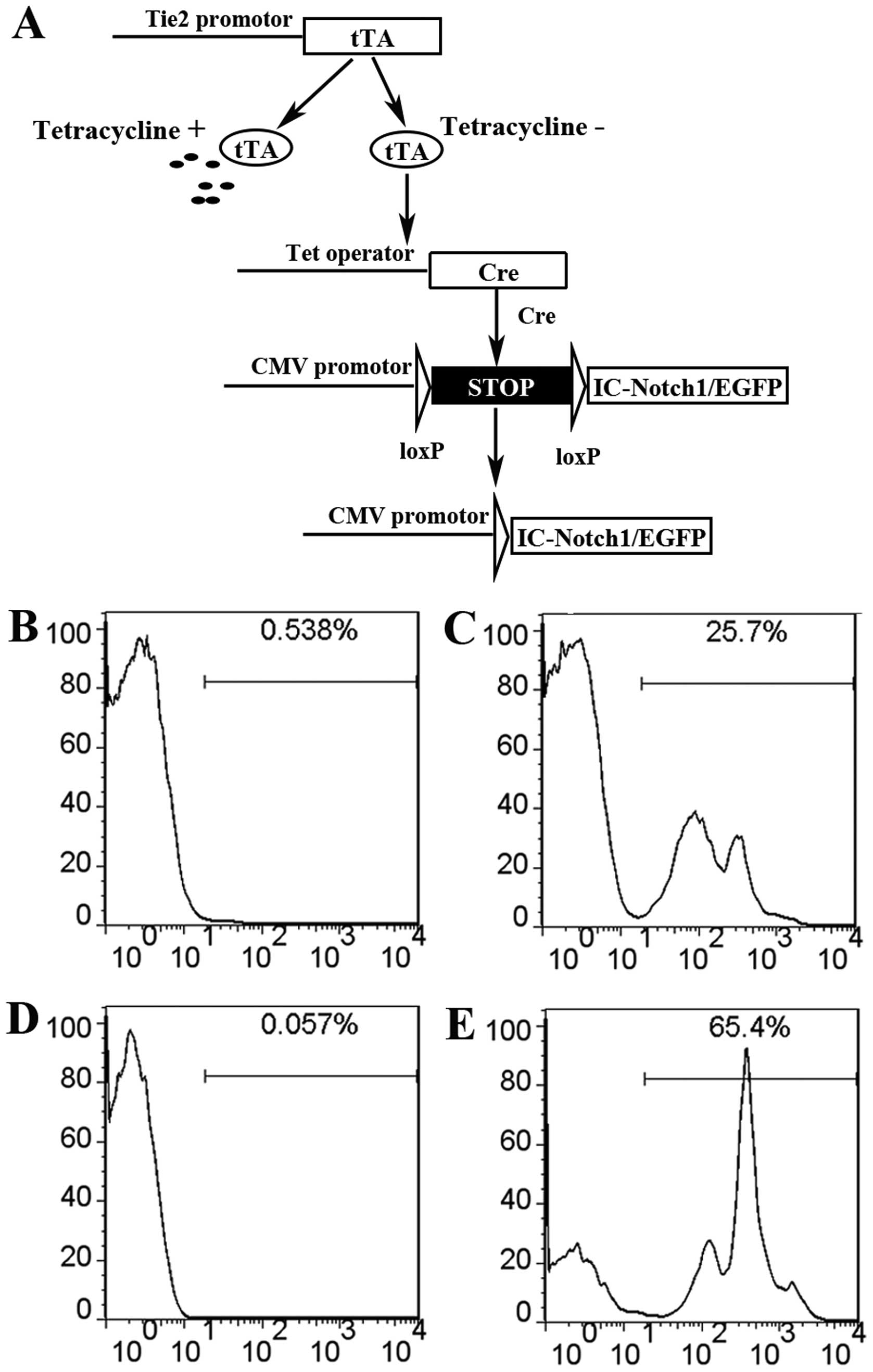
EGFP reporter driven by the CMV promoter (Fig. 2A). In this configuration the Cre
excision makes permanent genomic alterations to cells and their
progeny. We obtained a normal Mendelian ratio of live-born triple
transgenic ZEG-IC-Notch1/Tie2-tTA/tet-O-Cre pups, and these triple
transgenic mice continued to survive after removal of doxycycline.
