Introduction
Circadian rhythms are important mechanisms
regulating the daily oscillation of several biological processes
(1,2). These mechanisms provide the organisms
with survival advantages by organizing their behavior and
physiological adaptation to the cyclic changes in the environment
(3,4). Disruption of these rhythms may have
profound influence on human health (5–7) and
has been known as a risk factor in the development of human cancer
(7,8). Studies also found that the
proliferation of tumor cells followed the autonomous circadian
patterns that are out of phase from non-tumor cells (9–12).
The evidence suggested that the circadian clock may suppress tumor
formation at the systemic, cellular and molecular levels (13–15).
ARNTL (also known as BMAL1), an HLH-containing
transcription factor, is an important player in the control of
circadian rhythms (15). ARNTL
together with CLOCK regulates the transcription of several E-box
motif containing genes such as c-myc (16,17).
Importantly, expression of ARNTL has been demonstrated to be
downregulated in several human cancer including ovarian, head and
neck and in blood (18–20). Notably, ARNTL was also found to be
epigenetically silenced in leukemia. Re-expression of ARNTL in
leukemia cells inhibited tumor growth and restored the expression
pattern of circadian genes thus suggesting that it may be a tumor
suppressor.
Epigenetic modifications, including DNA methylation
play an important role in gene expression and cell fate commitment
(21,22). Previous reports including ours have
shown that tumor suppressor genes can be epigenetically silenced in
ovarian cancer (23–29). In order to identify genes that are
suppressed by promoter hypermethylation in ovarian cancer, we
performed meDIP-chip using Agilent 244K CpG island microarray in a
panel of ovarian cancer cell lines. Our result showed that
ARNTL is epigenetically silenced by promoter methylation and
histone modifications in a sub-set of ovarian cancer cells.
Restoration of ARNTL suppressed cancer cell growth and
restored the expression pattern of c-MYC thus suggesting
that it may be a tumor suppressor in ovarian cancer.
Materials and methods
Cell culture and epigenetic
treatment
Immortalized ovarian surface epithelia cells (IOSE)
(30) were maintained in a 1:1
mixture of medium 199 (Sigma, St. Louis, MO, USA) and 105 (Sigma)
supplemented with 10% fetal bovine serum (FBS; Gibco, Life
Technologies, Grand Island, NY, USA), 400 ng/ml hydrocortisone
(Sigma), 10 ng/ml EGF and 50 U/ml of penicillin/streptomycin (P/S;
Gibco). HeyC2 was maintained in Dulbecco’s modified Eagle’s medium
(DMEM; Gibco) supplemented with 10% FBS and 50 U/ml of P/S, 1X MEM
NEAA (Gibco) and 0.01 M of HEPES (Gibco). A2780 and its
cisplatin-resistant sublines CP70, MCP2 and MCP3 cells were
propagated in RPMI-1640 (Gibco) supplemented with 10% FBS and 50
U/ml of P/S. For demethylation treatment, cells were plated in a
60-mm plate and treated with 0.5 μM 5′-aza-2′-deoxycytidine
(5-aza-dC; Sigma) for 3 days. For treatment with the EZH2 inhibitor
(GSK126; Cayman Chemical, Ann Arbor, MI, USA), cells were either
treated with 10 μM GSK126 alone for 3 days or combined with
5-aza-dC for 3 days. In the control experiment, cells were treated
with DMSO. Culture medium and drugs were replaced every 24 h. Cells
were then harvested for RNA analysis.
Extraction of RNA and quantitative
RT-PCR
RNA extraction was performed using TRizol reagent
(Life Technologies) according to the manufacturer’s protocol. To
remove potential contaminating DNA from the complementary DNA, 1 μg
of total RNA was treated with DNase I (Amplification Grade;
Invitrogen) prior to reverse transcription. First strand cDNA
synthesis used MMLV High Performance Reverse Transcriptase
(Epicentre, Chicago, IL, USA). The expression of ARNTL in
ovarian cancer cell line was examined by RT-PCR analysis. Ten-fold
diluted cDNA (4 μl) was amplified in a total volume of 20 μl
containing 2X SYBR® Green Realtime PCR Master Mix
(Toyobo, Osaka, Japan), 0.2 μM of each primer (Table I). Samples were first denaturated
at 95°C for 10 min, followed by 40 cycles of 95°C for 15 sec, at
annealing temperature for 30 sec, and extension at 72°C for 30 sec,
followed by a melting curve. The relative gene expression level was
determined by comparing the threshold cycle of the test gene
against the Ct of GAPDH in a given sample. The qPCR
reactions were carried out using ABI StepOne™ Real-Time PCR systems
(Applied Biosystems, Foster city, CA, USA).
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Gene | Primer sequence
(5′-3′) | Annealing
temperature (°C) | Product size
(bp) |
|---|
| RT-PCR | | | |
| ARNTL RT-F1 |
CTGGAGCACGACGTTCTTTCTT | 60 | 123 |
| ARNTL RT-R1 |
GGATTGTGCAGAAGCTTTTTCG | | |
| COBRA-PCR | | | |
| ARNTL BSP F |
GGTGTTGTAGGAGTTTTTAAAGGGG | 60 | 342 |
| ARNTL BSP R |
ACRAACTTAAACTCCCCAAACCC | | |
| ARNTL cDNA
construct | | | |
| ARNTL F |
AGGGCTAGCGCCCACTAGGAGATGCTATGATTAAT | 60 | 1791 |
| ARNTL R |
AAGGGATCCGTAGTGTTTACAGCGGCCATG | | |
| ARNTL ChIP PCR | | | |
| ARNTL ChIP F |
TGGAAGGAAATGCAATGGAATC | 60 | 130 |
| ARNTL ChIP R |
CCCGAGGACTGCAAGTGTTC | | |
Protein extraction and western blot
analysis
Before protein extraction, 1×106 cells
were seeded into 100-mm dish. When the cells reached 90%
confluence, the medium was removed and the dish was washed with 1X
PBS at 4°C. The cells were then lysed with 100 μl of PRO-PREP
protein extraction solution (iNtRON Biotechnology, Seongnam, Korea)
according to the manufacturer’s protocol. Samples and pre-stained
marker were loaded to a 12.5% polyacrylamide gel for
electrophoresis. The proteins were then electrophoretically
transferred from the gel onto a PVDF membrane by the Mini
Trans-Blot® Electrophoretic Transfer Cell system
(Bio-Rad Laboratories, Hercules, CA, USA) at 400 mA for ~90 min.