To determine transgene activation in these triple transgenic mice,
the reporter EGFP expression in the hematopoietic organs were
examined by flow cytometry. We found that all the triple transgenic
mice have EGFP positive cells in thymus and spleen (Fig. 2B–E). However, the expression level
varies substantially from mouse to mouse. In thymus, the proportion
of GFP positive cells ranges from 17–55%. In spleen, the proportion
of GFP positive cells ranges from 12–70%. Mosaic expression was
also observed in endothelial cells of the triple transgenic mice
(25).
Postnatal IC-Notch1 activation induced
abnormalities in hematopoietic system
The ZEG-IC-Notch1/Tie2-tTA/tet-O-Cre triple
transgenic mice displayed no overt phenotype and no obvious
abnormalities in the organs examined macroscopically. Since Notch
signaling regulates T lymphocyte development, we examined
CD90/Thy1.2 antigen, which is expressed at high levels on
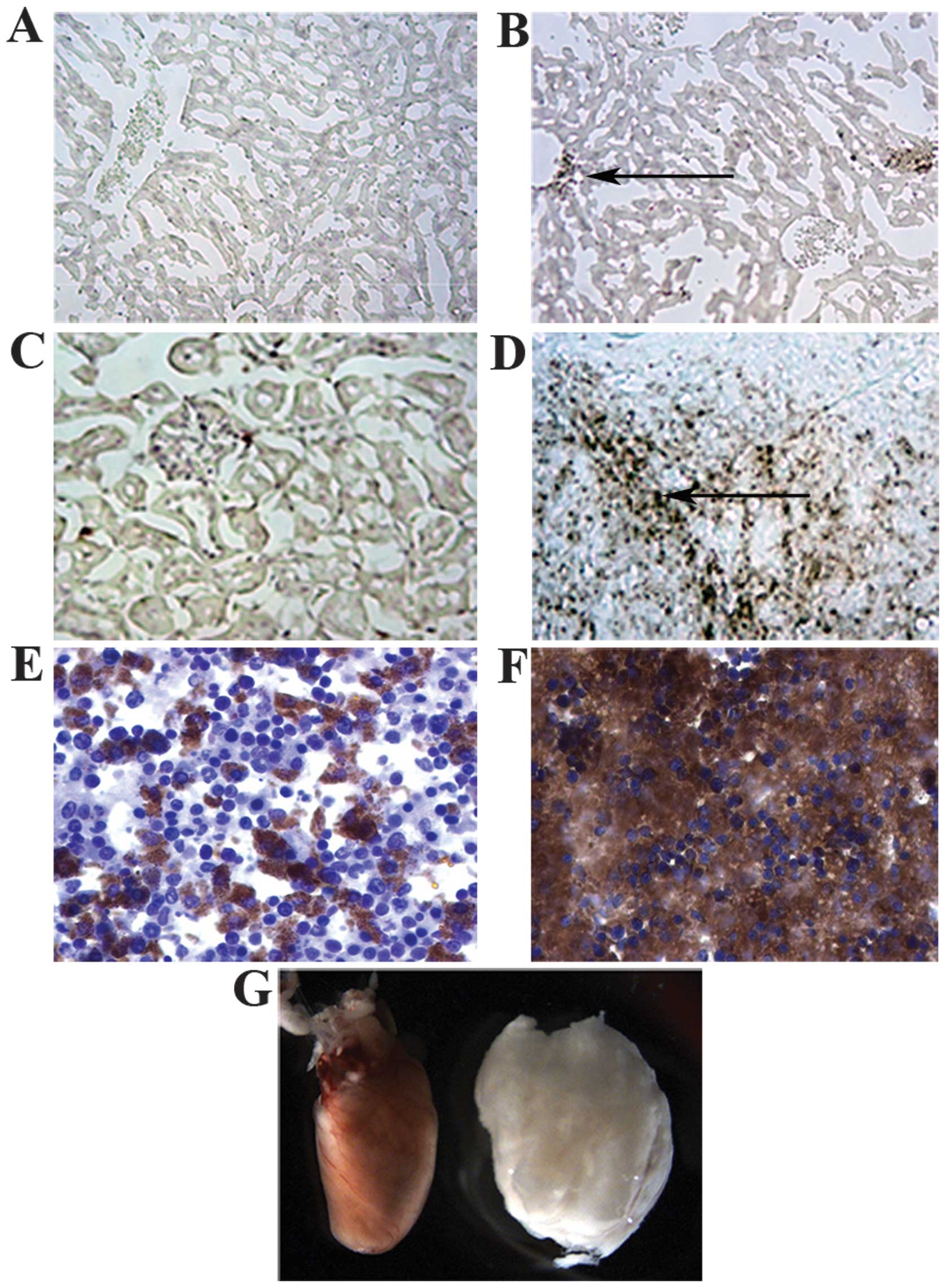
peripheral T-cells, in organs from IC-Notch1 expressing triple
transgenic mice. The CD90 positive cells were detected in the lymph
nodes, thymus and spleen of all the triple transgenic mice
examined. The liver and kidney, where T-cells do not normally
present, showed clusters of CD90 positive cells (Fig. 3A–D). Most CD90 positive cells were
adjacent to, but not limited to, small blood vessels. Furthermore,
33.3% of triple transgenic mice displayed neoplastic mass with many
T-cells in the kidneys at the age of 6 months. A group of IC-Notch1
expressing triple transgenic mice, their littermate controls and
ZEG/Tie2-tTA/tet-O-Cre control transgenic mice were maintained to
the age of 12 months and monitored for signs of disease.
Surprisingly, 88.89% (8/9) of IC-Notch1 expressing triple
transgenic mice displayed lymphoma-like neoplasm (Table III). The lymph nodes from the
control mice were invisible, but most of the lymphoma-like neoplasm
of triple transgenic mice showed a diameter of 0.4–0.8 cm and some
of them were even larger than the heart (Fig. 3G). The majority of lymphoma-like
neoplasms were superior mesenteric nodes, while a few of them
located at chest and posterior abdomen. Immunostaining showed that
most cells in the lymphoma-like neoplasms from IC-Notch1 expressing
transgenic mice were stained positive for CD90 while only a
fraction of cells in normal lymph node were CD90 positive (Fig. 3E and F). In addition, the
architecture of the lymphoma-like neoplasm was destroyed, and
replaced with a mass of fat blast T-cells and apoptotic cells.
Taken together, the IC-Notch1 expressing triple transgenic mice
developed lymphoma-like T-cell malignancies.
| Table IIIIncidence of LLN in 12-month old
mice. |
Table III
Incidence of LLN in 12-month old
mice.