After the transfer, the membrane was incubated with 5% non-fat milk
in 1X TBST for 1 h at room temperature. Then the first antibodys,
ARNTL antibody (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) or
actin antibody (Santa Cruz Biotechnology) was diluted with 5%
non-fat milk in 1X TBST and was added into the membrane followed by
incubation for overnight at 4°C. The membrane was then washed in 1X
TBST at room temperature 3 times. The secondary antibody conjugated
with horseradish peroxidase (IgG-HRP antibody) was also diluted
with 5% non-fat milk in 1X TBST and incubated with membrane at room
temperature for 1 h. The membrane was washed with 1X TBST at room
temperature 3 times. The Immobilon Western Chemiluminescent HRP
Substrate (Merck Millipore, Billerica, MA, USA) was prepared by
mixing equal volumes of the HRP substrate luminol reagent as well
as the HRP substrate peroxide solution and added onto the membrane.
Finally, the light-signal was detected by BioSpectrum®
2D Imaging System (UVP, Upland, CA, USA).
Bisulfite conversion and combined
bisulfite restriction analysis (COBRA)
DNA was bisulfite modified using EZ DNA methylation
kit (Zymo Research, Irvine, CA, USA) according to the
manufacturer’s protocol. For COBRA analysis, 4 μl of bisulfite
converted DNA was first amplified using specific primers (Table I) targeting promoter region of
ARNTL followed by digested with 20 U of BstUI (CGCG)
(New England Biolabs, Ipswich, MA, USA). In vitro methylated
DNA (IVD) (Merck-Millipore) was used as a positive control for
methylation and water was used as negative control. For the
un-digested control, water instead of BstUI was added.
Digested products were separated on 1.5% agarose gel for
visualization. The PCR reactions were carried out using the
Veriti® 96-Well Thermal Cycler (Applied Biosystems).
Plasmid constructs and transfection
The CDS of ARNTL was amplified by PCR using
specific primers (Table I) from
cDNA of IOSE cells which expressed ARNTL. The PCR products
were ligated into pIRES2-EGFP vector (Promega, Madison, WI, USA)
for sequencing confirmation. ARNTLpIRES was then digested
with NheI (New England Biolabs) and BamHI (New
England Biolabs), and inserted into multiple cloning site of
pIRES2-EGFP expression vector, which was predigested with
NheI and BamHI. ARNTL expression vector or
empty vector were transfected into CP70 and MCP2 cells using
Lipofectamine 2000 transfection reagent (Invitrogen) according to
the manufacturer’s protocol. The PCR reactions were carried out
using the Veriti® 96-Well Thermal Cycler (Applied
Biosystems).
Stable cell lines
Following transient transfection, the transfected
cells were cultivated with fresh culture medium containing 400
μg/ml geneticin (G418; Sigma) and replaced every 3 days. Each
single colony that formed was selected for further culture.
Colony forming assay
CP70 and MCP2 cells were plated at 1×106
cells/plate in a 100-mm culture dish one day before transfect
ARNTL. Transfection was performed as previously described.
On the second day after the transfection, cells were plated into 3
plates with fresh culture medium containing 400 μg/ml G418 (Sigma)
and replaced every 3 days. Surviving colonies were stained with
0.4% crystal violet (Sigma) in 50% methanol and visible colonies
were counted. The colony numbers were counted using Image-Pro 3D
Suite software version 5.1.1.38 for windows (Media Cybernetics,
Inc., Bethesda, MD, USA).
Cell proliferation assay
Cell growth was assessed by MTS assay, as previously
described. In brief, for MTS assays, 1×103 cells were
seeded in 96-well plates for 4 days with or without various
concentrations of cisplatin (Sigma). Cell growth was determined
using the CellTiter 96 Aqueous One Solution Cell Proliferation
Assay kit (Promega), according to the manufacture’s protocol.
Relative cell numbers were assessed using a 96-well ELISA plate
reader (Multiskan® FC Microplate Photometer; Thermo
Fisher Scientific Inc., Waltham, MA) with an absorbance set at 492
nm.
Soft agar assay for colony formation
base layer of agar (2.5 ml) (0.5% agar in culture
medium) was allowed to solidify in a 6-well flat bottomed plate
before the addition of 2 ml of cell suspensions containing
1×104 cells in 0.3% agar in culture medium. The
cell-containing layer was then solidified at room temperature for
20 min. Colonies were allowed to grow for 14–21 days at 37°C with
5% CO2 before imaging. Plates were stained with 1 mg/ml
iodonitrotetrazolium chloride (Sigma) overnight at 37°C. The
colonies which contained at least 50 cells were counted. The colony
numbers were counted using Image-Pro 3D Suite software version
5.1.1.38 for windows (Media Cybernetics).
Analysis of apoptosis by propidium iodide
staining and flow cytometry
Cells (1×105) were seeded in 60-mm plates
for 2 days with or without 1 mg/ml of cisplatin (Sigma). The
supernatant was carefully collected, and then wash in PBS. Followed
by centrifugation at 200 × g for 5 min at room temperature. The
supernatant was aspirated, the pellet resuspended in ~5 ml cold 1X
PBS, and then centrifuged for 5 min at 200 × g. Cells were fixed by
adding 4.5 ml of 70% (v/v) cold ethanol to the cell suspension
keeping the tubes on ice. The cells were stored in ethanol solution
at −20°C at least for 2 days. To remove the ethanol solution, the
supernatant was centrifuged at 400 × g for 5 min, the cells were
washed in 5 ml of PBS and centrifuged at 400 × g for 5 min. The
supernatant was removed and the cells were resuspended in 500 μl of
propidium iodide (1 mg/ml), then incubated for at least 30 min at
room temperature in the dark. Cells were analyzed by flow
cytometry.