| Mouse Genotype | No. of mice
examined | No. of mice with
LLN | Incidence rate
% |
|---|
| Control | 17 | 1 | 5.9 |
|
ZEG/Tie2-tTA/tet-O-Cre | 11 | 1 | 9.1 |
| ZEG-IC-Notch1/ | 9 | 8 | 88.9a |
|
Tie2-tTA/tet-O-Cre | | | |
p53 activity was suppressed in
lymphoma-like neoplasms from IC-Notch1 expressing triple transgenic
mice
The tumor suppressor protein p53 plays a critical
role in maintaining cellular homeostasis in response to various
cellular stresses (28–30). Stability of p53 protein is
negatively regulated by the ubiquitin ligase mdm2 (30). The alternative reading frame (ARF)
protein, which is a tumor suppressor transcribed from an alternate
reading frame of the INK4a/ARF locus, inhibits the activity of mdm2
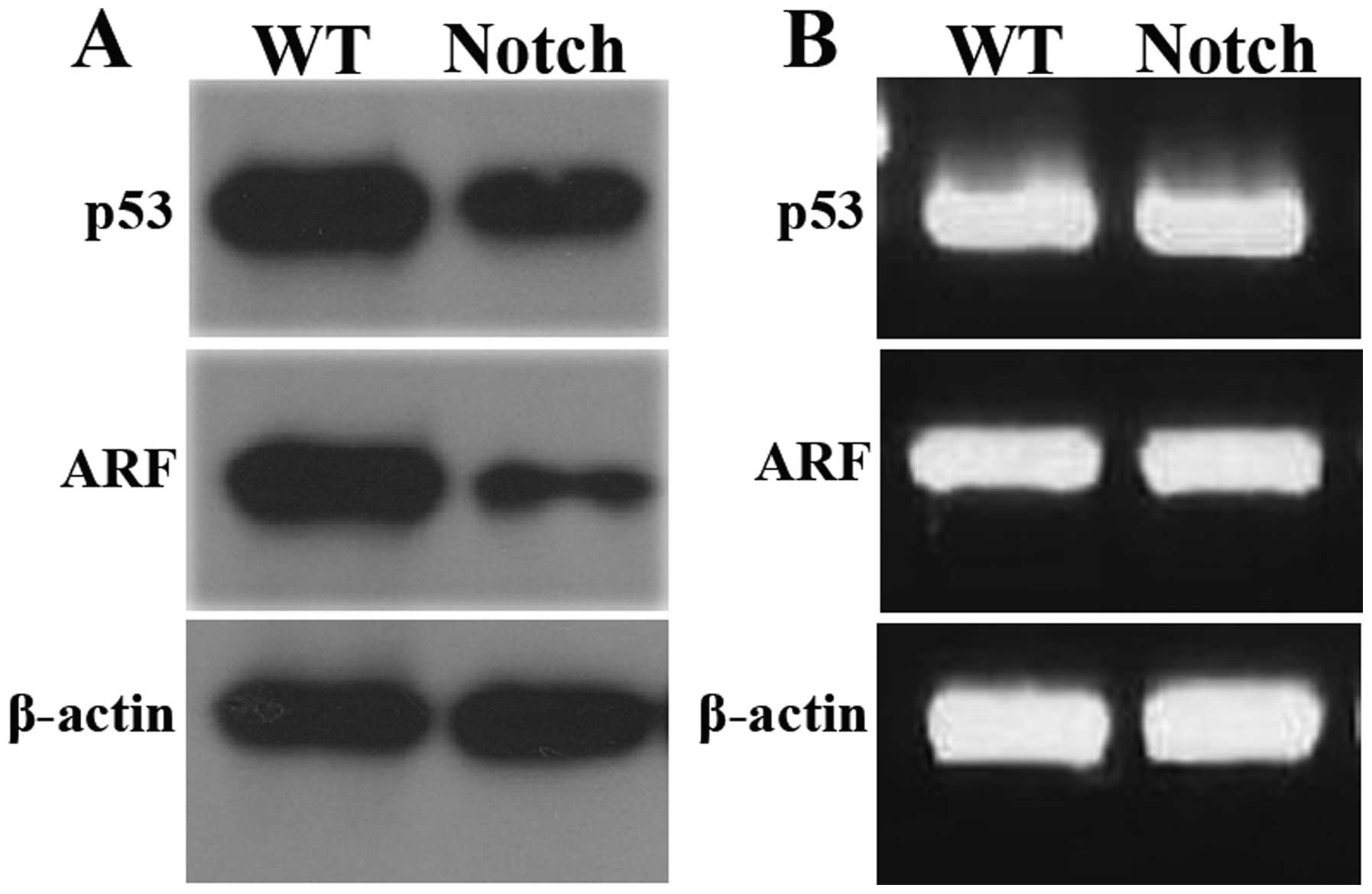
and activates p53 (31,32). We performed western blot analysis
using protein lysates from the lymphoma-like neoplasm of IC-Notch1
expressing triple transgenic mice, and the lymph nodes from WT mice
were used as control. As shown in Fig.
4A, the protein of p53 and ARF was significantly decreased in
lymphoma-like neoplasm. In addition, we examined the mRNA
expression of p53 and ARF by semi-quantitative PCR,
and no significant changes was found between the lymphoma-like
neoplasms and normal lymph nodes (Fig.
4B). These data suggested a post-transcriptional downregulation
of p53 and ARF in the lymphoma-like neoplasm of IC-Notch1
expressing triple transgenic mice.
Discussion
Notch signaling pathway regulates a spectrum of cell
fate decisions and differentiation processes of hematopoietic
system during fetal and postnatal development (1). The role of Notch signaling during
lymphopoiesis and T-cell neoplasia have been studied using
gain-of-function and conditional loss-of-function animal models for
the Notch1 ligands and receptors as well as their downstream
targets (33). However,
hematopoietic system is a developmental system (34,35),
and alteration of Notch signaling at different stages of
development may result in distinct types of diseases or prognosis.
Using conditional and inducible mouse model system, we were able to
examine the effects of constitutively active Notch1 signaling in
the hematopoietic system at early embryogenesis and adulthood. At
E9.5, IC-Notch1 activation does not significantly affect the
hematopoietic system. However, postnatal activation of IC-Notch1
induces T-cell malignancies, which may be mediated by
post-transcriptional downregulation of p53 and ARF. These results
contribute to the understanding of the role of Notch1 signaling in
leukemiagenesis/lymphomagenesis in adults.
The homogenous deletion of Notch1 or
constitutive activation of Notch1 under Tie2 promoter
in mice all induced early embryonic lethality between 9.5–11.5 with
a phenotype of disruption of vasculature (24,36).
Since Notch1 and Tie2 promoter are also expressed in
hematopoietic cells, defects in hematopoietic system might
contribute to the phenotype. In this study, we demonstrated that
circulation of hematopoietic cells and erythroid/myeloid cell fate
decision is not significantly affected by IC-Notch1 activation even
though the embryos are dying. The loss of function study in which
Tie2-Cre were crossed with floxed Notch1 mice also displayed that
hematopoietic systems are largely normal while it produced a
similar vascular defect phenotype as the Notch1 homogeneous
knockout embryos (37). These
results might partially be due to mosaic expression of Tie2-Cre
transgene as only a fraction of hematopoietic cells have the Cre
excision at this stage, however, hematopoietic development was not
inhibited in the embryos with global IC-Notch1 activation (15). In addition, another gain of
function study showed that expressing intracellular domain of
Notch4 at flk1 locus results in embryonic lethality at E9.5
without disturbing the hematopoietic development (19). Though aberrant Notch signaling
induces vascular defects and embryonic lethal phenotype, it may not
significantly affect the development of hematopoietic system before
mid-gestation.
By combination of Cre/loxP and tetracycline
inducible system, we successfully activated constitutive Notch1
signaling in hematopoietic cells of adult mice and observed T-cell
malignancy in the IC-Notch1 expressing triple transgenic mice.
Notch signaling is critical for normal T-cell development and
excessive Notch1 signaling is associated with T-cell leukemia
(38). In addition, high levels of
Notch1 protein expression were found in twelve primary human T-cell
ALCL samples as compared to B-cell lymphomas, and high levels of
cleaved Notch1 were seen in two human ALCL-derived cell lines
(13). Activation of Notch1
signaling may block T-cell differentiation, inhibit apoptosis and
promote proliferation (33).
Though the time point of gene alteration in hematopoietic cells is
important for development of malignancies, in vivo studies
that activate Notch signaling in adults have been limited. From
bone marrow transplant reconstitution models in which retrovirally
transduced HSC were transferred to lethally irradiated recipients,
constitutive expression of human IC-Notch1 led exclusively to
CD8+CD24+ or CD4+CD8+
T-cell leukemia/lymphomas (39).
Thus, postnatal activation of Notch1 signaling hematopoietic cells
may initiate and promote T-cell malignancies in normal adults.