Serum shock for detecting circadian
rhythmic activity
Cells were grown to confluence in 100-mm culture
dishes in RPMI-1640 media (Invitrogen) supplemented with 10% FBS,
followed by culture for 2 days with starvation medium (0.5% serum).
At time t=0, the medium was exchanged for 50% serum in RPMI-1640;
after 2 h, the medium was replaced with serum-free RPMI-1640. At
the indicated times (0, 4, 8, 12, 16 and 20 h) the dishes were
prepared for immediate RNA extraction.
Quantitative ChIP-PCR
cells (1×106) were cross-linked with 1%
fresh formaldehyde and then washed with cold 1X PBS in the presence
of the protease inhibitor. Cell were homogenized and the chromatin
was subjected to ChiP pull down using magnetic Dynabeads
(Invitrogen) and antibodies (against H3K4me3; Active Motif,
Carlsbad, CA, USA; against H3K27me3; Merck-Millipore; EZH2; Cell
Signaling Technology, Danvers, MA, USA; and IgG; Cell Signaling
Technology). The quantity of ChIP DNA was measured by
quantitative-PCR analysis using 200 pg of pull-down DNA amplified
by ARNTL promoter specific primer (Table I), and then quantified as a
percentage of the input signal. The PCR reactions were carried out
using the Veriti® 96-Well Thermal Cycler (Applied
Biosystems).
Statistical analysis
Comparison between groups were assessed by the
unpaired t-test. All statistical calculations were done using the
statistical package SPSS version 13.0 for windows (SPSS, Inc.,
Chicago, IL, USA). A P-value <0.05 was considered statistically
significant.
Results
ARNTL is frequently downregulated in
ovarian cancer cell lines and epigenetic treatments restore ARNTL
expression
To investigate genes that are hypermethylated in
ovarian cancer cell lines, we performed meDIP-Chip using Agilent
244K CpG island microarray in IOSE and a panel of ovarian cancer
cell lines. One of the targets that showed promoter
hypermethylation in a sub-set of ovarian cancer cell is
ARNTL (also known as BMAL1). To confirm our result,
we performed quantitative real-time RT-PCR to examine the mRNA
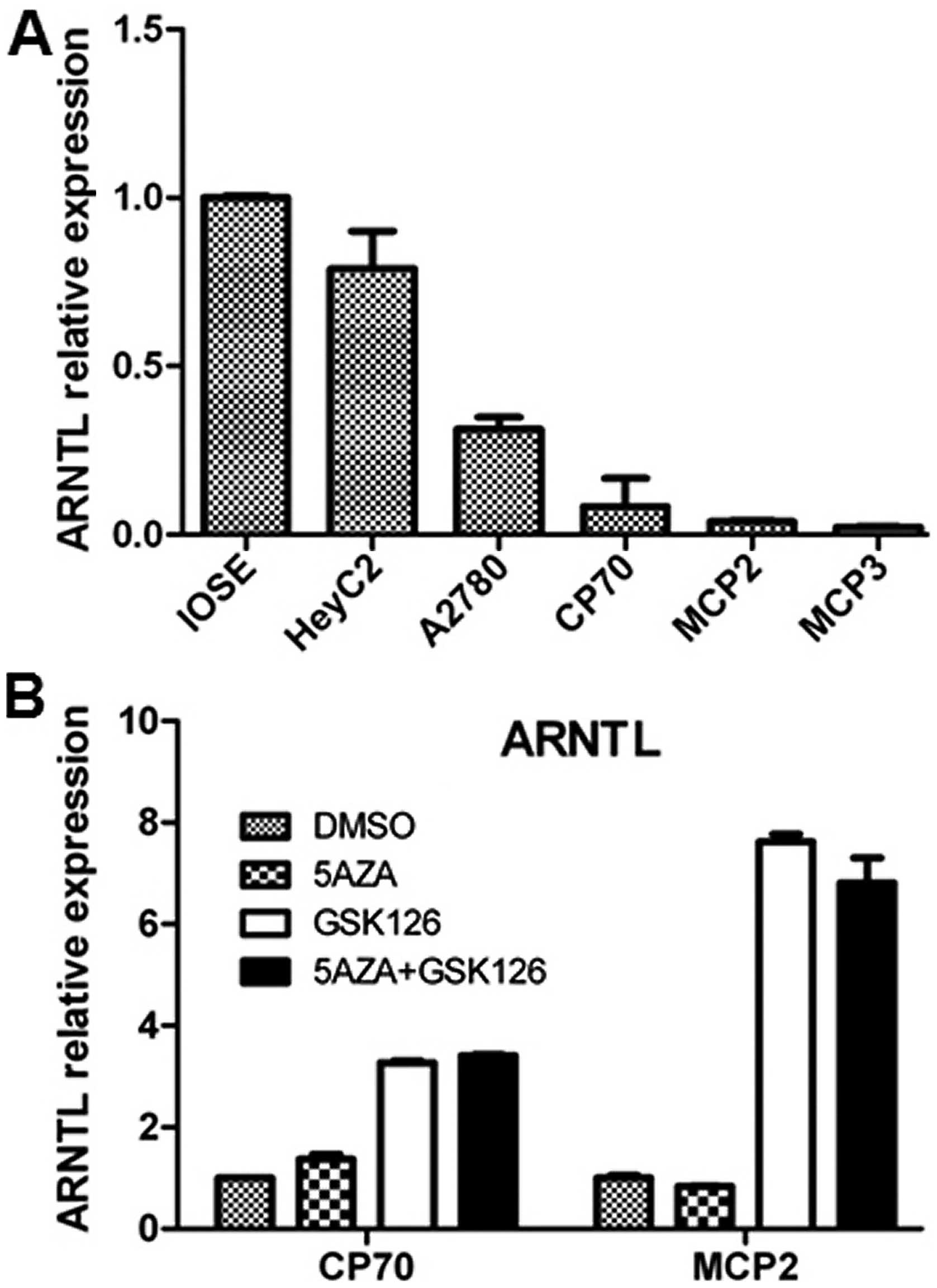
level of ARNTL in these cancer cell lines. Expression of
ARNTL was downregulated in A2780, CP70, MCP2 and MCP3
ovarian cancer cell lines as compared with IOSE (Fig. 1A). To examine if epigenetic
modifications contribute to this downregulation, we treated the
cells with DNA methylation inhibitor, 5-aza and/or the EZH2
inhibitor, GSK126 (31) in CP70,
MCP2 ovarian cancer cell lines. The resulted indicated that
treatment of 5-aza alone resulted in a partial re-expression of
ARNTL in CP70 cells, while treatment with GSK126 resulted in
a robust re-expression of ARNTL in these cells, thus,
suggesting that ARNTL silencing may occur through epigenetic
events (Fig. 1B).