In this study, we activated the entire Notch1
intracellular domain containing OPA and PEST sequence, which were
deleted in many commonly-used Notch1 transgenic mouse models
(8). The importance of the OPA
domain for transformation has been demonstrated by IC-Notch1
transgenic constructs showing the difference in sequence between
the fully leukemogenic with OPA and weakly leukemogenic constructs
without OPA (40). Removal of the
PEST sequences leads to decreased degradation of Notch1 protein and
subsequently increased Notch signaling (41). Other studies suggest that Numb, a
negative Notch regulator, binds PEST sequences and inhibits Notch
signaling (42,43). However, the presence of PEST
sequence is not essential for blocking IC-Notch1 induced
leukemiagenesis (44). Here we
demonstrated that activation of IC-Notch1 with PEST sequence still
induces T-cell malignancy.
ARF-mdm2-p53 pathway is an important tumor
suppressing mechanism that is activated in response to
inappropriately sustained proliferative signals (32,45).
Mutation of these genes leads to inactivation of p53 function and
development of lymphoma (46).
Attenuation of Notch1 expression causes a remarkably increase in
p53 protein levels and initiation of apoptotic programs in tumor
cells (47). Activation of Notch
signaling pathway reduces p53 protein through mdm2-dependent p53
degradation (47,48). In this study, we found a decrease
of p53 protein level but not the mRNA expression in lymphoma-like
neoplasms from IC-Notch1 expressing mice, implicating the
possibility of p53 protein degradation. The ARF tumor suppressor
binds to and inhibits the activity of mdm2, leading to p53
activation (49). In the tet-on
IC-Notch1 mouse model, Notch-induced lymphomas do not express ARF
protein (47). Though we observed
ARF protein expression in the lymphoma-like neoplasms from our
IC-Notch1 mouse model, the expression level is dramatically
decreased. In addition, the mRNA level of ARF remains unchanged,
suggesting that IC-Notch1 activation might repress ARF protein by a
post-transcriptional mechanism.
In summary, we activated IC-Notch1 in hematopoietic
cells under temporal control in transgenic mouse models using
tetracycline-induced Cre system. Our data suggested a new
perspective for understanding pathogenesis of T-cell malignancies
in adults. Modification of the time point of IC-Notch1 activation
in this model allows us to investigate the relationship between
Notch1 signaling and a range of hematopoietic diseases.
Acknowledgements
This study was supported by a grant from the Heart
and Stroke Foundation of Canada. We are grateful for the support
from Shandong Taishan Scholarship (to Ju Liu).
References
|
1
|
Artavanis-Tsakonas S, Rand MD and Lake RJ:
Notch signaling: cell fate control and signal integration in
development. Science. 284:770–776. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hansson EM, Lendahl U and Chapman G: Notch
signaling in development and disease. Semin Cancer Biol.
14:320–328. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baron M: An overview of the Notch
signalling pathway. Semin Cell Dev Biol. 14:113–119. 2003.
View Article : Google Scholar
|
|
4
|
Radtke F, Wilson A, Stark G, et al:
Deficient T cell fate specification in mice with an induced
inactivation of Notch1. Immunity. 10:547–558. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kumano K, Chiba S, Kunisato A, et al:
Notch1 but not Notch2 is essential for generating hematopoietic
stem cells from endothelial cells. Immunity. 18:699–711. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Milner LA and Bigas A: Notch as a mediator
of cell fate determination in hematopoiesis: evidence and
speculation. Blood. 93:2431–2448. 1999.PubMed/NCBI
|
|
7
|
Schmitt TM and Zúñiga-Pflücker JC:
Induction of T cell development from hematopoietic progenitor cells
by delta-like-1 in vitro. Immunity. 17:749–756. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Robey E, Chang D, Itano A, et al: An
activated form of Notch influences the choice between CD4 and CD8 T
cell lineages. Cell. 87:483–492. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Politi K, Feirt N and Kitajewski J: Notch
in mammary gland development and breast cancer. Semin Cancer Biol.