The transcription of ARNTL is associated
with promoter methylation
To further confirm if epigenetic modifications
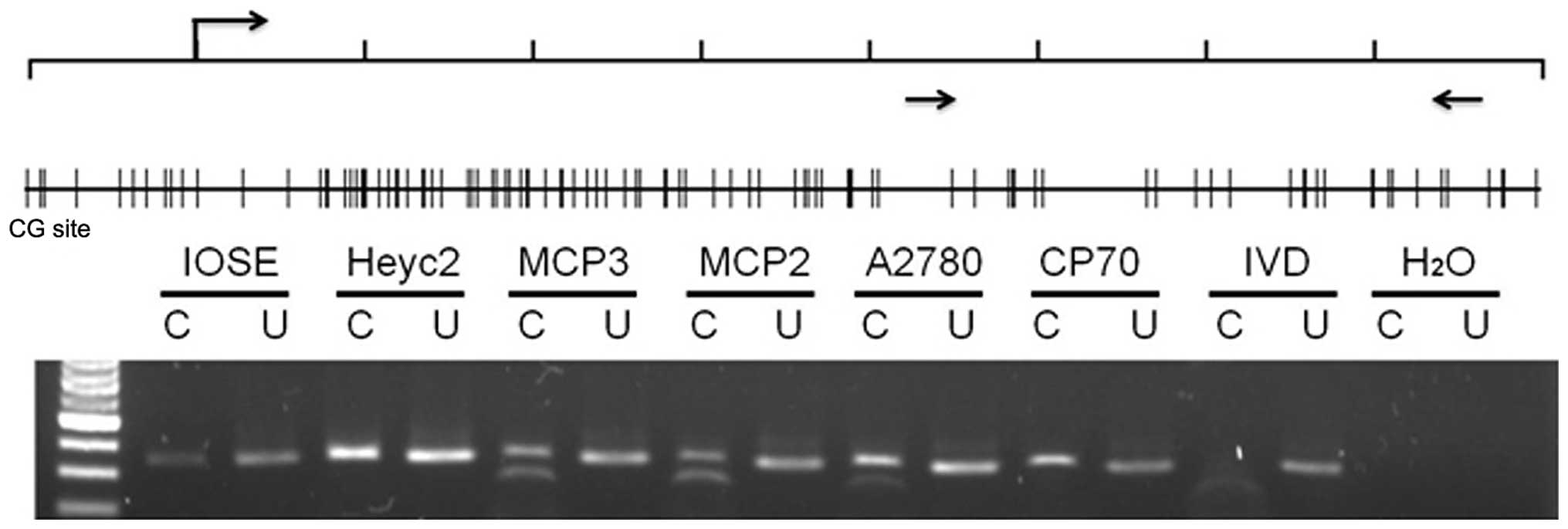
contribute to the silencing of ARNTL, we examined DNA
methylation in the promoter region of ARNTL in the ovarian
cancer cell lines. Combined bisulphite restriction analysis (COBRA)
was conducted to determine the methylation status of 342 bp CpG
island spanning (+421 bp to +763 bp) region around the
transcription start site in the promoter region of ARNTL.
Promoter methylation was obvious in MCP2, MCP3 and A2780 cells
(Fig. 2). However, promoter
methylation was not observed in IOSE and HeyC2 cells which
expressed ARNTL. Notably, methylation was not observed in
CP70 cells which did not express ARNTL.