14:341–347. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ellisen LW, Bird J, West DC, et al: TAN-1,
the human homolog of the Drosophila notch gene, is broken by
chromosomal translocations in T lymphoblastic neoplasms. Cell.
66:649–661. 1991.PubMed/NCBI
|
|
11
|
Weng AP, Nam Y, Wolfe MS, et al: Growth
suppression of pre-T acute lymphoblastic leukemia cells by
inhibition of notch signaling. Mol Cell Biol. 23:655–664. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Weng AP and Aster JC: Multiple niches for
Notch in cancer: context is everything. Curr Opin Genet Dev.
14:48–54. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jundt F, Anagnostopoulos I, Förster R,
Mathas S, Stein H and Dörken B: Activated Notch1 signaling promotes
tumor cell proliferation and survival in Hodgkin and anaplastic
large cell lymphoma. Blood. 99:3398–3403. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baumgärtner AK, Zettl A, Chott A, Ott G,
Müller-Hermelink HK and Starostik P: High frequency of genetic
aberrations in enteropathy-type T-cell lymphoma. Lab Invest.
83:1509–1516. 2003.PubMed/NCBI
|
|
15
|
Liu J and Lobe CG: Cre-conditional
expression of constitutively active Notch1 in transgenic mice.
Genesis. 45:259–265. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Holopainen T, Saharinen P, D’Amico G, et
al: Effects of angiopoietin-2-blocking antibody on endothelial
cell-cell junctions and lung metastasis. J Natl Cancer Inst.
104:461–475. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen L, Meng Q, Kao W and Xia Y: IκB
kinase β regulates epithelium migration during corneal wound
healing. PloS One. 6:e161322011.
|
|
18
|
Kisanuki YY, Hammer RE, Miyazaki J,
Williams SC, Richardson JA and Yanagisawa M: Tie2-Cre transgenic
mice: a new model for endothelial cell-lineage analysis in vivo.
Dev Biol. 230:230–242. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Uyttendaele H, Ho J, Rossant J and
Kitajewski J: Vascular patterning defects associated with
expression of activated Notch4 in embryonic endothelium. Proc Natl
Acad Sci USA. 98:5643–5648. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Puri MC and Bernstein A: Requirement for
the TIE family of receptor tyrosine kinases in adult but not fetal
hematopoiesis. Proc Natl Acad Sci USA. 100:12753–12758. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu J, Deutsch U, Fung I and Lobe C:
Conditional and inducible transgene expression in endothelial and
hematopoietic cells using Cre/loxP and tetracycline-off systems.
Exp Ther Med. (In press).
|
|
22
|
Sarao R and Dumont DJ: Conditional
transgene expression in endothelial cells. Transgenic Res.
7:421–427. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu J, Yuan L, Molema G, et al: Vascular
bed-specific regulation of the von Willebrand factor promoter in
the heart and skeletal muscle. Blood. 117:342–351. 2011. View Article : Google Scholar
|
|
24
|
Liu J, Dong F, Jeong J, Masuda T and Lobe
CG: Constitutively active Notch1 signaling promotes
endothelial-mesenchymal transition in a conditional transgenic
mouse model. Int J Mol Med. 34:669–676. 2014.
|
|
25
|
Liu J, Deutsch U, Jeong J and Lobe CG:
Constitutive notch signaling in adult transgenic mice inhibits
bFGF-induced angiogenesis and blocks ovarian follicle development.
Genesis. May 10–2014.(Epub ahead of print).
|
|
26
|
Schroeder T and Just U: Notch signalling
via RBP-J promotes myeloid differentiation. EMBO J. 19:2558–2568.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lam LT, Ronchini C, Norton J, Capobianco
AJ and Bresnick EH: Suppression of erythroid but not megakaryocytic
differentiation of human K562 erythroleukemic cells by notch-1. J
Biol Chem. 275:19676–19684. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dai C and Gu W: p53 post-translational
modification: deregulated in tumorigenesis. Trends Mol Med.