Histone modification of ARNTL promoter in
ovarian cancer cell lines
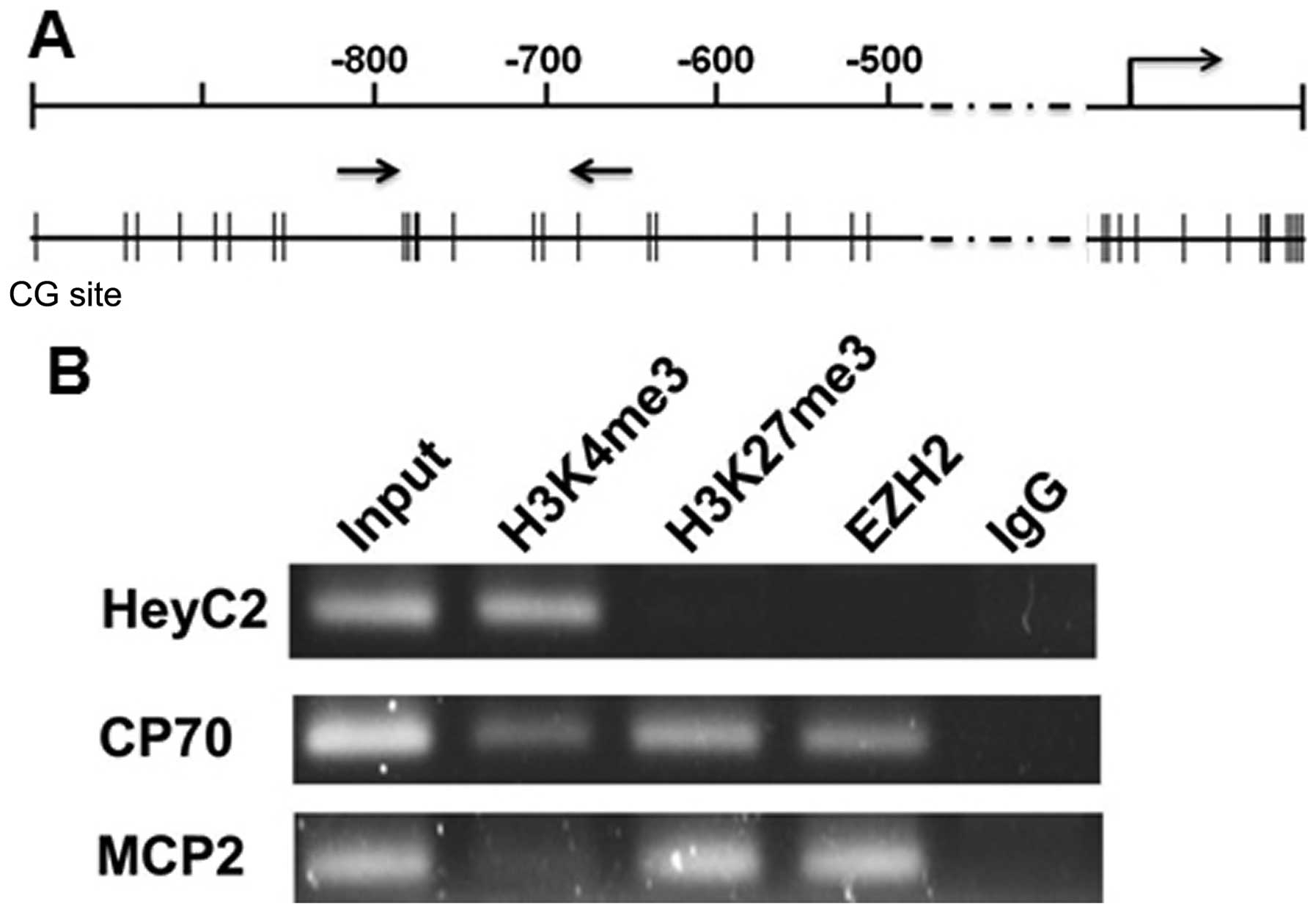
Beside DNA methylation, histone modifications also
control gene expression through their effects on chromatin
structure. We then analyzed the histone modifications for the
active and repressive chromatin marks in the ARNTL promoter.
Chromatin immunoprecipitation coupled with PCR (ChIP-PCR) was
performed using antibodies against trimethylated lysine 4 of
histone H3 (H3K4me3), trimethylated lysine 27 of histone H3
(H3K27me3) and EZH2. As expected, active histone mark
trimethyl-H3K4 but not repressive mark trimethyl-H3K27 was enriched
in ARNTL-expressing HeyC2 cells. On the contrary, trimethyl-H3K27
and EZH2 but not trimethyl-H3K4 were enriched at the promoter
region of CP70 and MCP2 cells (Fig.
3B). Hence, histone modifications were also involved in the
gene silencing mechanism of ARNTL in ovarian cancer cell
lines.
ARNTL inhibits cell proliferation and
colony formation
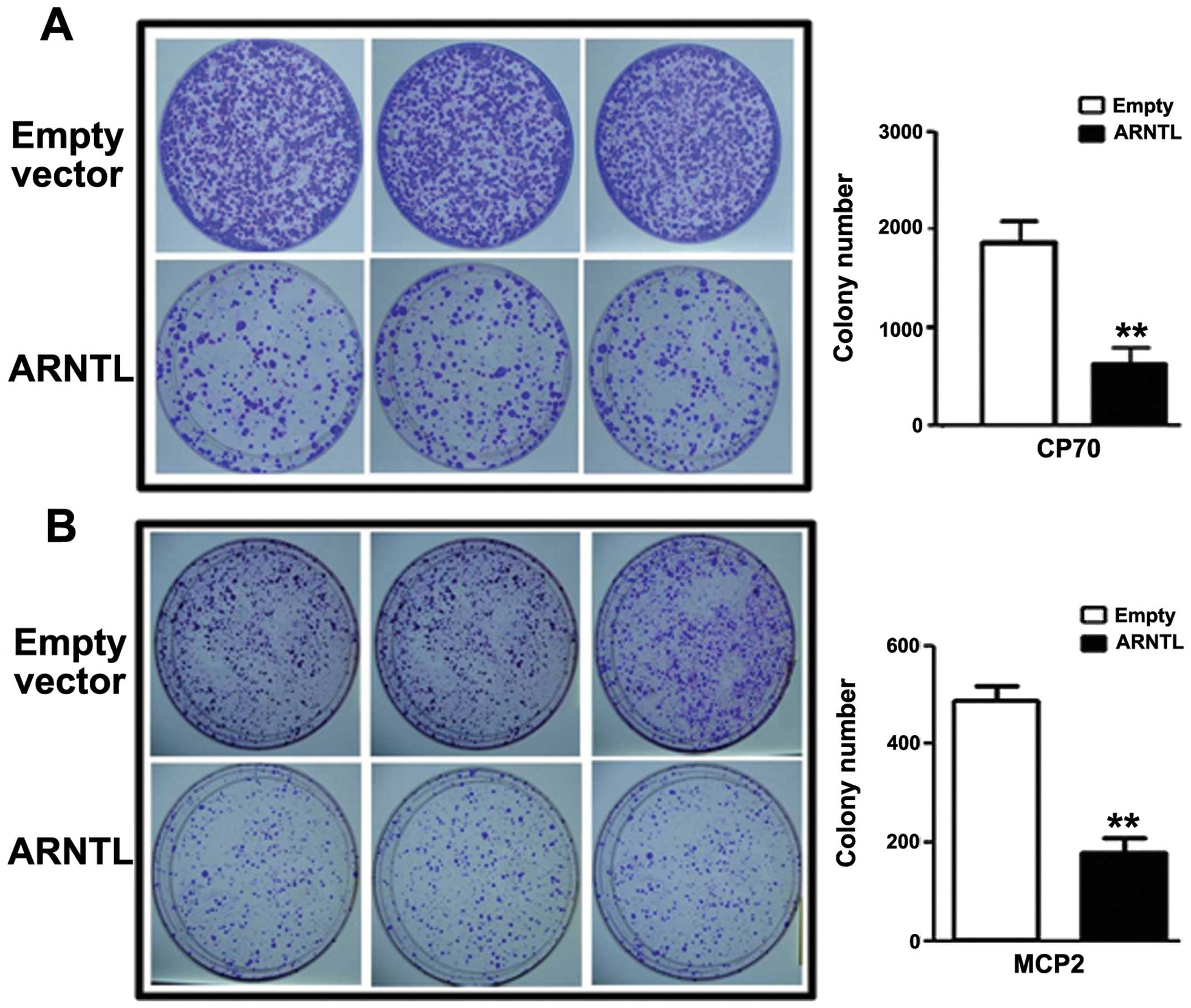
Having demonstrated that ARNTL is
epigenetically silenced by DNA methylation and histone
modifications, we then investigated the role of ARNTL in
ovarian cancer cell lines. By colony formation assay, CP70 or MCP2
cells transfected with ARNTL showed a significant decrease
in colony numbers as compared to control cells (Fig. 4).
ARNTL inhibits anchorage-dependent growth
and restores chemosensitivity
To investigate the effect of ARNTL to
anchorage-dependent growth and drug resistance, we selected cells
that stably overexpressed ARNTL in MCP2 ovarian cancer
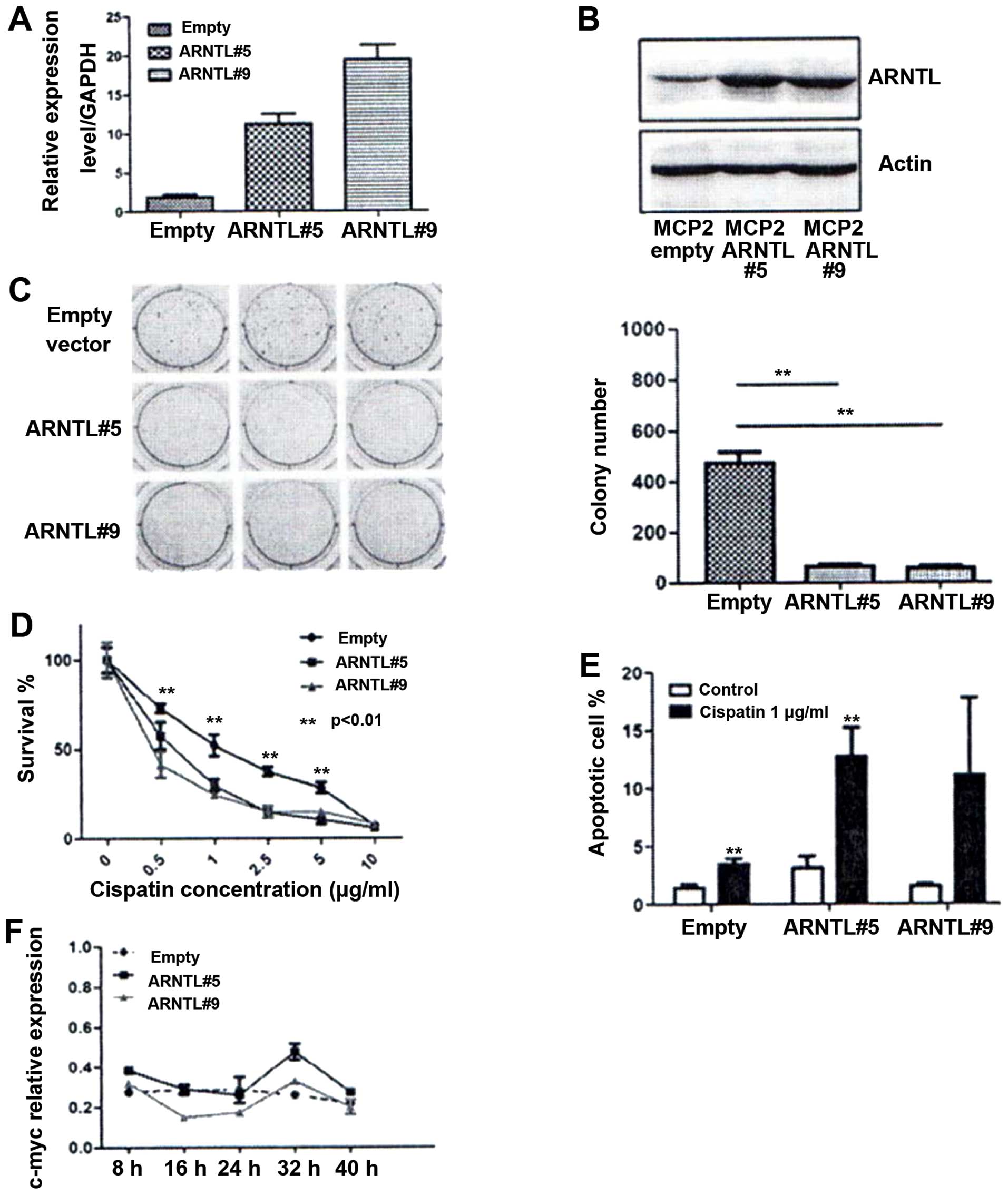
cells. The expression of ARNTL of the stable clones was
confirmed by quantitative RT-PCR and western blot analysis
(Fig. 5A and B). By soft-agar
assay, ARNTL-ovexpressed cells demonstrated a dramatic
decrease in colony numbers as compared to control cells (Fig. 5C). Finally, we examined if
ARNTL can affect chemosensitivity in these drug-resistant
MCP2 cells. Overexpression of ARNTL restored the
chemosensitivity to cisplatin in these cells (Fig. 5D). Taken together, these results
demonstrated that ARNTL may function as a potential tumor
suppressor in ovarian cancer.