16:528–536. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bode AM and Dong Z: Post-translational
modification of p53 in tumorigenesis. Nat Rev Cancer. 4:793–805.
2004. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kubbutat MH, Jones SN and Vousden KH:
Regulation of p53 stability by Mdm2. Nature. 387:299–303. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang Y, Xiong Y and Yarbrough WG: ARF
promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus
deletion impairs both the Rb and p53 tumor suppression pathways.
Cell. 92:725–734. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kamijo T, Weber JD, Zambetti G, Zindy F,
Roussel MF and Sherr CJ: Functional and physical interactions of
the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci USA.
95:8292–8297. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Radtke F, Wilson A, Mancini SJ and
MacDonald HR: Notch regulation of lymphocyte development and
function. Nat Immunol. 5:247–253. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Steinman RA: Cell cycle regulators and
hematopoiesis. Oncogene. 21:3403–3413. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Smith C: Hematopoietic stem cells and
hematopoiesis. Cancer Control. 10:9–16. 2003.PubMed/NCBI
|
|
36
|
Krebs LT, Xue Y, Norton CR, et al: Notch
signaling is essential for vascular morphogenesis in mice. Genes
Dev. 14:1343–1352. 2000.PubMed/NCBI
|
|
37
|
Limbourg FP, Takeshita K, Radtke F,
Bronson RT, Chin MT and Liao JK: Essential role of endothelial
Notch1 in angiogenesis. Circulation. 111:1826–1832. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zweidler-McKay PA and Pear WS: Notch and T
cell malignancy. Semin Cancer Biol. 14:329–340. 2004. View Article : Google Scholar
|
|
39
|
Pear WS, Aster JC, Scott ML, et al:
Exclusive development of T cell neoplasms in mice transplanted with
bone marrow expressing activated Notch alleles. J Exp Med.
183:2283–2291. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Washburn T, Schweighoffer E, Gridley T, et
al: Notch activity influences the alphabeta versus gammadelta T
cell lineage decision. Cell. 88:833–843. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Feldman BJ, Hampton T and Cleary ML: A
carboxy-terminal deletion mutant of Notch1 accelerates lymphoid
oncogenesis in E2A-PBX1 transgenic mice. Blood. 96:1906–1913.
2000.PubMed/NCBI
|
|
42
|
Gupta-Rossi N, Le Bail O, Gonen H, et al:
Functional interaction between SEL-10, an F-box protein, and the
nuclear form of activated Notch1 receptor. J Biol Chem.
276:34371–34378. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Oberg C, Li J, Pauley A, Wolf E, Gurney M
and Lendahl U: The Notch intracellular domain is ubiquitinated and
negatively regulated by the mammalian Sel-10 homolog. J Biol Chem.
276:35847–35853. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Campese AF, Bellavia D, Gulino A and
Screpanti I: Notch signalling at the crossroads of T cell
development and leukemogenesis. Semin Cell Dev Biol. 14:151–157.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sherr CJ: The INK4a/ARF network in tumour
suppression. Nat Rev Mol Cell Biol. 2:731–737. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Eischen CM, Weber JD, Roussel MF, Sherr CJ
and Cleveland JL: Disruption of the ARF-Mdm2-p53 tumor suppressor
pathway in Myc-induced lymphomagenesis. Genes Dev. 13:2658–2669.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Beverly LJ, Felsher DW and Capobianco AJ:
Suppression of p53 by Notch in lymphomagenesis: implications for
initiation and regression. Cancer Res. 65:7159–7168. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nair P, Somasundaram K and Krishna S:
Activated Notch1 inhibits p53-induced apoptosis and sustains
transformation by human papillomavirus type 16 E6 and E7 oncogenes
through a PI3K-PKB/Akt-dependent pathway. J Virol. 77:7106–7112.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Reed SM, Hagen J, Tompkins VS, Thies K,
Quelle FW and Quelle DE: Nuclear interactor of ARF and Mdm2
regulates multiple pathways to activate p53. Cell Cycle.
13:1288–1298. 2014. View Article : Google Scholar : PubMed/NCBI
|