Enhancement of apoptosis by cisplatin in
ARNTL overexpressing cells
To determine the mechanism of ARNTL in
restoring chemosensitivity in MCP2 cells, we investigated the
effect of ARNTL on apoptosis. Although ARNTL did not
increase the number of sub-G1 population under normal condition,
overexpression of ARNTL showed a marked increase in the
number of cells in the sub-G1 population under cisplatin treatment
(P<0.01; Fig. 5E). These
results indicated that ARNTL may restore the DNA
damage-induced apoptosis in MCP2 ovarian cancer cells.
Rhythmic activity of c-MYC in
ARNTL-overexpressing cells
Finally, we investigated the functional impact of
restoring ARNTL expression in ovarian cancer cells. This is
particularly interesting because the expression of several
mammalian cell cycle genes, such as c-myc (16,32,33),
is regulated in a circadian manner. Therefore, we synchronized the
ARNTL stable transfectants by serum starvation. RNA was then
extracted after serum restoration at the indicated time for gene
expression analysis. Notably, expression of c-myc displayed
a circadian rhythm in both of the ARNTL-expressing MCP2
clones (Fig. 5F), whereas a
relatively constant level of expression without any rhythmicity was
observed in the control cells.
Discussion
The relationship between circadian rhythm and cancer
has been previously discussed (13,33).
For example, mice deficient in the Per2 gene showed reduced
apoptosis in response to DNA damage and increased tumor formation
(32). Depletion of another core
circadian gene, NPAS2 failed to exhibit cell cycle arrest in
response to a mutagen in MCF7 breast cancer cells (34). Furthermore, these circadian
regulators were found to be epigenetically silenced in several
human cancer (35–37). These results suggested that genes
involved in circadian clock may be involved also in tumor
suppression and can be epigenetically silenced in human cancer.
In the present study, we found that another core
component of the circadian rhythm, ARNTL, is epigenetically
silenced by promoter methylation and histone modifications in
ovarian cancer cell lines. Overexpression of ARNTL
suppressed tumor growth, restored chemosensitivity and resumed the
rhythmic activity of c-myc in ovarian cancer cells. Our
results were consistent with a previous report that ARNTL
exhibited promoter hypermethylation and might act as a tumor
suppressor gene in hematologic malignancies (36). Although the role of ARNTL in cancer
is not fully understood, studies suggested that ARNTL may be a
regulator of the p53 tumor suppressor pathway (38). More recently, it was found that
ARNTL can suppress cancer invasion by inhibiting the AKT signaling
pathway (39). The molecular
mechanism of ARNTL in ovarian cancer warrants further
investigation.
Epigenetics involving complex silencing mechanisms
require cross-talk and coordination of multiple regulatory events.
Promoter DNA methylation plays a crucial role in gene inactivation
and its role in tumorigenesis has been established for decades
(21). Over the past few years,
increased attention has been given to histone methylation, in
particular the repressive H3K27me3-mediated gene silencing in
cancer (40,41). Previous studies showed that EZH2
may directly control DNA methylation for gene silencing (42). DNA methyltransferases can be
recruited to target loci for de novo methylation through
polycombmediated H3K27me3 within DNA methylation. the present study
showed that treatment of 5-aza resulted in a moderate re-expression
of ARNTL in CP70 cell, while treatment with GSK126 resulted
in a robust re-expression of ARNTL in CP70 and MCP2 cells thus
suggesting that ARNTL silencing may occur through a
repressive chromatin complex involving a polycomb repressor and DNA
methylation. It is suggested that H3K27me3-mediated gene silencing
may constitute a key epigenetic event repressing developmental
genes in early stage of tumorigenesis. The therapeutic effect of
using inhibitors against DNMT and EZH2 in the treatment of ovarian
cancer deserves further investigation.
In conclusion, the circadian gene ARNTL may
be a potential tumor suppressor in ovarian cancer. Epigenetic
silencing of ARNTL may be required for proliferation,
anchorage-independent ability and drug resistance in ovarian
cancer.
Acknowledgements
The present study was supported by the research
grants from the National Science Council, Taiwan
(NSC102-2320-B-194-006) and the NCCU Interdisciplinary Science
grant (103-03), National Chung Cheng University, Taiwan.
References
|
1
|
Sellix MT and Menaker M: Circadian clocks
in the ovary. Trends Endocrinol Metab. 21:628–636. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Harrington ME: Exercise strengthens
circadian clocks. J Physiol. 590:59292012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Green CB and Menaker M: Circadian rhythms.
Clocks on the brain. Science. 301:319–320. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Falcon J: Cellular circadian clocks in the
pineal. Prog Neurobiol. 58:121–162. 1999. View Article : Google Scholar
|
|
5
|
Stevens RG and Rea MS: Light in the built
environment: potential role of circadian disruption in endocrine
disruption and breast cancer. Cancer Causes Control. 12:279–287.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hansen J: Risk of breast cancer after
night- and shift work: current evidence and ongoing studies in
Denmark. Cancer Causes Control. 17:531–537. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schernhammer ES, Laden F, Speizer FE, et
al: Night-shift work and risk of colorectal cancer in the nurses’
health study. J Natl Cancer Inst. 95:825–828. 2003.
|
|
8
|
Canaple L, Kakizawa T and Laudet V: The
days and nights of cancer cells. Cancer Res. 63:7545–7552.
2003.PubMed/NCBI
|
|
9
|
Levin RD, Daehler MA, Grutsch JF, et al:
Circadian function in patients with advanced non-small-cell lung
cancer. Br J Cancer. 93:1202–1208. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gery S and Koeffler HP: Circadian rhythms
and cancer. Cell Cycle. 9:1097–1103. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Leonardi GC, Rapisarda V, Marconi A, et
al: Correlation of the risk of breast cancer and disruption of the
circadian rhythm (Review). Oncol Rep. 28:418–428. 2012.PubMed/NCBI
|
|
12
|
Yu EA and Weaver DR: Disrupting the
circadian clock: gene-specific effects on aging, cancer, and other
phenotypes. Aging (Albany, NY). 3:479–493. 2011.PubMed/NCBI
|
|
13
|
Rana S and Mahmood S: Circadian rhythm and
its role in malignancy. J Circadian Rhythms. 8:32010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Paschos GK and FitzGerald GA: Circadian
clocks and vascular function. Circ Res. 106:833–841. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Takahashi JS, Hong HK, Ko CH and McDearmon
EL: The genetics of mammalian circadian order and disorder:
implications for physiology and disease. Nat Rev Genet. 9:764–775.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hunt T and Sassone-Corsi P: Riding tandem:
circadian clocks and the cell cycle. Cell. 129:461–464. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ripperger JA and Schibler U: Rhythmic
CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp
transcription and chromatin transitions. Nat Genet. 38:369–374.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hsu CM, Lin SF, Lu CT, Lin PM and Yang MY:
Altered expression of circadian clock genes in head and neck
squamous cell carcinoma. Tumour Biol. 33:149–155. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tokunaga H, Takebayashi Y, Utsunomiya H,
et al: Clinicopathological significance of circadian rhythm-related
gene expression levels in patients with epithelial ovarian cancer.
Acta Obstet Gynecol Scand. 87:1060–1070. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang MY, Chang JG, Lin PM, et al:
Downregulation of circadian clock genes in chronic myeloid
leukemia: alternative methylation pattern of hPER3. Cancer Sci.
97:1298–1307. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Baylin SB and Jones PA: A decade of
exploring the cancer epigenome - biological and translational
implications. Nat Rev Cancer. 11:726–734. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Seisenberger S, Peat JR and Reik W:
Conceptual links between DNA methylation reprogramming in the early
embryo and primordial germ cells. Curr Opin Cell Biol. 25:281–288.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Caslini C, Capo-chichi CD, Roland IH,
Nicolas E, Yeung AT and Xu XX: Histone modifications silence the
GATA transcription factor genes in ovarian cancer. Oncogene.
25:5446–5461. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu Y, Fujii S, Yuan J, et al: Epigenetic
regulation of ARHI in breast and ovarian cancer cells. Ann NY Acad
Sci. 983:268–277. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chan MW, Wei SH, Wen P, et al:
Hypermethylation of 18S and 28S ribosomal DNAs predicts
progression-free survival in patients with ovarian cancer. Clin
Cancer Res. 11:7376–7383. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chan MW, Huang YW, Hartman-Frey C, et al:
Aberrant transforming growth factor beta1 signaling and SMAD4
nuclear translocation confer epigenetic repression of ADAM19 in
ovarian cancer. Neoplasia. 10:908–919. 2008.PubMed/NCBI
|
|
27
|
Chou JL, Su HY, Chen LY, et al: Promoter
hypermethylation of FBXO32, a novel TGF-beta/SMAD4 target gene and
tumor suppressor, is associated with poor prognosis in human
ovarian cancer. Lab Invest. 90:414–425. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yeh KT, Chen TH, Yang HW, et al: Aberrant
TGFbeta/SMAD4 signaling contributes to epigenetic silencing of a
putative tumor suppressor, RunX1T1 in ovarian cancer. Epigenetics.
6:727–739. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Su HY, Lai HC, Lin YW, Chou YC, Liu CY and
Yu MH: An epigenetic marker panel for screening and prognostic
prediction of ovarian cancer. Int J Cancer. 124:387–393. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gillan L, Matei D, Fishman DA, Gerbin CS,
Karlan BY and Chang DD: Periostin secreted by epithelial ovarian
carcinoma is a ligand for αVβ3 and αVβ5 integrins and promotes cell
motility. Cancer Res. 62:5358–5364. 2002.PubMed/NCBI
|
|
31
|
McCabe MT, Ott HM, Ganji G, et al: EZH2
inhibition as a therapeutic strategy for lymphoma with
EZH2-activating mutations. Nature. 492:108–112. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fu LN, Pelicano H, Liu JS, Huang P and Lee
CC: The circadian gene period2 plays an important role in tumor
suppression and DNA-damage response in vivo. Cell. 111:1055. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fu L and Lee CC: The circadian clock:
pacemaker and tumour suppressor. Nat Rev Cancer. 3:350–361. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hoffman AE, Zheng T, Ba Y and Zhu Y: The
circadian gene NPAS2, a putative tumor suppressor, is
involved in DNA damage response. Mol Cancer Res. 6:1461–1468.
2008.
|
|
35
|
Gery S, Komatsu N, Kawamata N, et al:
Epigenetic silencing of the candidate tumor suppressor gene
Per1 in non-small cell lung cancer. Clin Cancer Res.
13:1399–1404. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Taniguchi H, Fernandez AF, Setien F, et
al: Epigenetic inactivation of the circadian clock gene BMAL1 in
hematologic malignancies. Cancer Res. 69:8447–8454. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shih MC, Yeh KT, Tang KP, Chen JC and
Chang JG: Promoter methylation in circadian genes of endometrial
cancers detected by methylation-specific PCR. Mol Carcinog.
45:732–740. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mullenders J, Fabius AW, Madiredjo M,
Bernards R and Beijersbergen RL: A large scale shRNA barcode screen
identifies the circadian clock component ARNTL as putative
regulator of the p53 tumor suppressor pathway. PloS one.
4:e47982009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jung CH, Kim EM, Park JK, et al: Bmal1
suppresses cancer cell invasion by blocking the phosphoinositide
3-kinase-Akt-MMP-2 signaling pathway. Oncol Rep. 29:2109–2113.
2013.PubMed/NCBI
|
|
40
|
Varambally S, Dhanasekaran SM, Zhou M, et
al: The polycomb group protein EZH2 is involved in progression of
prostate cancer. Nature. 419:624–629. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kleer CG, Cao Q, Varambally S, et al: EZH2
is a marker of aggressive breast cancer and promotes neoplastic
transformation of breast epithelial cells. Proc Natl Acad Sci USA.
100:11606–11611. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Vire E, Brenner C, Deplus R, et al: The
Polycomb group protein EZH2 directly controls DNA methylation.
Nature. 439:871–874. 2006. View Article : Google Scholar : PubMed/NCBI
|